

Abstract
Clustered Kv1 K+ channels regulate neuronal excitability at juxtaparanodes of myelinated axons, axon initial segments, and cerebellar basket cell terminals (BCTs). These channels are part of a larger protein complex that includes cell adhesion molecules and scaffolding proteins. To identify proteins that regulate assembly, clustering, and/or maintenance of axonal Kv1 channel protein complexes, we immunoprecipitated Kv1.2 α subunits, and then used mass spectrometry to identify interacting proteins. We found that a disintegrin and metalloproteinase 22 (ADAM22) is a component of the Kv1 channel complex and that ADAM22 coimmunoprecipitates Kv1.2 and the membrane-associated guanylate kinases (MAGUKs) PSD-93 and PSD-95. When coexpressed with MAGUKs in heterologous cells, ADAM22 and Kv1 channels are recruited into membrane surface clusters. However, coexpression of Kv1.2 with ADAM22 and MAGUKs does not alter channel properties. Among all the known Kv1 channel-interacting proteins, only ADAM22 is found at every site where Kv1 channels are clustered. Analysis of Caspr-null mice showed that, like other previously described juxtaparanodal proteins, disruption of the paranodal junction resulted in redistribution of ADAM22 into paranodal zones. Analysis of Caspr2-, PSD-93-, PSD-95-, and double PSD-93/PSD-95-null mice showed ADAM22 clustering at BCTs requires PSD-95, but ADAM22 clustering at juxtaparanodes requires neither PSD-93 nor PSD-95. In direct contrast, analysis of ADAM22-null mice demonstrated juxtaparanodal clustering of PSD-93 and PSD-95 requires ADAM22, whereas Kv1.2 and Caspr2 clustering is normal in ADAM22-null mice. Thus, ADAM22 is an axonal component of the Kv1 K+ channel complex that recruits MAGUKs to juxtaparanodes.
Introduction
The excitable properties of neurons depend not only on the kinds of ion channels expressed in the plasma membrane but also on the location of these channels. Among the many different ion channels expressed in the nervous system, the Kv1 channels are an excellent example of channels that are restricted to distinct subcellular locations. Mutations or diseases that disrupt clustering, localization, or composition of Kv1 channels severely compromise nervous system function and lead to conduction block, episodic ataxia, and/or epilepsies (Rasband et al., 1998; Eunson et al., 2000; Nashmi et al., 2000; Manganas et al., 2001b).
In the intact nervous system, Kv1 channels are clustered in high density at (1) the basket cell terminals (BCTs) of cerebellar pinceau, where they regulate GABAergic inhibition of the Purkinje neuron efferent axon; (2) juxtaparanodes of myelinated axons, where they modulate action potential propagation and dampen repetitive firing of injured and developing myelinated axons; and (3) axon initial segments (AISs), where they regulate action potential waveform, synaptic efficacy, and threshold of cortical interneurons (Wang et al., 1993; Laube et al., 1996; Vabnick and Shrager, 1998; Zhang et al., 1999; Devaux et al., 2002; Kole et al., 2007; Goldberg et al., 2008; Ogawa et al., 2008).
Kv1 channels form macromolecular protein complexes with the cell adhesion molecules (CAMs) Caspr2 and TAG-1, the membrane-associated guanylate kinases (MAGUKs) PSD-93 and PSD-95, and the cytoskeletal scaffold 4.1B (Baba et al., 1999; Poliak et al., 1999, 2001; Traka et al., 2002; Horresh et al., 2008). Juxtaparanodal Kv1 channel clustering depends on CAMs, but not MAGUKs (Rasband et al., 2002; Poliak et al., 2003; Traka et al., 2003; Horresh et al., 2008). In contrast, AIS Kv1 channel clustering was reported to depend on MAGUKs rather than CAMs (Ogawa et al., 2008). At BCTs, neither Caspr2 nor PSD-95 is needed for channel clustering (Rasband et al., 2002). Thus, although these different axonal domains share a similar molecular organization, their mechanisms of assembly are unique.
Here, we identify a disintegrin and metalloproteinase 22 (ADAM22) as a component of the Kv1 channel protein complex. ADAM22 was previously reported to regulate synaptic transmission and to be a binding partner of leucine-rich glioma inactivated 1 (Lgi1), which is mutated in autosomal-dominant partial epilepsy with auditory features (Fukata et al., 2006). ADAM22-null mice have profound hypomyelination in the PNS (Sagane et al., 2005). We show here that ADAM22 is highly enriched in axons at juxtaparanodes, AISs, and BCTs. We use a variety of biochemical, cell biological, and genetic approaches to begin to elucidate the role of ADAM22 at these axonal locations.
Materials and Methods
Animals.
Sprague Dawley rats were purchased from Harlan. ADAM22-, Caspr-, Caspr2-, PSD-93-, PSD-95-, and PSD-95/PSD-93 double-null mice were described previously (Migaud et al., 1998; McGee et al., 2001; Sagane et al., 2005; Horresh et al., 2008). Animals were housed at the Center for Laboratory Animal Care at the Baylor College of Medicine, at the Erasmus MC University Medical Center, and at the Weizmann Institute of Science. All experiments were performed in accordance with the National Institutes of Health (NIH) guidelines for the humane treatment of animals.
Constructs.
The following plasmids were used: pGW-PSD-93EGFP (a gift from Dr. Bonnie Firestein, Rutgers, NJ), pGW-PSD95 (a gift from Dr. Morgan Sheng), and RBG4-Kv1.4 (Nakahira et al., 1996), and pSCT-Adam22 G20. The pSCT-Adam22 G20 construct drives expression from a CMV (cytomegalovirus) promoter of a mouse Adam22 full-length cDNA. The C-terminal intracellular domain corresponds to the previously described G20 splice variant (Sagane et al., 2005).
Antibodies.
Antibodies against Kv channel subunits antibody have been described previously (Rhodes et al., 1995). Mouse monoclonal anti-PSD-93 (N18/30) and anti-ADAM22 (cytoplasmic, N46/30; extracellular, N57/2) were obtained from the University of California Davis/NIH NeuroMab Facility and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis. The mouse monoclonal anti-PSD-95 (K28/43.2) has been described previously (Rasband et al., 2002). Anti-GAD-65 (GAD-6) was obtained from Developmental Studies Hybridoma Bank. Rabbit and Chicken anti-βIV spectrin antibodies were raised against synthetic peptides corresponding to amino acids 2237–2256 found in the SD (“specific” domain) of βIV spectrin. Rabbit Caspr antibody and mouse monoclonal pan-Neurofascin antibody (L11A/41.6) have been previously described (Schafer et al., 2004). Rabbit polyclonal anti-Caspr2 antibody was purchased from US Biological. Rabbit polyclonal anti-Lgi1 and anti-Lgi4 were purchased from Abcam. Chicken polyclonal anti-MAP2 antibody was purchased from EnCor Biotechnology. Rat monoclonal green fluorescent protein (GFP) antibody (GF090R) was purchased from Nacalai Tesque. NeuroTracer was purchased from Invitrogen. Secondary antibodies included Alexa 488- or 594-conjugated goat anti-mouse, Alexa 488 or 594-conjugated goat anti-rabbit, Alexa 488-conjugated goat anti-rat (Invitrogen), and AMCA (7-amino-4-methylcoumarin-3-acetic acid)-conjugated goat anti-chicken (Jackson ImmunoResearch).
Immunofluorescence.
Cultured hippocampal neurons were fixed with 1% paraformaldehyde (PFA) in PBS for 15 min at 4°C. Brains, spinal cords, or sciatic nerve were fixed by immersion with 4% PFA for 30 min at 4°C, cryoprotected with 20% sucrose, embedded in Tissue-Tek OCT mounting medium, and frozen on dry ice powder. Blocks were cut using a cryostat (Leica) to obtain 20 μm thickness for brain and spinal cord and 8 μm thickness for sciatic nerve sections, and the sections were placed on precoated slides (Thermo Fisher Scientific). Alternatively, some sections were cut using sliding blade microtome (Thermo Fisher Scientific). Cultured neurons or tissue sections were blocked with 10% normal goat serum in PBS with 0.3% Triton X-100 for 2 h at room temperature. Primary antibodies diluted in the blocking buffer were added at appropriate concentrations, incubated at room temperature overnight, and washed with PBS. Secondary antibodies were incubated at room temperature for 2 h and washed with PBS. In some cases, antigen retrieval was performed by incubating tissue sections with 0.2 mg/ml pepsin (Dako) in 0.2 m HCl for 10 min at 37°C. Sections were then rinsed and processed as described above. Fluorescence images were collected on an AxioImager (Carl Zeiss) fitted with an apotome for optical sectioning, and a digital camera (AxioCam; Carl Zeiss). AxioVision (Carl Zeiss) acquisition software was used for collection of images. In some images, brightness levels were subsequently adjusted using Photoshop (Adobe). No other processing of the images was performed.
Surface clustering assay.
Recombinant plasmids were cotransfected into COS7 cells using Lipofectamine LTX according to the manufacturer's instructions. The following plasmids were used: pGW-PSD-93EGFP, pGW-PSD95, RBG4-Kv1.4, and pSCT-Adam22 G20. After 18–24 h, transfected cells were fixed in 4% paraformaldehyde for 30 min at 4°C in PBS. After three washes with PBS, ADAM22 ectodomain-directed antibodies were added for 1 h at room temperature. After three washes with PBS, cytoplasmically directed antibodies were added for 1 h after permeabilization by 0.3% Triton X-100 with normal goat serum and washed with PBS. Secondary antibodies were incubated at room temperature for 2 h and washed with PBS. Fluorescence images were collected as described above.
Immunoprecipitation and mass spectrometry.
Whole rat brains were dissected and homogenized in ice-cold homogenization buffer (in mm: 320 sucrose, 1 EGTA, and 5 HEPES, pH 7.4). The homogenate was centrifuged at 1000 × g for 10 min. The supernatant was spun for 15 min at 13,000 × g, and the resulting pellet was resuspended in homogenization buffer. Detergent-resistant or solubilizing membranes were isolated by solubilizing brain membrane homogenates in 1% Triton X-100 lysis buffer (20 mm Tris-HCl, pH 8.0, 10 mm EDTA, 0.15 m NaCl, 10 mm iodoacetamide, 0.5 mm PMSF, 10 mm sodium azide, and a mixture of protease inhibitors: 2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, and 10 μg/ml benzamidine) at a concentration of 1 mg/ml protein for 1 h on a rotator at 4°C. The resulting lysate was centrifuged at 13,000 × g for 30 min to separate the soluble and insoluble fractions. For immunoprecipitations, we used the soluble fraction as the starting material. Rabbit polyclonal or mouse monoclonal antibodies were added overnight at 4°C and precipitated for 3 h at 4°C with protein A or protein G Tris-acryl (Pierce). After washing seven times with 1% Triton X-100 lysis buffer, Immunoprecipitated complexes were added to 2× concentrated reducing sample buffer, boiled, and then loaded and size fractionated on SDS-polyacrylamide gels. Polyacrylamide gels were silver stained using the SilverSnap silver staining kit (Pierce). For identification of proteins by mass spectrometry (MS), gels were stained with Colloidal Blue Staining kit (Invitrogen). For immunoblotting, proteins were electrophoretically transferred to nitrocellulose membranes, followed by immunoblotting using mouse and/or rabbit antibodies.
In-gel digestion.
Protein bands were excised from gels and digested in-gel with trypsin as described previously (Rosenfeld et al., 1992). The extracted digests were vacuum-evaporated and resuspended in 10 μl of 0.1% formic acid in water.
Reverse-phase liquid chromatography-MS/MS analysis.
The digests were separated by nanoflow liquid chromatography using a 100 μm × 150 mm reverse-phase Ultra 120 μm C18Q column (Peeke Scientific) at a flow rate of 350 nl/min in an Agilent 1100 HPLC system (Agilent Technologies). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. After equilibration of the column in 2% solvent B, approximately one-half of each digest (5 μl) was injected, and then the organic content of the mobile phase was increased linearly to 40% over 30 min and then to 50% in 3 min. The liquid chromatography eluate was coupled to a hybrid linear ion trap–Fourier transform mass spectrometer (LTQ-FT; Thermo Fisher Scientific) equipped with a nanoelectrospray ion source. Spraying was from an uncoated 15-μm-inner diameter spraying needle (New Objective). Peptides were analyzed in positive ion mode and in information-dependent acquisition mode to automatically switch between MS and MS/MS acquisition. MS spectra were acquired in profile mode using the ion cyclotron resonance analyzer in the m/z range between 300 and 2000. For each MS spectrum, the five most intense multiple charged ions over a threshold of 200 counts were selected to perform collisionally induced dissociation (CID) experiments. Product ions were analyzed on the linear ion trap in profile mode. The CID collision energy was automatically set to 25%. A dynamic exclusion window of 0.5 Da was applied that prevented the same m/z from being selected for 90 s after its acquisition. Peak lists were generated using Mascot Distiller, version 2.1.0.0 (Matrix Science). Parameters for MS processing were set as follow: peak half-width, 0.02; data points per dalton, 100. Parameters for MS/MS data were set as follows: peak half-width, 0.02; data points per dalton, 100. The peak list was searched against the rat subset of the NCBInr database as of January 2, 2008 (containing 64,988 entries) using in-house ProteinProspector, version 5.2.2 (a public version is available online). A minimal ProteinProspector protein score of 15, a peptide score of 15, minimal discriminant score threshold of 0.0, and a maximum expectation value of 0.1 were used for initial identification criteria. Carbamidomethylation and acrylamide modification of cysteine; acetylation of the N terminus of the protein; oxidation of methionine; and formation of pyro-Glu form N-term Gln were allowed as variable modifications. Peptide tolerance in searches was 40 ppm for precursor and 0.8 Da for product ions, respectively. Peptides containing two miscleavages were allowed. The number of modifications was limited to one per peptide. For identifications based on one or two peptide sequences with low expectation values, the MS/MS spectrum was reinterpreted manually by matching the observed fragment ions to a theoretical fragmentation obtained using MS Product (Protein Prospector) (Clauser et al., 1999).
Electrophysiology.
COS-1 cells were transiently transfected with 0.1 μg of RBG4-Kv1.2 plasmids using Lipofectamine 2000 (Invitrogen). A total of 1 μg of ADAM22 and PSD-95 (Kv1.2/ADAM22/PSD-95, 1:5:5) or RBG4 plasmid was cotransfected with Kv1.2. In both cases, 0.4 μg of Kvβ2 subunit was used to increase surface expression of Kv1.2 (Shi et al., 1996; Tiffany et al., 2000) and 0.2 μg of pEGFPc1 plasmid was used to identify the transfected cells under a fluorescence microscope. In independent experiments to determine the efficiency of cotransfection, when cells were cotransfected with GFP and Kv1.2, ADAM22, or PSD-95, >90% of cells expressed both proteins. When cells were cotransfected with GFP, Kv1.2, ADAM22, and PSD-95, followed by immunostaining for any three of the expressed proteins, >70% of cells were cotransfected. Thus, for any given GFP+ cell, >70% coexpress Kv1.2, ADAM22, and PSD-95.
Outward potassium currents were recorded by whole-cell voltage-clamp technique with an EPC-10 amplifier (HEKA). Data were sampled at 10 kHz (with the exception of C-type inactivation, which were sampled at 1 kHz) and filtered at 2.9 kHz using a digital Bessel filter. All currents were capacitance- and series-resistance compensated, and recorded at room temperature. Pipettes were pulled to give a final resistance of 2–3 MΩ when filled with pipette solution. External solution contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, pH 7.4. Tetrodotoxin at 300 nm was added to block Na+ currents. The pipette solution contained the following (in mm): 140 KCl, 2 MgCl2, 1 CaCl2, 5 EGTA, 10 glucose, and 10 HEPES, pH 7.2.
To see voltage-dependent current activation, cells were held at −100 mV and step depolarized to +70 mV in 10 mV increments for 200 ms with an interpulse interval of 5 s. Currents were leak-subtracted by standard P/n procedure. Conductances (G) were obtained from the peak current amplitude (I) at each test potential (V) using Ohm's law G = I/(V − EK) with a predicted Nernst potassium equilibrium potential EK of −84 mV. The normalized conductance were fitted to a standard single Boltzmann equation G = Gmax/(1 + exp[−(V − V1/2)/k]), where Gmax is the maximum conductance, V1/2 is the voltage of half-maximal conductance, and k represents the slope factor of the curve. For deactivation experiments, cells were first depolarized from −100 to +30 mV for 80 ms, and then step depolarized from −100 to 0 mV in 10 mV increments for 170 ms. C-type inactivation were recorded by depolarizing to +20 mV for 70 s.
Patchmaster software (HEKA) was used for acquisition and analysis of currents. Origin 7 software (OriginLab Corporation) was used for fitting and creating figures. All data are presented as mean ± SE, and statistically significant differences were determined using unpaired two-tailed Student's t tests (p < 0.05).
Results
ADAM22 associates with Kv1 channels and MAGUKs
To identify proteins that may regulate the function, assembly, and/or maintenance of Kv1 channel clusters, we used antibodies against Kv1.2 α subunits to coimmunoprecipitate interacting proteins from the Triton X-100-soluble fraction of a rat brain membrane homogenate. We then size-fractionated the immunoprecipitated proteins by one-dimensional SDS-PAGE. Silver staining of the gel revealed many prominent bands (Fig. 1A), including IgG and Kv1.2 α subunits (asterisk). Previously, we identified the MAGUKs PSD-93 and PSD-95 as Kv1 channel-interacting proteins, and these may correspond to the >90 kDa bands observed on our gel (Rasband et al., 2002; Horresh et al., 2008). In a parallel experiment, we stained the gel using colloidal blue and found a prominent band at ∼85 kDa. This band could also be observed on the silver-stained gel (Fig. 1A, arrowhead). We excised this band and performed mass spectrometry to determine the identity of this protein. Based on four unique peptides (mass spectra for two of these unique peptides are shown in Fig. 1B), we identified ADAM22 as a potential interacting protein (these four peptides cover 4.9% of the longest ADAM22 splice variant). For comparison, we also excised bands with molecular weights corresponding to Kv1.2 α subunits and Kvβ2 β subunits of the Kv1 K+ channel, and confirmed their identity (Kv1.2, 14 unique peptides covering 28% of the protein; Kvβ2, 22 unique peptides covering 53% of the protein).
Figure 1.

ADAM22 is part of the Kv1 channel protein complex. A, Silver-stained gel of a Kv1.2 immunoprecipitate. The asterisk indicates the Kv1.2 band, and the arrowhead indicates the ADAM22 band. B, Tandem mass spectra obtained from precursor ions with m/z = 629.8495+2 (top) and 719.3796+2 (bottom), corresponding to two peptides spanning the residues Leu-383 to Lys-393, and Ser-466 to Arg-479, respectively, of rat ADAM22. The observed sequence ions are labeled. Ions corresponding to neutral losses are marked with stars.
To confirm that ADAM22 is a constituent of the Kv1 K+ channel protein complex, we performed immunoprecipitation reactions using mouse monoclonal antibodies against Kv1.2, PSD-93, PSD-95, ADAM22, and pan-Neurofascin.
We found that with the exception of the control pan-Neurofascin antibody, each antibody could immunoprecipitate a macromolecular protein complex consisting of Kv1.2, PSD-93, PSD-95, Lgi1, and ADAM22 (Fig. 2A). The immunoblots of ADAM22 are consistent with the existence of multiple brain splice variants (Gödde et al., 2007). Among the major ADAM22 splice variants expressed and detected in brain, the lowest molecular weight form is the predominant Kv1.2-interacting species (Fig. 2A). The observation that ADAM22 could coimmunoprecipitate with PSD-95 and Lgi1 is consistent with one previous report (Fukata et al., 2006). However, the major ADAM22 splice variants that interact with PSD-93 and PSD-95 were of higher molecular weight than that detected in the Kv1.2 immunoprecipitation reaction (Fig. 2A), even though Kv1.2 could also coimmunoprecipitate the MAGUKs PSD-93 and PSD-95.
Figure 2.
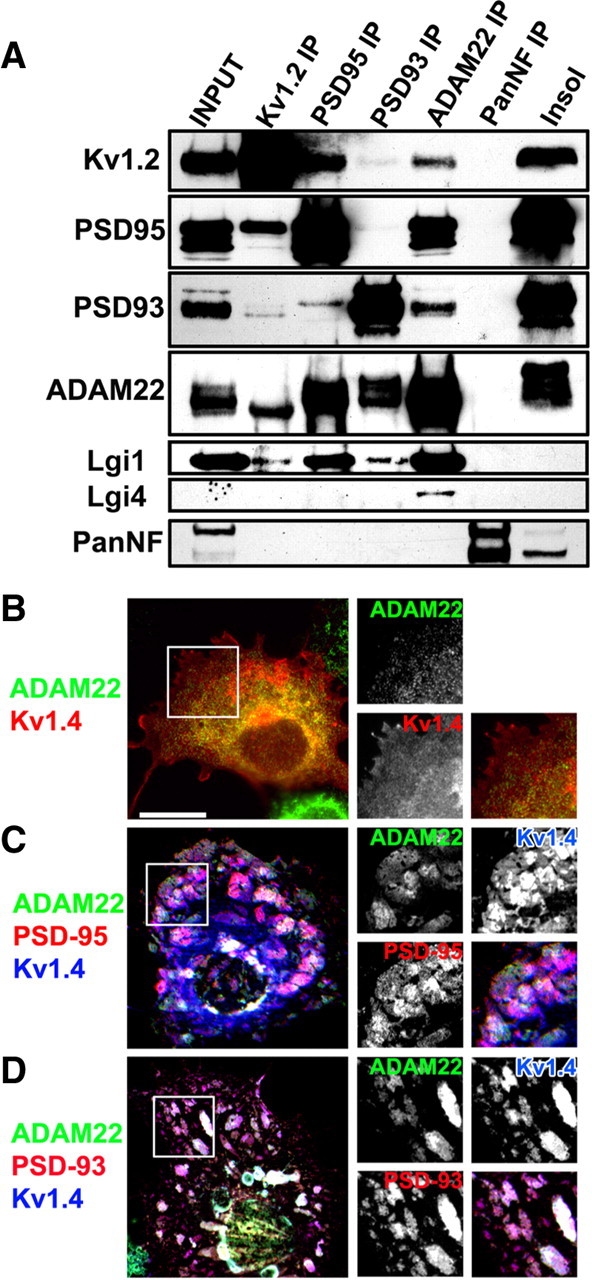
ADAM22 interacts with Kv1 channels, MAGUKs, and Lgi proteins. A, Immunoblot analysis of coimmunoprecipitation reactions using antibodies against Kv1.2, PSD-95, PSD-93, ADAM22, and pan-Neurofascin (PanNF). The input lane corresponds to the detergent soluble fraction, whereas the remainder of the protein is shown in the insoluble fraction (insol). B, Cotransfection of ADAM22 and Kv1.4 in COS7 cells shows no surface clustering. C, D, Cotransfection of ADAM22, Kv1.4, and PSD-95 (C) or PSD-93 (D) results in the formation of large surface clusters that can be detected using antibodies directed against an extracellular epitope of ADAM22. Scale bar, 20 μm.
As additional evidence that ADAM22 interacts with Kv1 channels and MAGUKs, we performed a surface-clustering assay (Kim et al., 1995) by coexpressing in COS cells ADAM22 with Kv1.4 channel α subunits alone (Fig. 2B), or ADAM22, Kv1.4, and the MAGUKS PSD-93 or PSD-95 (Fig. 2C,D). We used Kv1.4 because it colocalizes with Kv1.2 at juxtaparanodes and axon initial segments (Rasband et al., 2001; Ogawa et al., 2008) and has the same postsynaptic density-95/Discs large/zona occludens-1 (PDZ)-binding motif as Kv1.2, and because it is expressed more efficiently on the cell surface than Kv1.2 (Manganas et al., 2001a). When ADAM22 and Kv1.4 were expressed without MAGUKs, no surface clusters were detected (Fig. 2B) (for these experiments, we used an antibody directed against the ectodomain of ADAM22) (see Materials and Methods). However, when ADAM22 and Kv1.4 were coexpressed with PSD-93 or PSD-95, large clusters containing all three proteins formed on the cell surface (Fig. 2C,D). Together, these results suggest that ADAM22 can participate in a large macromolecular protein complex with Kv1 channels through binding to MAGUKs.
ADAM22 colocalizes with clustered Kv1 channels and MAGUKs
To further establish that ADAM22 is part of the Kv1 channel/MAGUK protein complex in vivo, we immunostained CNS and PNS tissues using antibodies against ADAM22, Kv1.2, PSD-93, and PSD-95. In myelinated nerve fibers of the CNS (Fig. 3A–C) and PNS (Fig. 3D), ADAM22 colocalizes at juxtaparanodes with Kv1.2, PSD-93, and PSD-95 (Horresh et al., 2008). Similarly, ADAM22 colocalizes with Kv1.2 at BCTs in the cerebellum (Fig. 3E, inset) where PSD-95 is highly enriched (Fig. 3F, inset) (Laube et al., 1996). In contrast, PSD-93 is expressed in Purkinje neurons rather than at BCTs (Fig. 3F). Finally, immunostaining of cultured hippocampal neurons revealed that ADAM22 could be detected at the AIS where it colocalizes with the cytoskeletal protein βIV spectrin, a highly specific marker of the AIS (Fig. 3G) (Berghs et al., 2000) and Kv1.1 (Fig. 3H). Immunostaining for other Kv1 channels and PSD-93 at the AIS revealed a similar colocalization (data not shown). However, in contrast to previous reports (Fukata et al., 2006), we did not detect enrichment of ADAM22 at synapses in cultured hippocampal neurons.
Figure 3.
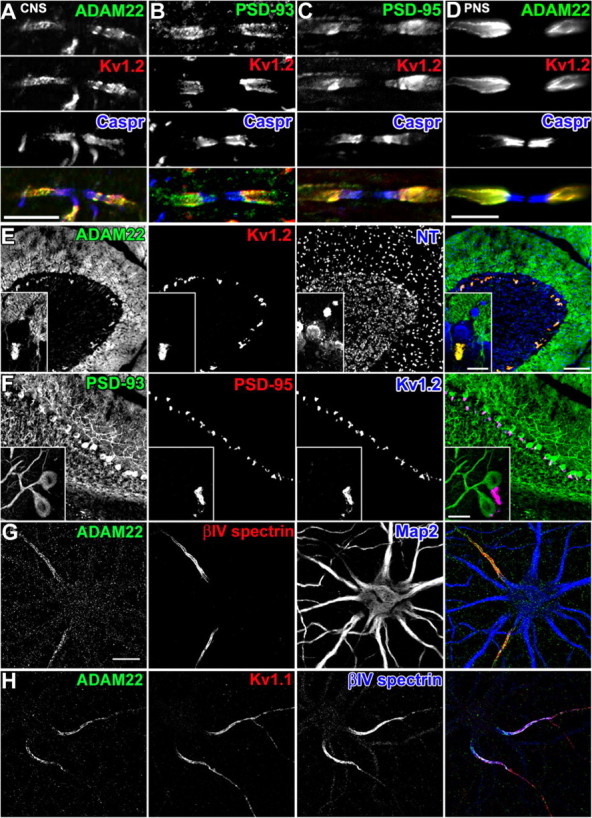
ADAM22 colocalizes with Kv1.2 and MAGUKs at juxtaparanodes, BCTs, and the AIS. A–D, Immunostaining for ADAM22 (A, D), PSD-93 (B), and PSD-95 (C) demonstrates colocalization with Kv1.2 (red) at juxtaparanodes in the CNS (A–C) and PNS (D). Caspr immunoreactivity (blue) defines the paranodal junctions. E, F, ADAM22 immunoreactivity colocalizes with Kv1.2 (E) and PSD-95 (F) at BCTs. PSD-93 (F) is enriched in somatodendritic domains of Purkinje neurons. In E, neurotracer (NT) fluorescence indicates the location of neuronal cell bodies and shows that the BCT lies below the Purkinje neuron (inset). G, In cultured hippocampal neurons, ADAM22 immunostaining (green) colocalizes with βIV spectrin (red) at the AIS and is excluded from somatodendritic domains indicated by MAP2 immunoreactivity (blue). H, ADAM22 immunoreactivity (green) colocalizes with Kv1.1 (red) and βIV spectrin (blue) at the AIS. Scale bars: A–D, 10 μm; E, F, 100 μm; E, F, inset; G, H, 20 μm.
A developmental analysis of juxtaparanode formation in the PNS showed that ADAM22 was present as early as postnatal day 4 (P4), which corresponded to the earliest time when Kv1 channels could be detected. Like Kv1 channels (Vabnick et al., 1999), ADAM22 was initially found to be present in paranodal regions, where it colocalized with Caspr immunostaining but then redistributed to juxtaparanodes (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Analysis of BCTs in the cerebellum showed that, although the βIV spectrin-labeled AIS of the Purkinje neuron was readily identifiable by P14, GAD-65+, Kv1.2+, and ADAM22+ BCTs were not detected until P16–P18 (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Together, these results demonstrate that ADAM22 expression and localization is spatially and temporally correlated with that for clustered Kv1 channels.
Kv1.2 channel properties are not altered by interaction with ADAM22 and PSD-95
The interaction of ion channels with accessory subunits can alter channel properties (Rettig et al., 1994). To determine whether PSD-95 and ADAM22 affect Kv1 channel properties, we measured K+ currents from COS cells expressing GFP and Kv1.2 or GFP and Kv1.2 together with ADAM22 and PSD-95 (Fig. 4A; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Currents in untransfected cells were negligible (Fig. 4A). GFP-positive transfected cells were selected for recording under a fluorescence microscope. We found that Kv1.2 currents, I–V curves, voltage dependence of activation, and activation and deactivation constants were unaffected by coexpression of Kv1.2 with ADAM22 and PSD-95 (Fig. 4). Similarly, inactivation time constants were unaffected by coexpression of Kv1.2 with ADAM22 and PSD-95 (Table 1). Together, these results suggest ADAM22 and PSD-95 binding to Kv1.2 does not alter the electrophysiological properties of the channel.
Figure 4.

Coexpression of ADAM22 and PSD-95 with Kv1.2 does not affect the voltage-dependent activation and deactivation kinetics. A, Current traces from COS cells transfected with Kv1.2 alone (0.1 μg) (top) or in combination with ADAM22 and PSD-95 (0.5 μg each) (bottom). Voltage steps were applied from holding potential −100 to +70 mV in 10 mV increments. B, I–V curve with Kv1.2 alone (filled squares) or with in combination of ADAM22 and PSD-95 (open circles). C, Voltage-dependent activation. The lines represent the single Boltzmann fits to the mean ± SE normalized data. D, Activation constant as a function of voltage. Current traces were fitted with a single exponential function from the time they reached half level to the maximum current. E, Deactivation constant. Cells were held at −100 mV and depolarized to 30 mV for 80 ms followed by test pulses to variable potentials from −100 to 0 mV.
Table 1.
Effects of ADAM22 and PSD-95 on Kv1.2-encoded K+ currents in COS cells
| COS cells | Activation |
Deactivationτ at −50, −20 mV (ms) | Inactivation (C-type) |
Steady-state inactivationV1/2; k | ||
|---|---|---|---|---|---|---|
| V1/2; k | τ at 10, 40, 70 mV (ms) | Imax/min | τ fast; τ slow(ms) | |||
| Kv1.2 | 7.9 ± 3.2 | 36.5 ± 5.4 | 15.3 ± 1.6 | 0.24 ± 0.03 | 3.6 ± 0.7 | −25.3 ± 4.2 |
| 11.5 ± 0.8 | 12.5 ± 3.6 | 23.0 ± 2.6 | 35.5 ± 1.8 | 8.5 ± 1.9 | ||
| (n = 17) | 4.17 ± 0.8 | (n = 14) | (n = 11) | (n = 6) | ||
| (n = 15) | ||||||
| Kv1.2 + ADAM22 + PSD-95 | 9.1 ± 5.5 | 40.7 ± 9.1 | 12.1 ± 1.8 | 0.28 ± 0.04 | 4.9 ± 2.6 | −32.9 ± 2.9 |
| 10.6 ± 0.5 | 10.0 ± 2.7 | 25.1 ± 3.6 | 40.0 ± 4.9 | 3.6 ± 0.8 | ||
| (n = 9) | 3.2 ± 0.6 | (n = 9) | (n = 5) | (n = 3) | ||
| (n = 9) | ||||||
Differential requirement for MAGUK-dependent clustering of ADAM22 at juxtaparanodes and BCTs
Although PSD-93 and PSD-95 coimmunoprecipitate and colocalize with Kv1 channels in neurons, and facilitate the clustering of Kv1 channels in transfected cells (Kim et al., 1995) (Fig. 2C,D), these MAGUKs are dispensable for clustering of Kv1 channels at juxtaparanodes (Horresh et al., 2008). Instead, juxtaparanodal Kv1 channel clustering depends on (1) paranodal junctions and (2) interactions with the cell adhesion molecule Caspr2. Thus, loss of paranodal junctions in Caspr−/− mice causes a redistribution of Kv1 channels into paranodal regions. Similarly, ADAM22 is also found colocalized with Kv1 channels at paranodes in Caspr−/− mice (Fig. 5, compare A, B). Caspr2−/− mice have a profound loss of both Kv1 channel and ADAM22 clustering (Fig. 5C). However, in the optic nerves of Caspr2−/− mice, we also found 32% of juxtaparanodes retained some ADAM22 and Kv1.2 (Fig. 5G), suggesting that other, Caspr2-independent mechanisms also exist in a subpopulation of myelinated axons that can facilitate Kv1.2 and ADAM22 clustering.
Figure 5.

MAGUKs are not required for clustering of ADAM22 at juxtaparanodes. A–G, Immunostaining of myelinated CNS axons for ADAM22 (green), Kv1.2 (red), and βIV spectrin or Caspr (blue) in WT, Caspr−/−, Caspr2−/−, PDS-93−/−, PSD-95−/−, and PSD-95/PSD-93−/− mice.
Since ADAM22 has a C-terminal “ETSI” PDZ binding motif and can be recruited into surface clusters with Kv1 channels when cotransfected with MAGUKs (Fig. 2C,D), rather than altering channel properties we considered whether PSD-93 and/or PSD-95 recruit ADAM22 to juxtaparanodes and BCTs. To test this possibility, we examined juxtaparanodes and BCTs from PSD-93-null, PSD-95-null, and PSD-93/PSD-95-double null mice. We found that, like Kv1 channels, deletion of one or both of these MAGUKs had no effect on the recruitment of ADAM22 to juxtaparanodes (Fig. 5D–F). Similarly, BCT clustering of Kv1 channels did not require PSD-93, PSD-95, or Caspr2 (Fig. 6) (Rasband et al., 2002); in contrast to PSD-95, neither PSD-93 nor Caspr2 are found at BCTs. However, in contrast to juxtaparanodes, we found that loss of PSD-95, but not PSD-93 or Caspr2, abolished the BCT clustering of ADAM22 (Fig. 6). Thus, MAGUKs play different roles for the same protein complex depending on the cellular context: at juxtaparanodes they are dispensable; at BCTs they are required for clustering of ADAM22.
Figure 6.

PSD-95 is required for clustering of ADAM22 at BCTs. A, Immunostaining of BCTs for ADAM22 (green) and Kv1.2 (red) in PSD-95−/−, PSD-93−/−, PSD-95/PSD-93 double −/−, and Caspr2−/− mice. B, High-magnification images showing immunostaining of BCTs for ADAM22 (green) and Kv1.2 (red), and Purkinje neuron AIS (βIV spectrin; blue) in PSD-95−/−, PSD-93−/−, PSD-93/PSD-95−/−, and Caspr2−/− mice. Scale bar, 10 μm.
ADAM22 is required for MAGUK clustering at juxtaparanodes
To determine whether ADAM22 contributes to the assembly of Kv1 channel protein complexes, we examined juxtaparanodes of P10 ADAM22-null mouse spinal cord and brainstem. ADAM22-null mice typically die at approximately P12 and have extensive peripheral hypomyelination (Sagane et al., 2005), making it difficult to examine the role of ADAM22 at BCTs and in Kv1 channel clustering in the PNS. Nevertheless, in both spinal cord and brainstem of P10 wild-type (WT) littermate controls and ADAM22-null mice, we found normally clustered Kv1 channels and Caspr2 (Fig. 7A,B, green). Importantly, immunostaining of ADAM22-null mice showed no juxtaparanodal staining, confirming the specificity of the ADAM22 antibody (Fig. 7A,B). However, in contrast to Caspr2 and Kv1 channels, immunostaining for PSD-93 and PSD-95 showed that these MAGUKs could not be detected at juxtaparanodes (Fig. 7C,D). Together, these results suggest that, although ADAM22 is not required for clustering of Kv1 channels or Caspr2, it is required for the recruitment of PSD-93 and PSD-95 to juxtaparanodes.
Figure 7.

ADAM22 is required for clustering of PSD-93 and PSD-95 at juxtaparanodes. Immunostaining of P10 WT and ADAM22-null (ADAM22−/−) myelinated spinal cord and brainstem axons. In the spinal cord, nodes are identified by βIV spectrin immunoreactivity (blue). A, ADAM22 (red), Kv1.2 (green), and βIV spectrin (blue). B, ADAM22 (red), Caspr2 (green), and βIV spectrin (blue). C, PSD-93 (red), Kv1.2 (green), and βIV spectrin (blue). D, PSD-95 (red), Kv1.2 (green), and βIV spectrin (blue). Scale bar, 5 μm.
ADAM22 is not required for clustering of Kv1 channels at the axon initial segment
In cultured hippocampal neurons, ADAM22 is found at the AIS where it colocalizes with Kv1 channels and markers of the AIS such as βIV spectrin (Fig. 3G,H) and ankyrinG (AnkG) (data not shown). However, we were unable to detect ADAM22 at the AIS in brain sections even after pepsin-mediated antigen retrieval, a method that was recently shown to be necessary to reveal AIS Kv1 channels in fixed tissues (Lorincz and Nusser, 2008b). To gain additional insight into the mechanisms of Kv1 channel clustering at the AIS, we performed pepsin-mediated antigen retrieval followed by immunostaining on brain sections from WT, Caspr-, Caspr2-, ADAM22-, PSD-93-, PSD-95-, and PSD-93/PSD-95-null mice. In every genotype analyzed, we found robust Kv1.2 channel immunoreactivity (green) that colocalized with AnkG (red) at the AIS (Fig. 8). These results are in contrast to our previous report that PSD-93 is required for clustering of Kv1 channels at the AIS (Ogawa et al., 2008) (see Discussion) and demonstrate that ADAM22 is not required for AIS clustering of Kv1.2.
Figure 8.

ADAM22 is not required for clustering of Kv1 channels at axon initial segments. Immunostaining of axon initial segments using antibodies against Kv1.2 (green) and AnkG (red) in WT, Caspr−/−, Caspr2−/−, ADAM22−/−, PSD-93−/−, PSD-95−/−, and PSD-93/PSD-95−/− mice. Scale bar, 10 μm.
Discussion
In many neurons, the restricted expression of axonal Kv1 channels regulates neurotransmitter release and action potential amplitude, duration, and firing rates. Kv1 channel surface expression and biophysical properties can be modulated by accessory cytoplasmic subunits (Rettig et al., 1994; Shi et al., 1996). Kv1 channels have also been shown to interact with a variety of cell adhesion molecules and cytoskeletal scaffolds; however, the functions of some of these proteins and the mechanisms responsible for clustering Kv1 channels at distinct axonal locations remain ill defined.
We used immunoprecipitation and mass spectrometry to identify ADAM22 as a new component of clustered Kv1 channel protein complexes. Intriguingly, among all the known Kv1 channel-interacting proteins, only ADAM22 is found at every single site where Kv1 channels are clustered (Table 2) (note that ADAM22 was detected at the AIS in culture, but we were unable to detect it at the AIS in brain sections) (see below). Furthermore, our immunoprecipitation reactions suggest that ADAM22 splice variants interact with different pools of Kv1 channel-associated proteins. For example, Kv1 channels interact with a lower molecular weight form of ADAM22 than does PSD-93. This observation may suggest that, although both Kv1 channels and PSD-93 can be detected at juxtaparanodes, they may interact with different pools of ADAM22 at these sites. Splice variant-specific antibodies will be needed to test this possibility.
Table 2.
Molecular composition of Kv1 channel protein complexes at sites where channels are clustered
| Juxtaparanode | Axon initial segment | Basket cell terminal | |
|---|---|---|---|
| Kv1.xa,b | ● | ● | ● |
| Kvβ2b,c | ● | ● | ● |
| Caspr2b | ● | ● | ○ |
| TAG-1b,d | ● | ● | ○ |
| PSD-93b | ● | ● | ○ |
| PSD-95b | ● | ○ | ● |
| 4.1Bb,e | ● | ○ | ○ |
| ADAM22 | ● | ● | ● |
●, Detected; ○, not detected.
ADAM22 is a transmembrane protein with an extracellular disintegrin and catalytically inactive metalloproteinase domain that is thought to participate in cell–cell and cell–matrix interactions (White, 2003). ADAM22 has been the subject of much interest since mutations in an extracellular ADAM22 ligand, Lgi1, causes autosomal-dominant partial epilepsy with auditory features. The basis of this epilepsy was proposed to result from disrupted binding of Lgi1 to ADAM22, an interaction that under normal conditions was reported to increase AMPA receptor-mediated synaptic transmission (Fukata et al., 2006). Since only one-half of families with this form of epilepsy were found to have mutations in Lgi1, ADAM22 was also investigated as an alternative candidate gene. However, corresponding mutations in ADAM22 have not been identified (Chabrol et al., 2007; Diani et al., 2008). Our antibodies against the cytoplasmic domain of ADAM22 failed to label excitatory synapses, suggesting either that ADAM22 is not found at these sites or a unique splice variant not detected by our antibody is found at excitatory synapses. Instead, we identified three main locations where ADAM22 is enriched: juxtaparanodes of myelinated axons, BCTs in the cerebellum, and the AIS of cultured hippocampal neurons. In every instance in which we found ADAM22 immunoreactivity, we also identified clustered Kv1 channels. It is possible that the postsynaptic targeting of ADAM22 may require additional extrinsic (e.g., Lgi1) factors not present in our cultures. Intriguingly, preliminary immunostaining experiments showed weak Lgi1 immunoreactivity at juxtaparanodes of PNS nerve fibers (M. N. Rasband, unpublished results). In any case, the identification of ADAM22 as primarily a postsynaptic protein of excitatory synapses must be reevaluated.
Experiments to define Kv1 channel-interacting proteins at juxtaparanodes have shown that many of these same proteins are also enriched at the AIS and BCTs (Table 2). Proteomic efforts to elucidate Kv1 channel-interacting proteins in brain identified PSD-93, PSD-95, Caspr2, Kvβ2, and Lgi1 (Rasband et al., 2002; Schulte et al., 2006). Interestingly, ADAM22 was also identified by Schulte et al. (2006) but was not investigated for its significance or biological function. However, the functional significance of Lgi1 as a putative component of the Kv1 channel protein complex remains controversial, since several reports indicate that Lgi1 is a secreted neuronal protein that interacts with ADAM22 (Senechal et al., 2005; Fukata et al., 2006; Sagane et al., 2008), whereas another suggests that it functions directly as a Kv1.1-associated subunit to inhibit channel inactivation by cytoplasmic, accessory Kvβ1 subunits (Schulte et al., 2006). Intriguingly, disruption of Lgi4, another ADAM22 binding partner, causes hypomyelination and deficits in axonal sorting (Bermingham et al., 2006), whereas ADAM22-null mice suffer from profound peripheral hypomyelination (Sagane et al., 2005). Since ADAM22 is clustered in axons after initiation of myelination, the role of ADAM22 in myelination is likely distinct from its role in Kv1 channel complexes.
What is the function of ADAM22 in axons? In addition to its contribution to early PNS myelination, ADAM22 assembles a PSD-93- and PSD-95-based scaffold at juxtaparanodes (Table 3) likely through its PDZ binding motif. Juxtaparanodal clustering of Kv1 channels, PSD-93, PSD-95, and ADAM22 strongly depends on neuron–glia interactions mediated by the cell adhesion molecules Caspr2 and TAG-1 (Poliak et al., 2003; Horresh et al., 2008), although some Kv1.2/ADAM22 protein complexes can assemble at juxtaparanodes without Caspr2. This observation suggests that there exist as-yet-unidentified compensatory and/or redundant mechanisms for Kv1 channel clustering in a subset of myelinated axons and that ADAM22 is recruited to juxtaparanodes through interactions with Kv1 channels rather than Caspr2 (Table 3). Future experiments will be needed to determine whether Kv1 channels interact directly with ADAM22 or whether other scaffolding proteins mediate this interaction. Our immunoprecipitation reactions showed that only a small amount of the total Kv1.2 and PSD-93 could coprecipitate each other. Recently, Horresh et al. (2008) demonstrated that not all juxtaparanodes have PSD-93, which could account for this difference. Alternatively, PSD-93/Kv1 channel interactions may be more susceptible than PSD-95/Kv1 channel interactions to disruption under the solubilization conditions used here.
Table 3.
Protein interactions that contribute to, or are required for, channel clustering
| Clustered protein | Juxtaparanode | Axon initial segment | Basket cell terminal |
|---|---|---|---|
| Kv1.x | (Caspr2) | (PSD-93) | ? |
| Caspr2 | TAG-1 | ? | N/A |
| PSD-93 | ADAM22 | ? | N/A |
| PSD-95 | ADAM22 | N/A | ? |
| ADAM22 | Kv1.2 | ? | PSD-95 |
Parentheses indicate that other unidentified mechanisms can also contribute to protein clustering. N/A, Not applicable.
By analogy with excitatory synapses, the PSD-93/PSD-95-based scaffolds recruited by ADAM22 may play important roles in recruiting other modulatory or accessory proteins that could influence Kv1 channel properties. However, we did not observe any changes in Kv1.2 K+ channel function when ADAM22 was coexpressed with PSD-95 and Kv1.2. Since these experiments were performed by transfection of cDNAs into COS cells, it is not possible to determine whether other accessory proteins, brought to the K+ channel through direct or indirect interactions with MAGUKs or ADAM22, are required for modulation of channel properties. A thorough proteomic analysis of axonal Kv1 channel complexes will be necessary to identify additional proteins that may influence Kv1 channel properties.
Opposite to the situation at juxtaparanodes, ADAM22 localization at BCTs requires PSD-95. BCTs, also referred to as cerebellar pinceau synapses, innervate the Purkinje neuron AIS and form junctions with morphological features similar to invertebrate septate junctions (Laube et al., 1996). Since Kv1.2, but not ADAM22, is found at BCTs in PSD-95-null mice, ADAM22 cannot be essential for Kv1 channel clustering at this specialized synapse. Nevertheless, ADAM22 may contribute to the function of BCTs by recruiting other proteins to the Kv1 channel complex, or by participating in the cell–cell interactions that stabilize this structure. Unfortunately, the early death of ADAM22-null mice before the formation of BCTs makes it very difficult to address this question.
The AIS and juxtaparanodes share a similar molecular composition (Table 2), although in contrast to cultured neurons, we did not detect AIS ADAM22 in brain sections. This may reflect differences in antigen presentation in brain compared with neurons in culture. For example, Lorincz and Nusser (2008a) recently showed that robust detection of Kv1 channels at the AIS of cortical neurons in fixed brain requires pepsin-mediated antigen retrieval, but these same channels are detected in cultured hippocampal neurons without any form of antigen retrieval (Ogawa et al., 2008). We previously reported that PSD-93 is required for Kv1 channel clustering at the AIS. However, application of this newer method for detection of Kv1 channels in ADAM22- and PSD-93-null brains showed strong Kv1 channel immunoreactivity. Intriguingly, acute silencing of PSD-93 expression by short hairpin RNA interference in culture blocked the clustering of Kv1 channels (Ogawa et al., 2008), suggesting that the chronic loss of PSD-93 in PSD-93−/− mice results in compensation or that there are redundant extrinsic interactions that can occur in the brain but not in cell culture. The idea that redundant mechanisms exist for Kv1 channel clustering may also be seen in Caspr2-null mice in which a subset of juxtaparanodes still retain Kv1 channel immunoreactivity.
In summary, we have identified ADAM22 as a component of clustered Kv1 channels in three distinct axonal domains: the AIS, BCTs in the cerebellum, and juxtaparanodes of myelinated nerve fibers. The results presented here demonstrate that a dichotomy exists between the juxtaparanode and the BCT: ADAM22 recruits MAGUKs to the juxtaparanode, but a MAGUK recruits ADAM22 to the BCT. Our results also emphasize the need for additional experiments to elucidate the molecular mechanisms responsible for recruitment of Kv1 channel complexes to the AIS, juxtaparanodes, and BCTs (Table 3). Thus, despite similar molecular organizations, within each axonal domain unique mechanisms are used to assemble ADAM22/Kv1 channel/MAGUK-containing protein complexes.
Footnotes
This work was supported by Mission Connect, The Dr. Miriam and Sheldon Adelson Medical Research Foundation, National Institutes of Health (NIH) Grants NS044916 (M.N.R.) and NS034383 (J.S.T.), and the National Multiple Sclerosis Society (E.P.). Mass spectrometry analysis was provided by the Bio-Organic Biomedical Mass Spectrometry Resource at University of California, San Francisco (San Francisco, CA) (A. L. Burlingame, Director), supported by the Biomedical Research Technology Program of the NIH–National Center for Research Resources (NCRR) (NIH–NCRR Grants P41RR001614 and RR019934). The University of California Davis/NIH NeuroMab Facility is supported by NIH Grant U24NS050606. M.N.R. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. Y.O. is supported by a postdoctoral fellowship from the National Multiple Sclerosis Society. We thank Drs. David Bredt, Seth Grant, and Koji Sagane for kindly providing PSD-93, PSD-95, and ADAM22-null mice, respectively.
References
- Baba H, Akita H, Ishibashi T, Inoue Y, Nakahira K, Ikenaka K. Completion of myelin compaction, but not the attachment of oligodendroglial processes triggers K+ channel clustering. J Neurosci Res. 1999;58:752–764. [PubMed] [Google Scholar]
- Berghs S, Aggujaro D, Dirkx R, Jr, Maksimova E, Stabach P, Hermel JM, Zhang JP, Philbrick W, Slepnev V, Ort T, Solimena M. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of Ranvier in the central and peripheral nervous system. J Cell Biol. 2000;151:985–1002. doi: 10.1083/jcb.151.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham JR, Jr, Shearin H, Pennington J, O'Moore J, Jaegle M, Driegen S, van Zon A, Darbas A, Ozkaynak E, Ryu EJ, Milbrandt J, Meijer D. The claw paw mutation reveals a role for Lgi4 in peripheral nerve development. Nat Neurosci. 2006;9:76–84. doi: 10.1038/nn1598. [DOI] [PubMed] [Google Scholar]
- Chabrol E, Gourfinkel-An I, Scheffer IE, Picard F, Couarch P, Berkovic SF, McMahon JM, Bajaj N, Mota-Vieira L, Mota R, Trouillard O, Depienne C, Baulac M, LeGuern E, Baulac S, et al. Absence of mutations in the LGI1 receptor ADAM22 gene in autosomal dominant lateral temporal epilepsy. Epilepsy Res. 2007;76:41–48. doi: 10.1016/j.eplepsyres.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (±10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- Devaux J, Gola M, Jacquet G, Crest M. Effects of K+ channel blockers on developing rat myelinated CNS axons: identification of four types of K+ channels. J Neurophysiol. 2002;87:1376–1385. doi: 10.1152/jn.00646.2001. [DOI] [PubMed] [Google Scholar]
- Diani E, Di Bonaventura C, Mecarelli O, Gambardella A, Elia M, Bovo G, Bisulli F, Pinardi F, Binelli S, Egeo G, Castellotti B, Striano P, Striano S, Bianchi A, Ferlazzo E, Vianello V, Coppola G, Aguglia U, Tinuper P, Giallonardo AT. Autosomal dominant lateral temporal epilepsy: absence of mutations in ADAM22 and Kv1 channel genes encoding LGI1-associated proteins. Epilepsy Res. 2008;80:1–8. doi: 10.1016/j.eplepsyres.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JB, Hanna MG, Kullmann DM, Spauschus A. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647–656. [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Gödde NJ, D'Abaco GM, Paradiso L, Novak U. Efficient ADAM22 surface expression is mediated by phosphorylation-dependent interaction with 14-3-3 protein family members. J Cell Sci. 2006;119:3296–3305. doi: 10.1242/jcs.03065. [DOI] [PubMed] [Google Scholar]
- Gödde NJ, D'Abaco GM, Paradiso L, Novak U. Differential coding potential of ADAM22 mRNAs. Gene. 2007;403:80–88. doi: 10.1016/j.gene.2007.07.033. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horresh I, Poliak S, Grant S, Bredt D, Rasband MN, Peles E. Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J Neurosci. 2008;28:14213–14222. doi: 10.1523/JNEUROSCI.3398-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Laube G, Röper J, Pitt JC, Sewing S, Kistner U, Garner CC, Pongs O, Veh RW. Ultrastructural localization of Shaker-related potassium channel subunits and synapse-associated protein 90 to septate-like junctions in rat cerebellar Pinceaux. Brain Res Mol Brain Res. 1996;42:51–61. doi: 10.1016/s0169-328x(96)00120-9. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Specificity of immunoreactions: the importance of testing specificity in each method. J Neurosci. 2008a;28:9083–9086. doi: 10.1523/JNEUROSCI.2494-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008b;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganas LN, Wang Q, Scannevin RH, Antonucci DE, Rhodes KJ, Trimmer JS. Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc Natl Acad Sci U S A. 2001a;98:14055–14059. doi: 10.1073/pnas.241403898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganas LN, Akhtar S, Antonucci DE, Campomanes CR, Dolly JO, Trimmer JS. Episodic ataxia type-1 mutations in the Kv1.1 potassium channel display distinct folding and intracellular trafficking properties. J Biol Chem. 2001b;276:49427–49434. doi: 10.1074/jbc.M109325200. [DOI] [PubMed] [Google Scholar]
- McGee AW, Topinka JR, Hashimoto K, Petralia RS, Kakizawa S, Kauer FW, Aguilera-Moreno A, Wenthold RJ, Kano M, Bredt DS. PSD-93 knock-out mice reveal that neuronal MAGUKs are not required for development or function of parallel fiber synapses in cerebellum. J Neurosci. 2001;21:3085–3091. doi: 10.1523/JNEUROSCI.21-09-03085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Shi G, Rhodes KJ, Trimmer JS. Selective interaction of voltage-gated K+ channel beta-subunits with alpha-subunits. J Biol Chem. 1996;271:7084–7089. doi: 10.1074/jbc.271.12.7084. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Jones OT, Fehlings MG. Abnormal axonal physiology is associated with altered expression and distribution of Kv1.1 and Kv1.2 K+ channels after chronic spinal cord injury. Eur J Neurosci. 2000;12:491–506. doi: 10.1046/j.1460-9568.2000.00926.x. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, Rasband MN. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. 2008;28:5731–5739. doi: 10.1523/JNEUROSCI.4431-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, Peles E. Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci. 2001;21:7568–7575. doi: 10.1523/JNEUROSCI.21-19-07568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. 1998;18:36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Zhen D, Arbuckle MI, Poliak S, Peles E, Grant SG, Trimmer JS. Clustering of neuronal potassium channels is independent of their interaction with PSD-95. J Cell Biol. 2002;159:663–672. doi: 10.1083/jcb.200206024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Rhodes KJ, Keilbaugh SA, Barrezueta NX, Lopez KL, Trimmer JS. Association and colocalization of K+ channel α- and β-subunit polypeptides in rat brain. J Neurosci. 1995;15:5360–5371. doi: 10.1523/JNEUROSCI.15-07-05360.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem. 1992;203:173–179. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N, Ino M, Oki T, Yamazaki K, Nagasu T. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 2005;6:33. doi: 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4:387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Bansal R, Hedstrom KL, Pfeiffer SE, Rasband MN. Does paranode formation and maintenance require partitioning of neurofascin 155 into lipid rafts? J Neurosci. 2004;24:3176–3185. doi: 10.1523/JNEUROSCI.5427-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE, Trimmer JS. Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron. 1996;16:843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- Tiffany AM, Manganas LN, Kim E, Hsueh YP, Sheng M, Trimmer JS. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K+ channel surface expression and clustering. J Cell Biol. 2000;148:147–158. doi: 10.1083/jcb.148.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Dupree JL, Popko B, Karagogeos D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J Neurosci. 2002;22:3016–3024. doi: 10.1523/JNEUROSCI.22-08-03016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Goutebroze L, Denisenko N, Bessa M, Nifli A, Havaki S, Iwakura Y, Fukamauchi F, Watanabe K, Soliven B, Girault JA, Karagogeos D. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol. 2003;162:1161–1172. doi: 10.1083/jcb.200305078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Shrager P. Ion channel redistribution and function during development of the myelinated axon. J Neurobiol. 1998;37:80–96. [PubMed] [Google Scholar]
- Vabnick I, Trimmer JS, Schwarz TL, Levinson SR, Risal D, Shrager P. Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci. 1999;19:747–758. doi: 10.1523/JNEUROSCI.19-02-00747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365:75–79. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- White JM. ADAMs: modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. 2003;15:598–606. doi: 10.1016/j.ceb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Messing A, Chiu SY. Specific alteration of spontaneous GABAergic inhibition in cerebellar Purkinje cells in mice lacking the potassium channel Kv1.1. J Neurosci. 1999;19:2852–2864. doi: 10.1523/JNEUROSCI.19-08-02852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]