

Abstract
M1 muscarinic acetylcholine receptors (mAChRs) may represent a viable target for treatment of disorders involving impaired cognitive function. However, a major limitation to testing this hypothesis has been a lack of highly selective ligands for individual mAChR subtypes. We now report the rigorous molecular characterization of a novel compound, benzylquinolone carboxylic acid (BQCA), which acts as a potent, highly selective positive allosteric modulator (PAM) of the rat M1 receptor. This compound does not directly activate the receptor, but acts at an allosteric site to increase functional responses to orthosteric agonists. Radioligand binding studies revealed that BQCA increases M1 receptor affinity for acetylcholine. We found that activation of the M1 receptor by BQCA induces a robust inward current and increases spontaneous EPSCs in medial prefrontal cortex (mPFC) pyramidal cells, effects which are absent in acute slices from M1 receptor knock-out mice. Furthermore, to determine the effect of BQCA on intact and functioning brain circuits, multiple single-unit recordings were obtained from the mPFC of rats that showed BQCA increases firing of mPFC pyramidal cells in vivo. BQCA also restored discrimination reversal learning in a transgenic mouse model of Alzheimer's disease and was found to regulate non-amyloidogenic APP processing in vitro, suggesting that M1 receptor PAMs have the potential to provide both symptomatic and disease modifying effects in Alzheimer's disease patients. Together, these studies provide compelling evidence that M1 receptor activation induces a dramatic excitation of PFC neurons and suggest that selectively activating the M1 mAChR subtype may ameliorate impairments in cognitive function.
Introduction
The muscarinic acetylcholine (ACh) receptors (mAChRs) play important roles in regulating higher cognitive function. Non-selective mAChR antagonists induce profound attention and memory deficits (Aigner et al., 1991; Fibiger et al., 1991; Miller and Desimone, 1993) and degeneration of forebrain cholinergic neurons is one of the earliest pathological changes observed in Alzheimer's disease (AD) (Bartus et al., 1982; Bartus, 2000). Furthermore, acetylcholinesterase inhibitors (AChEIs) have established efficacy in the treatment of AD symptoms (Birks, 2006; Muñoz-Torrero, 2008).
Of the five mAChR subtypes, the M1 receptor is viewed as the most important subtype for memory and attention mechanisms (Levey et al., 1991; Felder et al., 2000). Based on this, selective activators of the M1 receptor have been proposed as having potential utility in treatment of AD (Bodick et al., 1997; Gu et al., 2003; Caccamo et al., 2006, 2009; Jones et al., 2008). However, recent studies revealed that genetic deletion of the M1 receptor does not alter mAChR excitatory effects on hippocampal pyramidal cells (Rouse et al., 2000), impair hippocampal-dependent learning, or alter cognition-impairing effects of mAChR antagonists (Miyakawa et al., 2001; Anagnostaras et al., 2003). Interestingly, while hippocampal-dependent learning was intact, M1 receptor knock-out mice had specific deficits in forms of learning and memory that require activation of the prefrontal cortex (PFC) (Anagnostaras et al., 2003). Thus, the M1 receptor may play a role in regulating PFC function, and M1 receptor-selective activators could improve deficits in PFC-dependent learning in patients suffering from AD.
Unfortunately, lack of highly selective activators and antagonists of the M1 receptor has prevented detailed studies of the functional consequences of selective M1 receptor activation. The difficulty in developing highly selective M1 receptor agonists is due to the high sequence homology among the orthosteric binding sites of mAChR subtypes. However, an alternative strategy for achieving high subtype selectivity is targeting allosteric binding sites that are distinct from the ACh binding site (for review, see Conn et al., 2009a,b). We recently reported discovery of multiple positive allosteric modulators (PAMs) of the M1 receptor (Marlo et al., 2009). Furthermore, Ma et al. (2008) presented a preliminary report in which they showed evidence that benzylquinolone carboxylic acid (BQCA) is a potent and highly selective PAM at the human M1 receptor. Based on these preliminary findings, we synthesized a series of molecules related to BQCA and report that BQCA and related compounds are highly selective rat M1 receptor PAMs. These compounds do not interact with the ACh site, but dramatically increase the affinity of the M1 receptor for ACh and potentiate the response to orthosteric agonist. In addition, activation of the M1 receptor induces an inward current and increases excitatory synaptic currents in mPFC layer V pyramidal cells. Consistent with this, BQCA increases firing of mPFC neurons in vivo. Finally, BQCA reverses deficits in a PFC-dependent form of learning and memory in a transgenic mouse model of AD and promotes non-amyloidogenic APP processing in vitro. Together, these data suggest that the M1 receptor plays an important role in regulating excitatory drive to the PFC and that selective potentiation of activity at this receptor can reverse deficits in PFC-dependent cognitive function.
Materials and Methods
Materials
All tissue culture reagents, as well as fluo-4 AM, were obtained from Invitrogen. ACh chloride (ACh), carbachol (CCh), probenecid, pluronic F-127, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. Costar 96-well cell culture plates and V-bottom compound plates were purchased from Corning. Ninety-six-well poly-d-lysine coated assay plates were purchased from Becton Dickinson. l-[N-methyl-3H]scopolamine methyl chloride ([3H]-NMS) was purchased from GE Healthcare.
General medicinal chemistry methods
All NMR spectra were recorded on a 400 MHz Bruker NMR. 1H chemical shifts are reported in δ values in parts per million downfield from TMS as the internal standard in DMSO. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). 13C chemical shifts are reported in δ values in parts per million with the DMSO carbon peak set to 39.5 ppm. Low resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization. High resolution mass spectra were recorded on a Waters QToF-API-US plus Acquity system. Analytical thin-layer chromatography was performed on 250 mm silica gel 60 F254 plates. Analytical HPLC was performed on an Agilent 1200 analytical LCMS with UV detection at 214 and 254 nm along with ELSD detection. Preparative purification was performed on a custom Agilent 1200 preparative LCMS with collection triggered by mass detection. Solvents for extraction, washing and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical, Ryan Scientific, Maybridge, and BioBlocks, and were used without purification. All polymer-supported reagents were purchased from Biotage.
General procedure for library synthesis I
Each of seven glass vials containing 2 ml of DMF were loaded with ethyl 8-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (25 mg, 0.106 mmol, Maybridge BTB02003EA), K2CO3 (30 mg, 0.212 mmol, 2.0 equivalents), KI (2 mg, 0.011 mmol, 0.1 equivalents), and one of seven benzyl bromides (0.319 mmol, 3.0 equivalents). The reactions were stirred for 24 h at room temperature before receiving polystyrene-bound thiophenol (0.159 mmol, 1.5 equivalents) each, and then stirred for an additional 3 h. The reactions were then judged complete by LCMS, filtered, and separated into CH2Cl2 and H2O. The organics were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo yielding seven benzyl-substituted ethyl 8-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylates confirmed by analytical LCMS. Next, crude products (0.1 mmol) and LiOH (8 mg, 0.3 mmol, 3.0 equivalents) were dissolved in 3 ml of THF:H2O (9:1) in glass vials. The reactions were microwave irradiated at 120°C for 10 min and then separated into EtOAc and H2O, which was acidified to pH 4 drop-wise using 1N HCl. Organics were dried over MgSO4, filtered, and concentrated in vacuo yielding seven benzyl-substituted 8-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acids confirmed by LCMS. Purification using mass-directed HPLC afforded the seven compounds (25–85% total yield) as TFA salts with >98% purity.
General procedure for library synthesis II
Each of seven glass vials containing 2 ml of DMF were loaded with ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylate (25 mg, 0.115 mmol, Ryan Scientific 6J-050), K2CO3 (32 mg, 0.230 mmol, 2.0 equivalents), KI (2 mg, 0.012 mmol, 0.1 equivalents), and one of seven benzyl bromides (0.345 mmol, 3.0 equivalents). The reactions were stirred for 24 h at room temperature before receiving polystyrene-bound thiophenol (0.173 mmol, 1.5 equivalents) each, and then stirred for an additional 3 h. The reactions were then judged complete by LCMS, filtered, and separated into CH2Cl2 and H2O. The organics were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo yielding seven benzyl-substituted ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylates confirmed by analytical LCMS. Next, crude products (0.1 mmol) and LiOH (8 mg, 0.3 mmol, 3.0 equivalents) were dissolved in 3 ml of THF:H2O (9:1) in glass vials. The reactions were microwave irradiated at 120°C for 10 min and then separated into EtOAc and H2O, which was acidified to pH 4 drop-wise using 1N HCl. Organics were dried over MgSO4, filtered, and concentrated in vacuo yielding seven benzyl-substituted 4-oxo-1,4-dihydroquinoline-3-carboxylic acids confirmed by LCMS. Purification using mass-directed HPLC afforded the seven compounds (25–85% total yield) as TFA salts with >98% purity.
General procedure for library synthesis III
Each of seven glass vials containing 2 ml of DMF were loaded with ethyl 5,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (25 mg, 0.099 mmol, Ryan Scientific 6J-020), K2CO3 (27 mg, 0.198 mmol, 2.0 equivalents), KI (2 mg, 0.099 mmol, 0.1 equivalents), and one of seven benzyl bromides (0.297 mmol, 3.0 equivalents). The reactions were stirred for 24 h at room temperature and atmosphere before receiving polystyrene-bound thiophenol (0.149 mmol, 1.5 equivalents) each, and then stirred for an additional 3 h. The reactions were then judged complete by LCMS, filtered, and separated into CH2Cl2 and H2O. The organics were washed with brine, dried over MgSO4, and concentrated in vacuo yielding seven benzyl-substituted ethyl 5,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylates confirmed by analytical LCMS. Next, crude products (0.1 mmol) and LiOH (8 mg, 0.3 mmol, 3.0 equivalents) were dissolved in 3 ml of THF:H2O (9:1) in glass vials. The reactions were microwave irradiated at 120°C for 10 min and then separated into EtOAc and H2O, which was acidified to pH 4 drop-wise using 1N HCl. Organics were dried over MgSO4 and concentrated in vacuo yielding seven benzyl-substituted 5,8-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acids confirmed by LCMS. Purification using mass-directed HPLC afforded the seven compounds (25–85% total yield) as TFA salts with >98% purity.
Sodium 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate
To stirred solution of 200 ml of DMF in a glass flask was added ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylate (3.40 g, 15.66 mmol, Ryan Scientific 6J-050), K2CO3 (4.33 g, 31.32 mmol, 2.0 equivalents), KI (260 mg, 1.57 mmol, 0.1 equivalents), and 4-methoxybenzyl bromide (4.70 g, 23.49 mmol, 1.5 equivalents). After 48 h of stirring at room temperature and atmosphere, the reaction was monitored by LCMS and judged complete. The reaction was then partitioned into CH2Cl2 and H2O, and the organics were washed with brine, dried over MgSO4, and concentrated in vacuo. Purification by diethyl ether washing (6 × 50 ml) afforded the intermediate product ethyl 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (4.99 g, 14.83 mmol, 95%) as an off-white solid at >98% purity by LCMS. To a glass vial containing ethyl 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (4.99 g, 14.83 mmol) in 90 ml THF:H2O (5:1) was added LiOH (1.07 g, 44.49 mmol, 3.0 equivalents). The reaction was microwave irradiated at 120°C for 10 min and then partitioned into CH2Cl2 and H2O. The solution was reacidified to pH 5 drop-wise using 2N HCl. The organics were dried over MgSO4, filtered, concentrated in vacuo, and analyzed by LCMS. The crude product was purified by diethyl ether washing (6 × 50 ml) and additional H2O wash (1 × 100 ml) to afford the intermediate product 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.20 g, 10.35 mmol, 70%) as an off-white crystalline solid at >98% purity by LCMS. To a stirred solution of 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (1.89 g, 6.11 mmol) in 25 ml of DMF in a glass flask at 0°C was added NaH (143 mg, 5.99 mmol, 0.98 equivalents). The reaction was allowed to warm to room temperature and stirred for 1 h before concentration in vacuo. The crude product was washed with diethyl ether (3 × 30 ml) to afford the title compound (1.80 g, 5.44 mmol, 89%) as a white solid at >98% purity by LCMS. 1H NMR (400 MHz, D2O): δ = 9.07 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 8.4 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 8.8 Hz, 2H), 5.35 (s, 2H), 3.67 (s, 3H). 13C NMR (100 MHz, D2O, externally referenced to DMSO-d6): δ = 176.3, 172.2, 158.0, 147.4, 138.5, 132.3, 127.5, 127.2, 126.9, 125.5, 124.5, 117.5, 116.9, 113.8, 55.8, 54.7. HRMS calculated for C18H14NO4Na2 [M + 2Na] 354.0718, found 354.0716.
Cell culture
For calcium mobilization assays, all recombinant Chinese hamster ovary (CHO-K1) cell lines stably expressing rat M1, human M3, or human M5 receptors were plated at a seeding density of 50,000 cells/100 μl/well in 96-well plates. CHO-K1 cells stably coexpressing human M2/Gqi5 and rat M4/Gqi5 were plated at a seeding density of 60,000 cells/100 μl well. Cells were incubated in antibiotic-free medium overnight at 37°C/5% CO2 and assayed the following day.
Calcium mobilization assay
Cells were loaded with calcium indicator dye (2 μm Fluo-4 AM) for 45 min at 37°C. Dye was removed and replaced with the appropriate volume of assay buffer, pH 7.4 (1× HBSS (Hanks' balanced salt solution), supplemented with 20 mm HEPES and 2.5 mm probenecid). All compounds were serially diluted in assay buffer for a final 2× stock in 0.6% DMSO. This stock was then added to the assay plate for a final DMSO concentration of 0.3%. ACh (EC20 concentration or full dose–response curve) was prepared at a 10× stock solution in assay buffer before addition to assay plates. Calcium mobilization was measured at 25°C using a FLEXstation II (Molecular Devices). Cells were preincubated with test compound (or vehicle) for 1.5 min before the addition of the agonist, ACh. Cells were then stimulated for 50 s with a submaximal concentration (EC20) or a full dose–response curve of ACh. The signal amplitude was first normalized to baseline and then as a percentage of the maximal response to ACh.
Radioligand binding studies
All binding reactions were performed essentially as described by Shirey et al. (2008) using 25 μg of membrane protein prepared from rM1 receptor expressing CHO cells and 0.1 nm [3H]-NMS (GE Healthcare) in a final volume of 1 ml. Non-specific binding was determined in the presence of 1 μm atropine.
Ancillary pharmacology assays
Before conducting in vivo experiments, BQCA was submitted to Millipore's GPCR (G protein-coupled receptor) Profiler Service where it was evaluated for agonist, antagonist, and allosteric potentiator activity against a panel of 16 GPCRs in a functional screening paradigm.
JetMilling
BQCA was JetMilled under Zero Air, to afford uniform nanoparticles, before vehicle formulation and in vivo studies using a Model 00 Jet-O-Mizer with a High-Yield Collection Module from Fluid Energy Processing & Equipment Company.
Electrophysiology
Animals.
All animals used in these studies were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experimental protocols were in accordance with all applicable guidelines regarding the care and use of animals. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AALAC) approved facility with ad libitum access to food and water. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Brain slice electrophysiology.
Brain slices were prepared from Sprague Dawley rats (Charles River), wild-type C57BL/6Hsd (Harlan) or M1 receptor knock-out (KO) mice (Taconic, with permission from J. Wess, National Institutes of Health–National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD); all animals were postnatal day 16–26. Animals were anesthetized with isoflurane. Brains were rapidly removed and submerged in ice-cold modified oxygenated artificial CSF (ACSF) composed of 126 mm choline chloride, 2.5 mm KCl, 8 mm MgSO4, 1.3 mm MgCl, 1.2 mm NaH2PO4, 26 mm NaHCO3, 10 mm d-glucose, 5 μm glutathione, and 0.5 mm sodium pyruvate. Coronal brain slices (295–300 μm) containing the mPFC were made using a Leica VT1000S or 3000 Vibratome. Slices were incubated in oxygenated ACSF at 32°C for 30–60 min and then maintained at 20−22°C (room temperature) for 1–6 h until they were transferred to a recording chamber. The recording chamber was continuously perfused at 30 ± 0.2°C with oxygenated ACSF containing 126 mm NaCl, 2.5 mm KCl, 3.0 mm CaCl2, 2.0 mm MgSO4, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm d-glucose.
Spontaneous and miniature EPSCs were recorded from layer V pyramidal cells in whole-cell voltage-clamp mode using either an Axon Multiclamp 700B amplifier (Molecular Devices) or a Warner 501A amplifier (Warner Instruments) and visualized with an Olympus BX50WI upright microscope (Olympus) coupled with a 40× water-immersion objective and Hoffman optics. Borosilicate glass (World Precision Instruments) patch pipettes were prepared using a Flaming-Brown micropipette puller (Model P-97; Sutter Instruments) and filled with 123 mm potassium gluconate, 7 mm KCl, 1 mm MgCl2, 0.025 mm CaCl2, 10 mm HEPES, 0.1 mm EGTA, 2 mm ATP, and 0.2 mm GTP at a pH of 7.3 and osmolarity of 285–295 mOsM. Filled patch pipettes had resistances of 2–4 MÙ. EPSCs were recorded at a holding potential of −70 mV; GABAA receptor-mediated inhibitory currents were undetectable under these conditions. The voltage-clamp signal was low-pass-filtered at 5 kHz, digitized at 10 kHz, and acquired using a Clampex9.2/DigiData1332 system (Molecular Devices). All drugs were bath-applied. Compounds were made in a 100× or 1000× stock and diluted into oxygenated ACSF immediately before use. After a stable baseline was recorded for 5–10 min, the effect of each compound on baseline sEPSC or mEPSC amplitude and frequency was examined. Miniature EPSCs and inward currents were recorded in the presence of 1 μm tetrodotoxin, a concentration which completely blocked action potential firing upon depolarizing current injections in current-clamp mode.
Statistical analysis.
EPSCs were detected and analyzed using the Mini Analysis Program (Synaptosoft). The peak amplitude and interevent interval of sEPSCs and mEPSCs from 2 min episodes during control and drug application were used to generate cumulative probability plots. The mean values of EPSC amplitude and interevent interval from the 2 min episode were grouped (mean ± SEM) and compared using a paired t test. Inward current data analysis was performed using Clampfit software (v9.2, Molecular Devices). All electrophysiology data were quantified and graphed using GraphPad Prism (GraphPad Software) and Excel (Microsoft). Cumulative probability plots were made using Origin (v6, Microcal Origin). Statistical analysis was performed using the student's paired or unpaired t test, and statistical significance was set at p < 0.05. Averaged data are presented as mean ± SEM.
APP processing.
PC12 N21 cells stably expressing human sequence Swedish mutation Amyloid Precursor Protein (APP) and human M1 muscarinic receptor were maintained as described (Jones et al., 2008). For amyloid processing experiments, cells were plated at 50,000 in 12-well trays 3–4 d before the experiment. On the day of the experiment, the culture medium was replaced with 450 μl of DMEM containing the indicated concentration of BQCA or dimethylsulfoxide (DMSO). Following a 10 min pretreatment, carbachol was added in 50 μl of DMEM to the indicated final concentrations, and the medium was conditioned for 4 h at 37°C. Western blot analysis of APP metabolites in conditioned media and cell extracts, and sandwich ELISA determination of Amyloid-β40 levels in conditioned media were performed as described (Jones et al., 2008). Statistical analysis was performed using Graphpad Prism 4.0 software.
In vivo mPFC unit activity. Multichannel single unit recordings were obtained from extracellular electrode arrays (NeuroLinc) chronically implanted in the medial prefrontal cortex (mPFC) of 300–400 g Sprague Dawley rats performing an auditory detection task for food reward. For recording sessions, animals were fitted with a HST/16V-G20 miniature headstage 20× preamplifier (Plexon) and spike event data (1.1 ms data window) was captured by a Cheetah 32-channel acquisition system (Neuralynx) for off-line processing. Individual data sessions consisted of a 30 min preinjection baseline followed by three 30 min postinjection (vehicle or BQCA, 20 mg/kg) epochs. Single neurons were isolated off-line using a manual spike sorter (Mclust; A. D. Redish, University of Minnesota, Minneapolis, MN). A sorted file was only considered to emanate from the activity of a single neuron if bins within ±1.1 ms (considered absolute refractory period) of the autocorrelogram contained counts <1% of the overall mean of the autocorrelogram. In addition, cells with properties characteristic of fast-spiking interneurons (spike width <250 ms and firing rate >6 Hz) were eliminated from analysis. Following off-line clustering, the mean firing rate for each neuron within an epoch was calculated by averaging rates across all 10 s pretone intervals within an epoch (∼50 tone presentations/30 min epoch). The average firing rate in an epoch was expressed as a percentage of the preinjection baseline rate and data were compared across treatment conditions with respect to changes in mean rate across the three 30 min postinjection epochs.
Behavioral studies
Subjects.
Forty Tg2576 mice on the 129S6 background were obtained when they were 10–12 weeks of age from Taconic. Tg2576 APPsw mice overexpressed a 695 amino acid splice form (Swedish mutation K670N M671L) of the human amyloid precursor protein (APP695) that results in an fivefold increase in Aβ 1–40 and a 14-fold increase in Aβ 1–42 with increasing age. In this study, 10 hemizygous males and 12 of their wild type male littermates and 9 hemizygous females and 9 of their wild type female litter mates were individually housed, maintained on a 12 h light/dark cycle (lights on at 8:00 A.M.), with ad libitum food and water. At ∼12 months of age evaluation of reversal learning began. The mice were divided into groups and counter-balanced for genotype and treatment type, either BQCA or vehicle. Experiments were performed during the light cycle.
Before the start of testing, subjects were placed on a restricted food diet of ∼1–2 g of food per day, contingent on their performance on the food motivated tasks. A weight basis of 85% of their prefood deprivation weight was used as a guideline to avoid excessive weight loss. Water was available ad libitum during all phases of testing. All experimental procedures were approved by the Wake Forest University School of Medicine Animal Care and Use Committee and were conducted in compliance with guidelines set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Apparatus.
The reversal-learning test was adapted from a rat set-shifting paradigm. Subjects were trained to dig in identical terra cotta pots to retrieve a food reward. A 1/20th piece of a Reese's Peanut Butter Chip (The Hershey Company) was the food reward. Pots were 1 ¾” in diameter and 1 ½” deep. A square of vinyl window screen was glued inside the pot to form a cavity underneath the pot in which to place a food reward that was unobtainable by the subject to serve as a control for odor. Essential Oils (New Directions Aromatics) were applied to the rim of the pot to produce a long-lasting odor and media placed inside the pots to a depth that produced vigorous digging for the subject to reach the food reward. Each odor had unique pots assigned to it and the pots were filled with the corresponding media and placed in a plastic sealable container where they were returned after each use.
Testing was performed in the subject's home cage placed inside an ordinary 24 inches long × 16 inches wide × 16 inches deep cardboard box to shield the subject from seeing movement within the room. A Plexiglas holder was fabricated to insert and remove two pots at a time from the testing cage. The two pots were separated by a Plexiglas partition. A 1000 ml plastic beaker was painted black and placed over the subject at the end of the cage closest to the experimenter to create a holding area. When the holder with the two pots was placed at the opposite end of the cage, the subject was then released from the black beaker. Upon completion of the dig, the subject was recovered to the holding area with the black beaker. Between all discriminations, the interdiscrimination delay was ∼3 min.
Habituation and shaping.
After 3 d of food deprivation, subjects in their home cages were habituated to the test apparatus (holding beaker and pot holder) and then shaped to dig a reward after release from the holding beaker. Two pots were filled with Alpha-Dri (Shepherd Specialty Papers) and a reward was randomly placed at the very bottom of either pot to encourage the subject to dig vigorously to find it. The subjects were released from the holding area and allowed to dig in the pots. When the reward was found, the subject was recovered to the holding area using the black beaker. A pot was rebaited randomly and the trial rerun until a total of 10 digs was recorded. If the subject did not reach 10 digs, this habituation procedure was repeated the following day. No subject required more than 2 d of habituation.
Testing paradigm.
The reversal learning digging task was used previously in Tg2576 mice (Zhuo et al., 2007, 2008). The reversal learning testing was performed with olfactory discriminations as this has proven, in our hands, to be the more difficult of discriminations compared to using media as the stimulus. One hour before testing, BQCA or vehicle was administered subcutaneously to the subjects at 30 mg/kg. The first four trials of the discriminations (exploratory trials) allowed the subject to explore both pots to find the reward. If the reward was found, a correct response was recorded and the subject recovered to the beaker. If digging first occurred in the non-reward pot, an error was recorded and the subject was allowed to search the other pot for the reward. If the subject remained motionless for 1 min, a “no dig” was recorded, the trial discontinued and the next trial started. In the subsequent trials after the initial four, a correct dig was recorded when the subject retrieved the reward and an incorrect dig “error” was recorded if the subject dug vigorously in the incorrect pot. Vigorous digging was defined as the subject having its head and shoulders within the pot and using its paws to vigorously move the media. The subject was limited to 40 trials to reach criteria. No subjects were eliminated due to the 40 trial limit. Analysis was based on the total number of trials the subject took to reach the criteria of six correct trials in a row including the first four exploratory trials but not counting correct trials within the exploratory trials as part of the six correct trials.
Media and odor for the compound discriminations were established in pairs to reduce the degrees of freedom (see supplemental Table 2, available at www.jneurosci.org as supplemental material). For example, in a simple discrimination (SD) using odor as the relevant dimension, aniseed odor would always be on one pot while benzoin odor would always be on the other pot. Alpha-Dri medium was always used as the irrelevant dimension in both pots in the simple discriminations. In the compound discrimination (CD), two separate pairs of pots were used and presented to the subjects in pairs in a random order. For example, the first pair of pots would have the exemplar, jamaroosa root, paired with soft sorbent in one pot and myrrh paired with soft snow in the other. The second pair of pots would have the exemplar, jamaroosa root, paired with soft snow in one pot and myrrh paired with soft sorbents in the other.
Two shaping SD's were run the first day of testing. Each subject was allowed one discrimination with medium as the relevant dimension and one discrimination with odor as the relevant dimension. Once a pair of dimensions had been used during the shaping SD's, they were not presented to the subjects again in the testing paradigm. (For an example of experimental design, see supplemental Table 3, available at www.jneurosci.org as supplemental material).
On the second day of testing, a simple odor discrimination was performed first. Upon reaching criteria for the SD, a simple discrimination reversal (SDR) was performed so that the pot with the odor that was not rewarded now became the rewarded pot. Following that, a compound discrimination was performed. An irrelevant dimension (different media) was added at this point that had no predictive power on the location of the reward. Upon reaching criteria on the compound discrimination, a compound discrimination reversal (CDR) was performed so that the pot with the odor that was not rewarded now became the pot with the odor with the reward. Data for each phase of the digging test (e.g., simple discrimination, compound discrimination) were analyzed using a χ2 analysis and subsequent odds ratio calculation to identify the relative likelihood of choice errors on the discrimination tests in the presence of BQCA within the two groups. Each task phase was analyzed independently. Since most subjects had excellent performance with no errors, only those subjects making 1 or more errors were selected for analysis.
Pharmacokinetics and brain/plasma exposure profiling.
Male Sprague Dawley rats (Harlan, Indianapolis) weighing 225–250 g, were injected intraperitoneally with the micro-suspension (containing 10% Tween 80) of BQCA at the dose of 10 mg/kg. The blood and whole brain tissue samples were collected at 0.5, 1, 2, 4 and 8 h. Blood samples were collected through cardiac puncture in EDTA vacutainer tubes. The plasma was separated by centrifugation and stored at −80°C until analysis. The animals were decapitated and the whole brain tissue were removed and immediately frozen on dry ice.
Brain tissue was weighed and homogenized in 5 ml of ice-cold phosphate buffered saline using a Sonic Dismembrator Model 100 (Fisher Scientific) at maximal speed for 2 min. Homogenate samples (500 μl) were treated with 2.0 ml of an ice-cold solution of acetonitrile containing 0.1% formic acid and VU178 (internal standard), 100 ng/ml, and vortexed for 1 min. Plasma samples (100 μl) were combined with 500 μl of ice-cold solution of the internal standard (100 ng/ml) in acetonitrile with 0.1% formic acid and vortexed. The samples were centrifuged at 14,000 rpm for 5 min using a Spectrafuge 16M Microcentrifuge (Labnet). The supernatants were evaporated and the residues were reconstituted in 100 μl of 80:20 acetonitrile/water, filtered through 0.2 μm nylon filter and injected onto LC-MS-MS.
LC separation was performed on Waters Acquity UPLC BEH C18 (1.7 μm 1.0 × 50 mm) column at a flow rate of 0.6 ml/min flow rate. The gradient started with 80% solvent A (0.1% formic acid in water) and 20% solvent B (0.1% formic acid in acetonitrile), and held for 1 min. The mobile phase composition was increased to 100% B by 2 min and held for 1 min, before it was returned to the initial conditions. The samples were analyzed in a run time of 6 min. Mass spectrometry was performed using a ThermoFinnigan TSQ Quantum Ultra (Thermo Scientific) mass spectrometer in positive ion mode. Xcalibur (version 2.0) software was used for instrument control and data collection. The ESI source was fitted with a stainless steel capillary (100 μm, i.d.). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion-transfer capillary tube temperature was 300°C. The spray voltage, tube lens voltage, pressure of sheath gas, and auxiliary gas were optimized to achieve maximal response using the test compounds infused with the mobile phase A (50%) and B (50%) at a flow rate of 0.6 ml/min. Collision-induced dissociation (CID) was performed on the test compounds and internal standards under 1.0 mTorr of argon. Selected reaction monitoring (SRM) was performed using the transitions from m/z 310 to 121 at 17 eV for BQCA and m/z 310 to 223 at 25 eV for the internal standard. The unknown concentrations were determined against calibration curves constructed by spiking known amounts of test compounds into the blank brain homogenate and plasma samples. A linear response was achieved from 10 ng/ml to 2 μg/ml in plasma and 10 ng/ml to 1 μg/ml in brain homogenates. PK parameters were calculated by non-compartmental analysis of individual concentration-time data using WinNonLin, version 5.2.1 (Pharsight).
Results
A panel of 21 compounds related to BQCA have a range of activities as positive allosteric modulators at the rat M1 mAChR
Ma et al. (2008) recently presented a preliminary report in which they found that BQCA is a selective PAM of the human M1 muscarinic receptor (hM1). However, GPCR PAMs can display species specificity, and the effects of BQCA were not evaluated on the rat M1 receptor (rM1). Thus, to determine whether BQCA and related compounds have properties needed for use in rodent studies, we synthesized BQCA (see Note added in proof) and a panel of 20 structurally related analogs to identify compounds that can act as selective PAMs for the rM1 receptor. Effects of BQCA and related compounds were evaluated by measuring effects on calcium mobilization elicited by a submaximal concentration (EC20) of ACh (Fig. 1). Libraries I, II, and III each consisted of seven compounds possessing the same N-benzyl substitutions based on either an 8-fluorinated quinolone carboxylic acid (Ia-Ig), a quinolone carboxylic acid (IIa-IIg, including BQCA), or a 5,8-difluorinated quinolone carboxylic acid (IIIa-IIIg) template, respectively (Fig. 1a). The activity of test compounds was initially assessed by incubating CHO-K1 cells stably expressing the rM1 receptor with fixed concentrations of each compound at 10, 1, or 0.3 μm (Fig. 1b–d) for 1.5 min before the addition of an EC20 concentration of ACh. From the panel, four compounds that exhibited robust potentiator activity at 0.3 μm were selected for further evaluation based on their structural diversity. As can be seen in the representative trace, 1 μm BQCA has no effect when added alone, but greatly enhances the response to an EC20 concentration of ACh when compared to vehicle. A maximal response to ACh is also shown for comparison (Fig. 1e). To determine the potency of each of these compounds, full concentration response curves (CRCs) were generated by preincubating rM1 CHO-K1 cells with increasing concentrations of test compound, followed 1.5 min later by the addition of an EC20 concentration of ACh (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material). All four compounds had similar potencies at the rM1 receptor, with EC50 values in the 200–400 nm range. As a second measure of their ability to potentiate the rM1 receptor-mediated calcium response to ACh, rM1 receptor expressing CHO-K1 cells were preincubated with a fixed concentration (3 μm) of the test compound (or vehicle) and then stimulated with increasing concentrations of ACh to generate a series of ACh CRCs. Each of the four test compounds elicited a robust potentiation of the ACh response, as manifest by a leftward shift in the ACh CRC (9.5–18.6-fold shift) (supplemental Fig S1b, available at www.jneurosci.org as supplemental material).
Figure 1.
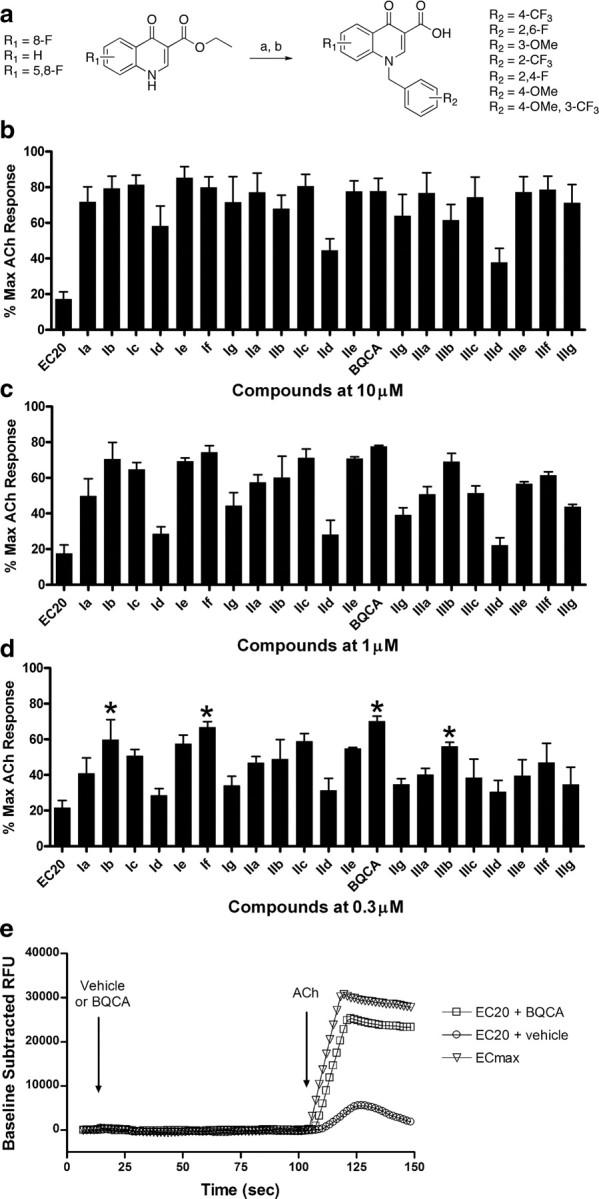
Twenty-one M1 receptor PAMs were synthesized and evaluated at the rM1 mAChR for their ability to potentiate an EC20 concentration of ACh. a, Synthesis: K2CO3, KI, R-Br, DMF, 24 h at room temperature (a), LiOH, 9:1 THF:H2O 10 min 120°C μwave (b). Calcium mobilization was measured using a Flexstation II, as described in Materials and Methods. b–d, Test compounds were evaluated at fixed concentrations of 10 μm (b), 1 μm (c), or 0.3 μm (d) in the presence of an EC20 concentration of ACh. Four compounds, denoted by an asterisk (*), were selected for further evaluation based on their structural diversity and ability to potentiate an EC20 concentration of ACh at 0.3 μm. Data were normalized as a percentage of the maximal response to 10 μm ACh and represent the mean ± SEM of 3 independent experiments. e, A representative calcium trace from one experiment shows the effect of 1 μm BQCA on the rM1 receptor response to an EC20 concentration of ACh; the response to an EC20 and ECmax concentration of ACh in the presence of vehicle are also shown for comparison.
BQCA is a potent and selective positive allosteric modulator of the rat M1 receptor in vitro
Of the molecules tested in this panel screen, BQCA was among the most potent and efficacious at potentiating rM1 receptor-mediated responses. This is consistent with its activity at the human receptor (Ma et al., 2008). Based on this and its favorable physiochemical properties, we chose to pursue studies focusing exclusively on BQCA. First, we evaluated the potency of BQCA as a positive allosteric modulator of the rM1 receptor by measuring calcium mobilization in CHO-K1 cells stably expressing this receptor. Cells were incubated with increasing concentrations of BQCA for 1.5 min before the addition of an EC20 concentration of ACh, yielding a concentration response curve for BQCA with an EC50 value of 267 ± 31 nm (Fig. 2a). We next determined the effect of increasing fixed concentrations of BQCA on the ACh CRC. rM1 CHO-K1 cells were preincubated with a fixed concentration (0.3, 1, and 3 μm) of BQCA and subsequently stimulated with increasing concentrations of ACh. BQCA induced a dose-dependent leftward shift in the ACh CRC with a maximal shift of 21-fold observed with 3 μm BQCA (Fig. 2b).
Figure 2.

BQCA is a potent positive allosteric modulator of the rM1 receptor in vitro. a, Potency of BQCA was evaluated at the rM1 receptor by measuring calcium mobilization in CHO-K1 cells stably expressing the rM1 receptor. Increasing concentrations of test compound was added to cells, followed 1.5 min later by addition of an EC20 concentration of ACh. BQCA robustly potentiated the response to ACh with an EC50 value of 267 ± 31 nm. b, The ability of BQCA to potentiate the response of the rM1 receptor to ACh is also manifest by a dose-dependent leftward shift in the ACh CRC. ACh alone stimulated calcium mobilization with an EC50 value of 2.42 ± 0.337 nm (■). In the presence of increasing fixed concentrations of BQCA (0.3–3 μm), a robust leftward shift in the ACh CRC was induced, resulting in the following EC50 values (fold shift in the ACh curve is shown in parentheses); 0.3 μm (♦) = 0.762 ± 0.56 nm (3.3×), 1.0 μm (▾) = 0.221 ± 0.079 nm (12×), 3 μm (▴) = 0.123 ± 0.026 nm (21×). Data were normalized as a percentage of the maximal response to 10 μm ACh and represent the mean ± SEM of 3–4 independent experiments. BQCA does not compete for orthosteric antagonist binding and it induces a robust leftward shift in ACh affinity at the rM1 receptor. c, At concentrations up to 10 μm, BQCA (■) did not displace the orthosteric radioligand, [3H]-NMS (0.1 nm) in competition binding studies. However, the orthosteric antagonist, atropine (▴), potently inhibited [3H]-NMS binding with a Ki of 1.35 ± 0.022 nm. d, In the presence of vehicle alone, an increasing concentration of ACh displaces [3H]-NMS (0.1 nm) binding with a Ki of 1700 ± 96.4 nm (■). In the presence of increasing fixed concentrations (0.3–3.0 μm) of BQCA, the potency of ACh to displace [3H]-NMS binding is shifted leftward, yielding Ki values of 348 ± 43.4 nm (0.3 μm, ▴), 163 ± 22.9 nm (1.0 μm, ▾), and 56.1 ± 4.99 nm (3.0 μm, •), which represent fivefold, 10.6-fold and 30.6-fold shifts in ACh potency, respectively. Data represent the mean ± SEM of 3 independent experiments, performed in duplicate.
We previously reported that novel selective PAMs of the rM4 receptor, exemplified by VU10010 and VU152100, have no detectable affinity at the orthosteric ACh binding site of the rM4 receptor but allosterically increase affinity of ACh for the rM4 receptor (Brady et al., 2008; Shirey et al., 2008). To determine whether BQCA shares this property with the rM4 PAMs, we assessed the ability of this compound to compete for binding with the orthosteric radioligand, [3H]-NMS (0.1 nm) to the orthosteric site using membranes prepared from cells expressing the rM1 receptor. BQCA had little effect on [3H]-NMS binding, with no displacement of radioligand observed at concentrations up to 10 μm (Fig. 2c). In contrast, the orthosteric antagonist, atropine, potently inhibited [3H]-NMS binding with a Ki value of 1.35 ± 0.022 nm (Fig. 2c). The effect of BQCA on the affinity of ACh for the rM1 receptor was also evaluated by assessing the ability of increasing concentrations of ACh to displace [3H]-NMS (0.1 nm) binding in the absence or presence of fixed concentrations of the M1 receptor potentiator (0.3, 1.0, and 3.0 μm). BQCA induced a robust concentration-dependent leftward shift in the concentration response curve of ACh-induced displacement of [3H]-NMS binding to the rM1 receptor, with a 30-fold shift observed at the highest concentration tested (3.0 μm). This shift reveals that BQCA induces a reduction in the ACh Ki from 1700 ± 96.4 nm (vehicle) to 348 ± 43.4 nm (0.3 μm), 163 ± 22.9 nm (1.0 μm), and 56.1 ± 4.99 nm (3.0 μm), respectively (Fig. 2d). Together, these data strongly suggest that BQCA acts at a site on the M1 receptor that is distinct from the orthosteric binding site and that it may enhance M1 receptor activation by increasing the affinity for ACh.
BQCA is functionally selective for the M1 mAChR subtype
One of the primary difficulties in developing novel selective ligands for muscarinic receptors has been the failure to identify compounds that can distinguish between the highly conserved orthosteric binding site shared by the five members of this GPCR subfamily. Development of ligands that bind to allosteric sites, both potentiators and direct acting agonists, has proven to be a practical way to circumvent this issue (for review, see Conn et al., 2009a,b). Thus, it was important to determine whether BQCA is selective for the M1 mAChR relative to other mAChR subtypes. We evaluated the effect of BQCA on the ACh CRC in calcium mobilization assays at each of the other mAChR subtypes. As shown in Figure 2b, preincubation of rM1 receptor-expressing CHO-K1 cells with 3 μm BQCA results in a robust leftward shift in the CRC for ACh. However, at this same concentration, BQCA had no effect on the ACh concentration response curves generated in CHO-K1 cells stably expressing the hM2, hM3, rM4, or hM5 receptors (Fig. 3a–d). To further assess selectivity of BQCA for the M1 receptor relative to other class A GPCR targets that may also harbor similar allosteric sites, we took advantage of the GPCR Profiler service offered by Millipore to determine the effect of this compound on the functional response of 15 other closely related GPCRs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). A two-addition protocol afforded the ability to detect potential agonist, potentiator, and antagonist activity of BQCA at these other GPCR subtypes. When applied alone in the first addition, BQCA (12.5 μm) had no agonist activity at any receptor tested (data not shown). However, consistent with our internal studies, BQCA induced robust potentiation at the hM1 receptor, but had no activity in this assay at the hM4 receptor. Moreover, BQCA had no effect at any of the other GPCRs tested (supplemental Fig. 2a–p, available at www.jneurosci.org as supplemental material). This included a lack of PAM activity or antagonist activity (either allosteric or orthosteric) at any of these other GPCRs, which would have resulted in a rightward shift in the concentration response curve. Together, these data suggest that BQCA is highly selective for the M1 mAChR subtype and has no detectable activity at closely related family A GPCRs that were tested.
Figure 3.
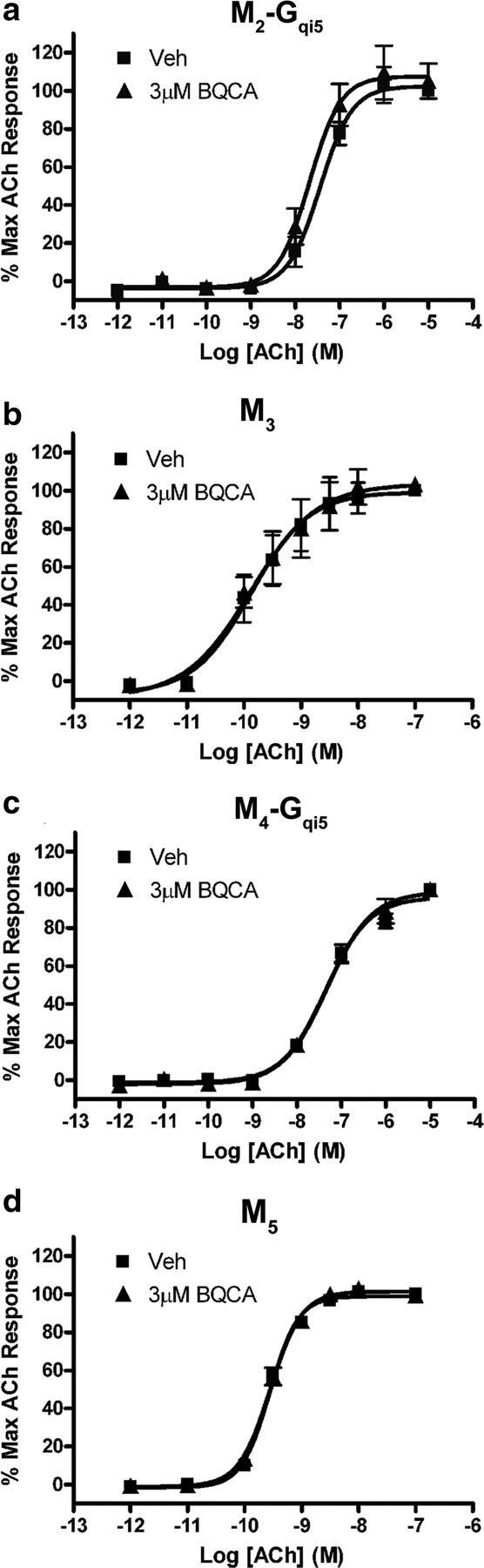
The presence of BQCA has no effect on the ACh concentration-response curve at any other mAChR subtype. a–d, No shift in the ACh CRC was observed in the presence of 3 μm BQCA in CHO K1 cells stably expressing M2-Gqi5 (a), M3 (b), M4-Gqi5 (c), or M5 (d) receptors. Calcium mobilization was measured in response to increasing concentrations of ACh following preincubation of cells with either vehicle (■) or 3 μm BQCA (▴), as described in Materials and Methods. Data represent the mean ± SEM of three independent experiments.
Activation of the M1 receptor induces an inward current in rat mPFC layer V pyramidal cells and this effect is potentiated by BQCA
Prefrontal cortical function is required for higher executive function, memory storage and retrieval, and cognition (Miller and Cohen, 2001). Recent studies suggest that M1 receptor signaling may play an important role in activation of the prefrontal cortex by lower brain regions (Anagnostaras et al., 2003). Based on this, it was postulated that activation of the M1 receptor could increase excitability of mPFC pyramidal cells or increase excitatory synaptic drive to these neurons. To examine the effects of M1 receptor activation on mPFC pyramidal cells, layer V pyramidal neurons were visually identified and membrane currents measured using patch-clamp recordings in acute coronal slices. Cell type was confirmed by examining firing properties upon depolarizing current injection. Typical resting membrane potentials of these pyramidal neurons were −55 to −65 mV under the conditions used. Holding current was measured in cells voltage-clamped at −70 mV during baseline recording, drug application, and wash. Bath application of CCh induced a robust, concentration-dependent inward current as shown in Figure 4 (10 μm CCh, 16.55 ± 1.93 pA, n = 4; 100 μm CCh, 53.14 ± 5.92 pA, n = 4). Although this CCh–induced inward current is in agreement with previously reported studies (Krnjeviæ, 2004; Carr and Surmeier, 2007), it is not known whether this response is mediated by the M1 receptor or another mAChR subtype. However, previous studies suggest that the M1 receptor may not be responsible for induction of inward currents in hippocampal CA1 pyramidal cells (Rouse et al., 2000). Before evaluating the effect of BQCA on this current, we determined the effect of VU0255035, the first highly selective M1 receptor antagonist that was recently reported (Sheffler et al., 2009), on the CCh-induced inward current. The M1 receptor antagonist, VU0255035 (10 μm), had no effect on holding current alone but significantly blocked the current induced by 100 μm CCh (p = 0.0202, unpaired t test). These results suggest that the CCh-induced inward current in rat mPFC layer V pyramidal cells is largely mediated by activation of the M1 receptor. If this is the case, we would predict that the M1 receptor PAM BQCA should potentiate the CCh-induced inward current. Interestingly, BQCA induced a small change in holding current when applied alone (21.54 ± 2.42 pA, n = 5). In addition, BQCA significantly increased the inward current induced by 10 μm CCh (55.07 ± 6.28 pA upon coapplication, n = 5, compared to 10 μm CCh alone, p = 0.0210). These data are consistent with the hypothesis that activation of the M1 receptor induces an inward current in mPFC layer V pyramidal cells and that M1 receptor PAMs can induce a marked potentiation of this response.
Figure 4.

CCh-induced inward current in mPFC layer V pyramidal cells is reduced by M1 receptor antagonist VU0255035 and potentiated by BQCA. a, Sample traces from single experiments showing the change in holding currents upon drug treatments. b, Activation of mAChRs by CCh causes a dose-dependent inward current as measured in voltage-clamp mode as a change in holding current (16.55 ± 1.93 pA in the presence of 10 μm CCh, n = 4; 53.14 ± 5.92 pA with 100 μm CCh, n = 4). M1 receptor PAM BQCA also caused an inward current when applied alone, and significantly increased the effect of 10 μm CCh (21.54 ± 2.42 pA change with 10 μm BQCA, n = 5; 55.07 ± 6.28 pA when BQCA was coapplied with 10 μm CCh, n = 5, as compared to 10 μm CCh alone, p = 0.0210). The effect of 100 μm CCh was also significantly inhibited by 10 μm M1 receptor antagonist VU0255035 (11.78 ± 1.90 pA in the presence of antagonist, n = 3, compared to 100 μm CCh alone, p = 0.0202). Sample traces represent experiments from single cells, and bars show duration of drug exposure. All changes in holding current were compared to baseline control and are represented as mean ± SEM. Asterisks indicate significant differences from control (*p < 0.05; unpaired t test).
mAChR activation increases mPFC spontaneous EPSC amplitude and frequency
It is also possible that activation of the M1 receptor could increase activity of excitatory synaptic inputs to the mPFC and that this could contribute to the postulated role of this receptor in increasing PFC activity from in vivo studies in M1 receptor knock-out mice (Anagnostaras et al., 2003). Thus, we determined the effect of mAChR activation on spontaneous EPSCs (sEPSCs) in mPFC pyramidal cells. Application of CCh caused a dramatic, concentration-dependent increase in the frequency of spontaneous EPSCs; the effect of a maximal concentration of 100 μm CCh on one representative cell is shown in Figure 5a. Cumulative probability plots of amplitude and interevent interval from the same cell demonstrate significant shifts in the presence of 100 μm CCh that are reversible upon wash (Fig. 5b). The concentration-response relationships for CCh effects on sEPSC amplitude and frequency are shown in Figure 5c. A concentration of 10 μm CCh was without effect (97.5 ± 4.4% control for amplitude, 90.1 ± 12.4% control for frequency, n = 6); however, 30 and 100 μm CCh increased both amplitude and frequency (30 μm amplitude, 108.3 ± 3.9%, frequency, 455.0 ± 101.9% control, n = 7; 100 μm amplitude, 154.3 ± 46.2%, 887.6 ± 268.5% control for frequency, n = 5). The effects of 30 μm CCh on both amplitude and frequency were completely blocked by the non-selective muscarinic antagonist, atropine (5 μm, 102.9 ± 7.8% and 104.4 ± 19.6% control, respectively, n = 3) indicating that the effect of CCh was due to activation of mAChRs.
Figure 5.

mAChR activation increases mPFC spontaneous EPSC amplitude and frequency. a, Representative traces from one cell showing the effect of a maximal concentration of 100 μm CCh. b, Change in cumulative probability plots of sEPSC amplitude (top panel) and interevent interval (bottom panel) upon addition and wash of 100 μm CCh from one representative cell. c, Averaged amplitude and frequency show that CCh treatment induces a dose-dependent increase in both sEPSC amplitude and frequency which is reversible upon washout and is inhibited by 5 μm atropine (Amplitudes: 10 μm CCh, 97.5 ± 4.4%, n = 6, p = 0.2727; 30 μm CCh, 108.3 ± 3.9%, n = 7, p = 0.0498; 100 μm CCh, 154.3 ± 46.2%, n = 5, p = 0.2393; 5 μm atropine/30 μm CCh, 102.9 ± 7.8% of control, n = 3, p = 0.5365. Thirty micromolars μm CCh vs 5 μm atropine/30 μm CCh, p = 0.4478. Frequencies: 10 μm CCh, 90.1 ± 12.4%, p = 0.9364; 30 μm CCh, 455.0 ± 101.9%, p = 0.0139; 100 μm CCh, 887.6 ± 268.5%, p = 0.0314; 5 μm atropine/30 μm CCh, 104.4 ± 19.6% of control, p = 0.6260. Thirty micromolars CCh vs 5 μm atropine/30 μm CCh, p = 0.0458). All changes in amplitude and frequency were compared to baseline control and are represented as mean ± SEM. Asterisks indicate significant differences from control or between conditions (*p < 0.05; paired or unpaired t test).
The effect of CCh on sEPSC amplitude and frequency is inhibited by VU0255035
To further evaluate the role of the M1 receptor in the effect of CCh on sEPSCs, slices were treated with the selective M1 receptor antagonist VU0255035 (5 μm) for 2 min before addition of 30 μm CCh (Fig. 6a,b). VU0255035 alone decreased sEPSC amplitude (92.9 ± 3.4% control, n = 11), and amplitude was further decreased by coapplication with 30 μm CCh (87.1 ± 3.5% control) (Fig. 6c). Antagonist alone had no effect of sEPSC frequency (114.1 ± 25.8% control) but caused a significant decrease in frequency in the presence of CCh (62.5 ± 10.1% control) (Fig. 6c). These data suggest that the CCh-induced increase in sEPSC amplitude and frequency is mediated by activation of the M1 receptor. The reversal of the CCh effect on sEPSC frequency in the presence of the M1 receptor antagonist suggests that blocking the M1 receptor unmasks an inhibitory action of CCh that may be mediated by another mAChR subtype, possibly M2 or M4 receptors.
Figure 6.

The effect of CCh on sEPSC amplitude and frequency is inhibited by VU0255035. a, Sample traces from one cell showing lack of effect of 30 μm CCh in the presence of the M1 receptor antagonist VU0255035. b, Cumulative probability plots of sEPSC amplitude and frequency from the same cell. c, Averaged (n = 11) amplitude and frequency. The increase in sEPSC amplitude and frequency induced by 30 μm CCh was reversed by 5 μm VU0255035 (amplitudes: 5 μm VU0255035, 92.9 ± 3.4%, p = 0.0338; 5 μm VU0255035/30 μm CCh, 87.1 ± 3.5%, p = 0.0026. Antagonist alone vs. with CCh, p = 0.0350. Frequencies: 5 μm VU0255035, 114.1 ± 25.8%, p = 0.4543; 5 μm VU0255035/30 μm CCh 62.5 ± 10.1% of control, p = 0.0357. Antagonist alone vs. with 30 μm CCh, p = 0.0161). All changes in amplitude and frequency were compared to baseline control and are represented as mean ± SEM. Asterisks indicate significant differences from control or between drug conditions (*p < 0.05; **p < 0.01; ***p < 0.0001; paired or unpaired t test).
BQCA increases sEPSCs and potentiates the effect of a subthreshold concentration of CCh on sEPSC frequency
Our results thus far suggest that the M1 receptor is responsible for the CCh-induced increase in sEPSC frequency; therefore, this response should be potentiated by BQCA. To test this hypothesis, slices were treated with BQCA alone for 2 min (10 μm) before addition of 10 μm CCh. Sample traces and cumulative probability plots are shown in Figure 7, a and b. Treatment with BQCA alone did not significantly affect sEPSC amplitude, but increased the frequency of events (108.3 ± 6.6% control, 277.0 ± 97.7% control, respectively, n = 11) (Fig. 7c). Coapplication of BQCA and 10 μm CCh induced a further increase in sEPSC frequency (994.5 ± 301.5% control), which differed significantly from the effect of 10 μm CCh (p = 0.0045, unpaired t test) (see Fig. 5c) or BQCA alone (p = 0.0116, paired t test).
Figure 7.

BQCA increases sEPSCs and potentiates a subthreshold concentration of CCh to increase sEPSC frequency. a, Representative traces from one cell showing the effect of 10 μm BQCA alone and in the presence of 10 μm CCh. This concentration of CCh had no significant effect on sEPSC amplitude or frequency when applied alone. b, Change in cumulative probability plots from one representative cell of sEPSC amplitude (top panel) and interevent interval (bottom panel) upon addition of 10 μm BQCA with and without 10 μm CCh. c, Averaged (n = 11) amplitude and frequency. Neither drug condition elicited a significant effect on sEPSC amplitude (10 μm BQCA, 108.3 ± 6.6%, p = 0.5013; 10 μm BQCA/10 μm CCh, 136.0 ± 15.3% control, p = 0.0524) but significantly increased frequency (10 μm BQCA, 277.0 ± 97.7%, p = 0.0229; 10 μm BQCA/10 μm CCh, 994.5 ± 301.5% control, p = 0.0045; the effect of combined drug treatment vs 10 μm BQCA alone was significantly different, p = 0.0116). The effect of 10 μm CCh on frequency in the absence and presence of BQCA was also significantly different (p = 0.0177). All changes in amplitude and frequency were compared to baseline control and are represented as mean ± SEM. Asterisks indicate significant differences from control or between drug conditions (*p < 0.05; **p < 0.01; paired or unpaired t test).
BQCA has no effect on sEPSCs in slices from M1 receptor KO mice
To confirm that the actions of BQCA were mediated by M1 receptor activation, recordings of sEPSCs in mPFC layer V neurons were made using slices from mice lacking the M1 receptor and compared to wild-type (WT) controls. Consistent with our studies in rat slices, CCh caused a concentration-dependent increase in sEPSC amplitude and frequency in WT mice (Fig. 8a, left panel; d, black bars). While 3 μm CCh had no effect on amplitude or frequency, 30 μm CCh significantly increased both parameters (Amplitudes: 3 μm CCh, 102.6 ± 11.7% of control, n = 3; 30 μm CCh, 143.1 ± 22.0%, n = 5. Frequencies: 3 μm CCh, 83.2 ± 47.1%, 30 μm CCh, 398.3 ± 56.2%). In contrast to effects in rat slices, BQCA had no effect alone in WT slices (10 μm BQCA, 97.3 ± 11.3% control amplitude, 99.8 ± 11.3% control frequency, n = 5), but induced robust increases in both amplitude and frequency when coapplied with 3 μm CCh (137.2 ± 16.7% control amplitude, 500.5 ± 212.3% control frequency). In slices from M1 receptor KO mice, the response to CCh was markedly reduced. In M1 KO mice, CCh decreased sEPSC amplitude at both concentrations tested and induced a more modest increase in sEPSC frequency that did not achieve statistical significance (Amplitudes: 3 μm CCh, 79.4 ± 14.9%, n = 4; 30 μm CCh, 80.7 ± 5.2%, n = 4. Frequencies: 3 μm CCh, 186.3 ± 187.4%, 30 μm CCh, 271.7 ± 310.4%). Importantly, the response to BQCA was completely absent in slices from M1 receptor KO mice. Thus, BQCA had no effect when applied alone or when coapplied with 3 μm CCh (Amplitudes: 10 μm BQCA, 96.9 ± 10.6%; BQCA/CCh, 84.5 ± 25.3%, n = 5. Frequencies: 10 μm BQCA, 101.3 ± 26.1%; BQCA/CCh, 86.6 ± 17.3%). Responses to coapplication of BQCA and CCh differed significantly between WT and M1 receptor KO for both sEPSC amplitude and frequency (p = 0.0046 for amplitude; p = 0.0025 for frequency, unpaired t test). These results confirmed that the actions of BQCA are due to its action at M1 receptors.
Figure 8.
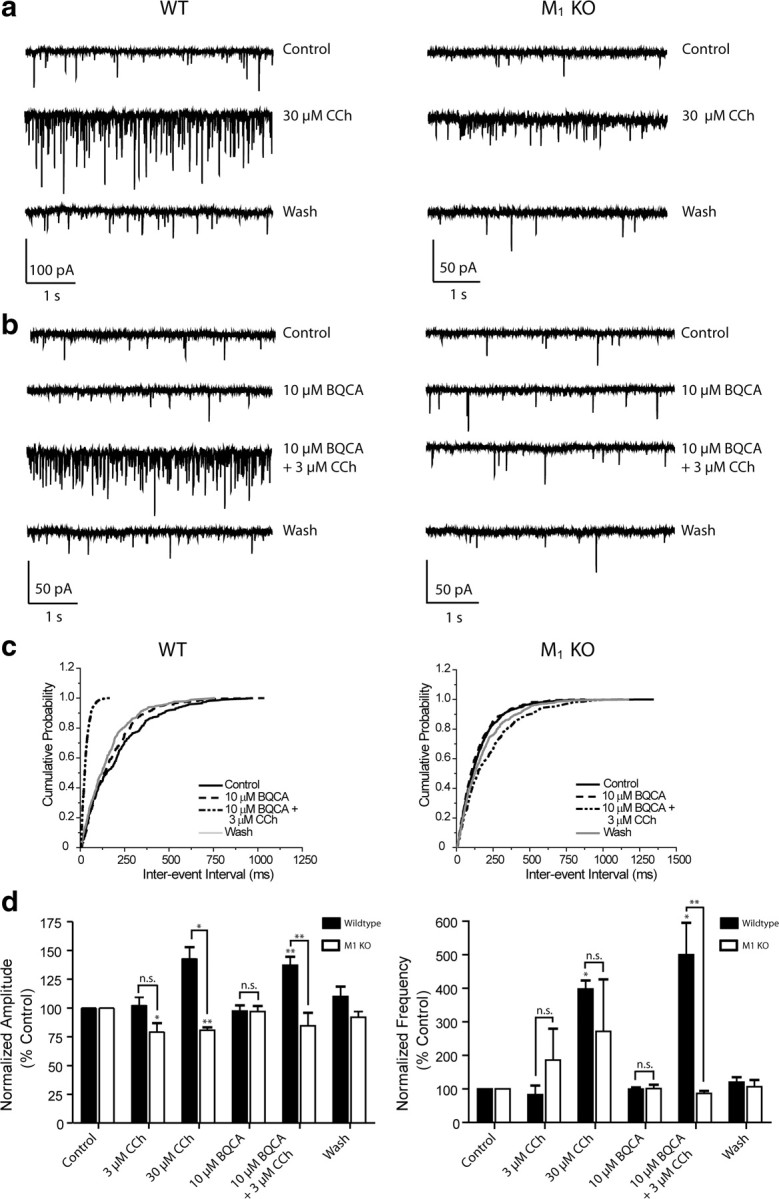
BQCA has no effect and does not potentiate the CCh effect on sEPSCs in M1 receptor KO mice. a, Sample traces from individual cells in slices made from wild-type (left panels) and M1 receptor KO mice (right panels) showing the robust effects of 30 μm CCh on both sEPSC amplitude in frequency compared to the milder effect in the M1 receptor KO slice (top panels). b, Bottom panels illustrate the lack of effect of 10 μm BQCA in both the WT and M1 receptor KO and contrast the increase in amplitude and frequency with the addition of BQCA and 10 μm CCh to the lack of effect in the M1 receptor KO. c, Cumulative probability plots of the interevent intervals from the two cells shown in bottom panels above. d, Averaged amplitude and frequency of sEPSCs measured in wild-type (black bars: 3 μm CCh, n = 3; 30 μm CCh, n = 5; 10 μm BQCA and 10 μm BQCA + 3 μm CCh, n = 5) and M1 receptor KO slices (white bars: 3 μm CCh, n = 4; 30 μm CCh, n = 4; 10 μm BQCA and 10 μm BQCA + 3 μm CCh, n = 5). In wild-type slices, 3 μm CCh had no significant effect on amplitude or frequency (102.6 ± 11.7%, p = 0.6381 and 83.2 ± 47.1% of control, p = 0.4423, respectively) whereas 30 μm CCh increased both (143.1 ± 22.0%, p = 0.0306 for amplitude, 398.3 ± 56.2%, p = 0.0342 for frequency). BQCA had no effect on amplitude or frequency but potentiated the response to 3 μm CCh (Amplitudes: 10 μm BQCA, 97.3 ± 11.3%, p = 0.7642 compared to control; 10 μm BQCA/3 μm CCh, 137.2 ± 16.7%, p = 0.0052 compared to control. Frequencies: 10 μm BQCA, 99.8 ± 11.3%, p = 0.7261 compared to control; 10 μm BQCA/3 μm CCh, 500.5 ± 212.3%, p = 0.0209 compared to control. In M1 receptor KO slices, 3 μm CCh decreased amplitude but had no significant effect on frequency (79.4 ± 14.9%, p = 0.0490 and 186.3 ± 187.4% of control, p = 0.7656, respectively). Thirty micromolars CCh also significantly decreased amplitude and increased frequency although the effect on frequency was less dramatic than that seen in WT controls (80.7 ± 5.2%, p = 0.0092 for amplitude, 271.7 ± 310.4%, p = 0.6010 for frequency). While the difference between the 30 μm CCh effect on amplitude was significant between genotypes (p = 0.0229), the effect on frequency was not (p = 0.4756). BQCA had no effect alone in KO slices (amplitude, 96.9 ± 10.6%, p = 0.4925; frequency, 101.3 ± 26.1%, p = 0.7286) and there was no difference in this lack of effect between genotypes (amplitude, p = 0.9596; frequency, p = 0.9133). When applied with 3 μm CCh, BQCA had no significant effect on amplitude or frequency (amplitude, 84.5 ± 25.3%, p = 0.3383; frequency, 86.6 ± 17.3%, p = 0.1388 compared to KO control), and both effects were significantly different those in WT slices (p = 0.0046 for amplitude, p = 0.0025 for frequency). All changes in amplitude and frequency were compared to baseline control and are represented as mean ± SEM. Asterisks indicate significant differences from control or between drug conditions (*p < 0.05; **p < 0.01; paired or unpaired t test).
CCh and BQCA have no effect on miniature EPSC amplitude and frequency in rat mPFC layer V pyramidal cells
To determine whether the actions of CCh and BQCA require action potential-dependent EPSCs, we determined the effects of these compounds on miniature EPSCs (mEPSCs). mEPSCs were recorded in voltage-clamp mode at a holding potential of −70 mV and in the presence of 1 μm tetrodotoxin (TTX) to block voltage-gated sodium channels. At this concentration, TTX completely eliminates action potential firing and action potential-mediated synaptic activity [data not shown; see also Morisset and Urban (2001)]. Under these conditions, neither CCh nor BQCA elicited any effect on mEPSC amplitude or frequency. Sample traces from one cell in a slice to which 100 μm CCh was applied in the presence of TTX show a clear lack of effect (supplemental Fig. S3a, available at www.jneurosci.org as supplemental material). Cumulative probability plots of amplitude and frequency during control, CCh treatment, and wash from the same cell overlap (supplemental Fig. S3b, available at www.jneurosci.org as supplemental material). Pooled amplitude and frequency for all drug treatments are quantified in supplemental Fig. S3c, available at www.jneurosci.org as supplemental material (10 μm CCh, n = 5; 100 μm CCh, n = 4; 10 μm BQCA with and without 10 μm CCh, n = 4). The only significant effect was that of 10 μm CCh, which slightly decreased mEPSC amplitude (88.6 ± 3.8% control). The effects of M1 receptor activation on spontaneous EPSCs thus require action potential firing.
BQCA has excellent brain penetration and increases the firing rate of mPFC neurons in vivo in rats
The studies outlined above suggest that BQCA could be an excellent tool for probing M1 receptor function. Furthermore, based on these and previous studies, it is possible that BQCA could enhance mPFC activity and enhance PFC-dependent cognitive function. However, before using BQCA for in vivo studies, it was critical to determine whether this compound had a pharmacokinetic (PK) profile suitable from systemic dosing and whether it crossed the blood brain barrier. Thus, we performed a PK analysis of BQCA after systemic dosing. BQCA was measured at multiple time points in both plasma and brain after intraperitoneal (i.p.) injection in rats (supplemental Fig. S4, Table 1, available at www.jneurosci.org as supplemental material). BQCA is slowly but very significantly absorbed into systemic circulation with maximum concentration (∼10 μg/ml) being achieved 2 h after i.p. administration. The compound is rapidly taken up into the brain and achieves a maximal brain concentration between 30 min and 1 h after dosing. Furthermore the brain concentration is maintained at a stable level for ∼4 h. While the brain concentrations are significantly lower when compared to plasma concentrations (supplemental Fig. S4, Table 1, available at www.jneurosci.org as supplemental material), this provides an acceptable PK profile and brain penetration to allow use for in vivo studies of effects of BQCA on CNS function.
Table 1.
BQCA reverses impairments in discrimination learning in Tg2576 mice
| Group | Simplediscrimination | Simplediscriminationreversal | Compounddiscrimination* | Compounddiscriminationreversal** |
|---|---|---|---|---|
| Wild-type + vehicle | 2/9 (22%) | 5/9 (55%) | 4/9 (44%) | 3/9 (33%) |
| Wild-type + BQCA | 3/12 (25%) | 6/12 (50%) | 5/12 (42%) | 4/12 (33%) |
| Tg2576 + vehicle | 1/8 (12%) | 4/8 (50%) | 5/8 (62%) | 7/8 (87%) |
| Tg2576 + BQCA | 2/11 (18%) | 5/11 (45%) | 1/11 (9%) | 3/11 (27%) |
The frequency of errors on discrimination learning between wild-type and Tg2576 mice in the presence of vehicle or BQCA (number of subjects with errors ≥1/total number of subjects in group expressed as a percentage).
*χ2 = 23.19, p < 0.0001;
**χ2 = 13.03, p < 0.001.
Having established the PK profile and CNS penetration of BQCA, we performed in vivo electrophysiology studies to test the hypothesis that the electrophysiological effects observed on mPFC neurons in vitro can lead to increases in activity of mPFC neurons in behaving animals. To accomplish this, multiple single-unit recordings were obtained from the mPFC of rats trained to perform an auditory detection task for food reward. A total of 57 cells (vehicle, n = 20; BQCA, n = 37) with waveform and firing rate characteristics consistent with those of putative pyramidal cells were obtained from 6 rats in the presence of either vehicle or drug (20 mg/kg). Figure 9a shows the average percentage change, relative to a 30 min preinjection epoch, in the spontaneous firing rate of mPFC cells following drug or vehicle administration. Consistent with the acute cortical slice data, BQCA caused an elevation in spontaneous firing rate significantly different from vehicle (two-way ANOVA: drug vs vehicle, p < 0.005). Significant elevations in firing rate were observed within the first thirty-min epoch following injection and were maintained across the entire hour and a half recording period. Unit recordings were highly stable over the course of the recordings as illustrated by action potential traces recorded during baseline (solid black trace) and at the end (60–90 min) of the postdrug recording period (dashed gray trace) (Fig. 9b) as well as monitoring of normalized peak spike amplitude over the time of the experiment (Fig. 9c).
Figure 9.

BQCA increases the firing rate of mPFC neurons in vivo in rats. Multiple single-unit recordings were obtained from the medial prefrontal cortex of conscious, freely moving rats. A total of 57 cells (vehicle, n = 20, BQCA, n = 37) with waveform and firing rate characteristics consistent with those of putative pyramidal cells were obtained from 6 rats in the presence of either vehicle or drug (20 mg/kg). The mean firing rate for each neuron within an epoch was calculated as described in Materials and Methods and expressed as a percentage of the preinjection baseline rate. a, Treatment with BQCA (■) resulted in an elevation in spontaneous firing rate that was significantly different from vehicle (▴) (2-way ANOVA; BQCA vs vehicle; p < 0.005) within the first 30 min epoch and was maintained across the entire trial period. b, Unit recordings were highly stable over the course of the recordings as illustrated by action potential traces recorded during baseline (solid black trace) and at the end (60–90 min) of the postdrug recording period (dashed gray trace). c, Normalized peak spike amplitude over the course of the experiment; each point is a spike, and the gap in recording corresponds to time of BQCA or vehicle injection.
Acute administration of BQCA restores impairment in reversal learning in Tg2576 mice
Recent studies have revealed that mice overexpressing a familial AD mutant form of the amyloid precursor protein (Tg2576 mice) are impaired on compound discrimination reversal learning compared to littermate controls (Zhuo et al., 2007, 2008). Interestingly, reversal learning is a PFC-dependent form of learning, suggesting that this mouse model of AD leads to disruption of at least one form of PFC-dependent cognition. Based on the finding that M1 receptor KO mice have deficits in PFC function and that BQCA increases PFC activity, it is possible that this M1 receptor-selective PAM could reverse deficits in compound discrimination reversal learning observed in Tg2576 mice. In agreement with previously published reports, we found that Tg2576 mice exhibit impaired performance in a compound discrimination reversal learning task (Fig. 10). Acute administration of BQCA improved the performance of the Tg2576 mice on the compound discrimination and the compound discrimination reversal task by reducing the odds that errors would be committed, χ2 = 23.19 and χ2 = 13.03, 1, respectively (p < 0.001) (Table 1, Fig. 10c,d). On the compound discrimination, the odds that vehicle-treated Tg2576 mice made errors were 6.89 times greater than the BQCA-treated Tg2576 mice. Similarly, on the compound discrimination reversal, the odds of the vehicle-treated Tg2576 mice to make errors were 3.22 times greater than the BQCA-treated Tg2576 mice. There prevalence of errors on the simple discrimination or the simple discrimination reversal tasks did not significantly differ across groups or treatments. Overall, the results indicate that BQCA improves compound reversal learning which is consistent with hypothesis that M1 activation may enhance PFC-dependent cognitive function. Additionally, BQCA may also have more widespread effects on cognition, indicated by the reduction of errors on the compound discrimination in BQCA-treated Tg2576 mice, and may be of even broader utility in enhancing other domains of cognitive function.
Figure 10.
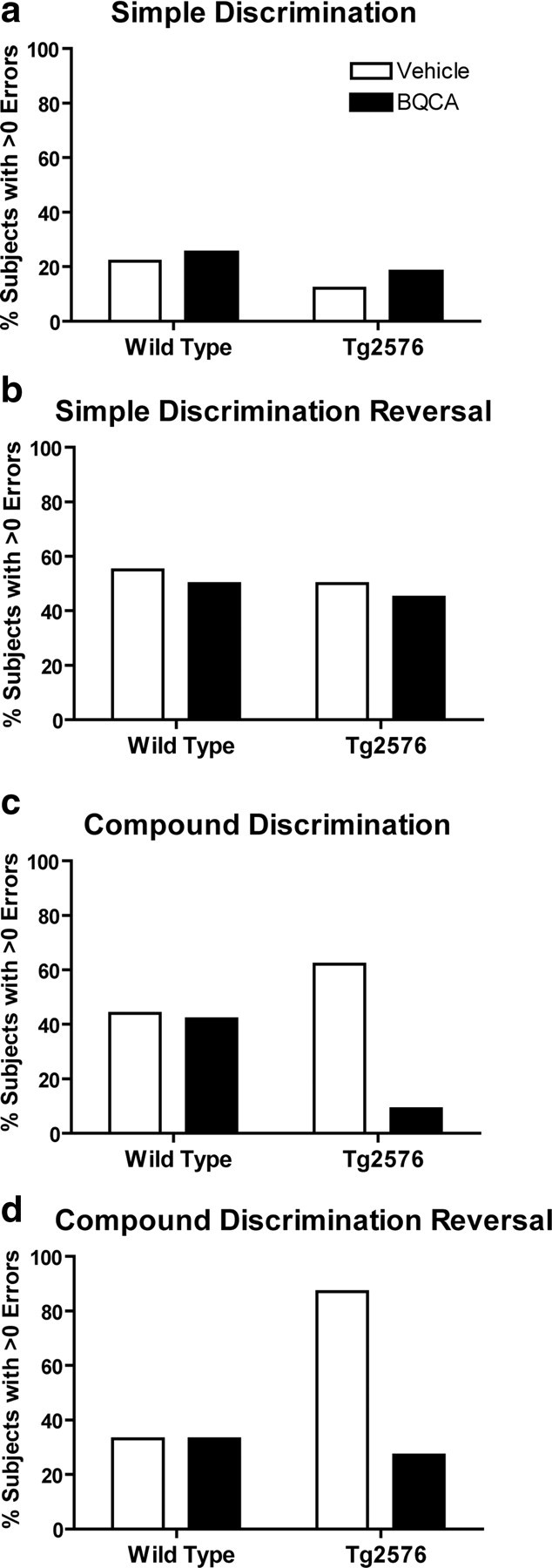
Effects of acute administration of BQCA on discrimination learning in Tg2576 mice. Errors to reach criterion on a discrimination task were assessed in wild-type and Tg2576 mice at 12 months of age. Mice were injected subcutaneously 1 h before testing with either saline vehicle or 30 mg/kg BQCA. Shown are the frequency of errors on discrimination learning between wild-type and Tg2576 mice in the presence or absence of BQCA. Data are expressed as the number of subjects with errors >0/total number of subjects in the group and expressed as a percentage. BQCA significantly reduced the odds of Tg2576 mice making errors on the compound discrimination and the compound discrimination reversal. Chi-square, p < 0.001 for both compound discrimination and compound discrimination reversal.
BQCA regulates non-amyloidogenic APP processing
The data presented above suggest that BQCA has efficacy in improving at least one form of cognitive function in an animal model of AD. In addition to providing symptomatic relief, it has been postulated that increasing M1 receptor activity could also have disease modifying effects in AD patients (Fisher, 2008; Caccamo et al., 2009). The amyloid precursor protein (APP) undergoes proteolytic cleavage in two competing pathways (for review, see Thinakaran and Koo, 2008). In the amyloidogenic pathway, sequential cleavage by β-secretase and γ-secretase releases the Aβ peptide, which forms the core of amyloid plaques found in AD and is implicated in numerous models of neurotoxicity. Alternatively, in the non-amyloidogenic pathway, APP is cleaved by α-secretase within the Aβ sequence, preventing Aβ generation. Interestingly, previous studies suggest that activation of M1 promotes APP processing through the non-amyloidogenic pathway (Caccamo et al., 2006; Jones et al., 2008). If BQCA can promote non-amyloidogenic processing of APP, this could provide a mechanism for slowing accumulation of Aβ and potentially slow progression of AD.
To determine whether BQCA can potentiate the APP processing effect of a low concentration of the mAChR agonist CCh, we treated PC12 cells overexpressing human APP and the M1 receptor with an approximate EC20 concentration (50 nm) of CCh in the presence of increasing concentrations of BQCA and measured the levels of APP metabolites in the conditioned media and cell extracts. BQCA caused a dose-dependent increase in the shedding of APPsα, the N-terminal ectodomain of APP released by α-secretase cleavage (Fig. 11a,b). The highest concentration of BQCA tested (30 μm) increased APPsα levels to 244% of vehicle-treated cells (p < 0.05). BQCA treatment also resulted in the accumulation of CTFα (C83), the corresponding carboxy-terminal fragment generated by α-secretase (Fig. 11a,c) (increased to 245% of vehicle, p < 0.05). Finally, consistent with the observed increases in non-amyloidogenic APP fragments, 30 μm BQCA treatment resulted in a 30% decrease (p < 0.01) in the secretion of the β-secretase derived Aβ40 peptide (Fig. 11d). Together, these results indicate that BQCA can effectively regulate non-amyloidogenic APP processing, suggesting that M1 receptor PAMs have the potential to provide both symptomatic and disease modifying effects in AD patients.
Figure 11.

BQCA regulates non-amyloidogenic APP processing. a, Western blot analysis of APP metabolites from conditioned media and cell lysates demonstrates increased generation of APPsα and CTFα with increasing concentrations of BQCA as compared to the submaximal concentration of 50 nm CCh. Ten micromolars CCh is shown as a maximum concentration. β-actin is shown as a loading control. b, Quantitation of APPsα band intensity from conditioned media demonstrates a dose-dependent effect of BQCA on the shedding of APPsα (repeated-measures ANOVA, p = 0.0271), and pairwise comparisons revealed significant differences at all concentrations of BQCA compared to 50 nm CCh alone (p values for paired t tests are shown). c, Quantitation of CTFα band intensity from cell lysates shows a dose-dependent effect of BQCA on the production of CTFα (repeated-measures ANOVA, p = 0.0017) and a significant difference (paired t test) between 30 μm BQCA plus 50 nm CCh as compared to 50 nm CCh alone. d, ELISA measurements from conditioned media demonstrate that BQCA decreases the secretion of Aβ40 peptide in a dose-dependent manner (repeated-measures ANOVA, p = 0.0019), with significant differences between 50 nm CCh alone and the two highest concentrations of BQCA (paired t tests). Mean values are shown from three or four independent experiments performed in duplicate. All values are normalized to vehicle-treated cells.
Discussion
The M1 receptor has long been viewed as an exciting potential target for increasing cognitive function in patients suffering from AD and other CNS disorders (Langmead et al., 2008a; Wess et al., 2007; Fisher, 2008; Caccamo et al., 2009). Despite major efforts to develop highly selective M1 receptor agonists over the past two decades, this receptor has proven intractable using traditional approaches, thus preventing M1 receptor agonists from advancing to clinical use for treatment of AD and other disorders. Also, lack of agents that selectively activate this receptor has made it impossible to develop a full understanding of the functional effects of selectively increasing M1 receptor activity in the CNS. Discovery and characterization of BQCA and its structural analogs provide a major advance in establishing the utility of M1 receptor PAMs as an alternative approach to increasing activity of this receptor in a highly subtype-selective manner. Unlike traditional agonists, these small molecules do not bind to the orthosteric ACh binding site, but instead act at a distinct site to potentiate activation of the receptor by its natural ligand, ACh. This is directly analogous to the use of benzodiazepines as selective GABA-A receptor PAMs, which provide an effective and safe approach to the treatment of anxiety and sleep disorders without inducing the potentially lethal effects of direct-acting GABA-A receptor agonists (Möhler et al., 2002). While allosteric modulators of ion channels are well established as research tools and therapeutic agents, they have not been a traditional focus of drug discovery efforts for GPCRs. However, BQCA adds to recent major advances in developing highly selective allosteric modulators of mAChRs (Brady et al., 2008; Chan et al., 2008; Ma et al., 2008; Shirey et al., 2008; Marlo et al., 2009) and other GPCRs (May et al., 2007; Conn et al., 2009a). However, BQCA is distinct from recently discovered allosteric agonists of the M1 receptor (Spalding et al., 2002; Sur et al., 2003; Jones et al., 2008; Langmead et al., 2008b), in that this compound has no intrinsic agonist activity, but rather potentiates the response to ACh.
Studies with BQCA, along with the new M1 receptor-selective antagonist VU0255035, provide important support for the hypothesis that the M1 receptor may increase activation of the PFC and may enhance PFC-dependent cognitive function (Anagnostaras et al., 2003). Non-selective mAChR agonists, such as CCh, induce an inward current in PFC pyramidal cells, and the present data provide strong evidence that this response is mediated by activation of the M1 receptor. In addition, activation of the M1 receptor increases the frequency of spontaneous excitatory synaptic events in mPFC layer V pyramidal cells. While the source of glutamatergic afferents giving rise to these sEPSCs has not been established, this is consistent with the hypothesis that the M1 receptor plays an important role in increasing excitability and excitatory drive to mPFC pyramidal cells.
Interestingly, mAChR activation induces direct excitatory effects in hippocampal CA1 pyramidal cells that are similar to those observed in mPFC pyramidal cells. However, while CA1 pyramidal cells express high levels of the M1 receptor (Levey et al., 1991), previous studies suggest that the M1 receptor is not the mAChR subtype responsible for excitatory effects on these cells (Rouse et al., 2000). Thus, the precise physiological roles of the M1 receptor are likely to vary in different brain regions and neuronal populations. The finding that M1 receptor activation has excitatory effects and increases excitatory synaptic activity in mPFC pyramidal cells is interesting in the context of the recent finding that M1 receptor KO mice display clear deficits in PFC-dependent learning (Anagnostaras et al., 2003), whereas hippocampal-dependent learning is largely unaffected in M1 receptor KO mice (Anagnostaras et al., 2003) and in animals treated with the M1 receptor-selective antagonist VU0255035 (Sheffler et al., 2009).
One of the most important implications of these studies is that they raise the possibility that highly selective M1 receptor PAMs may provide a novel approach for treatment of AD and other CNS disorders that may involve impaired cholinergic signaling. Clinical studies using both direct and indirect-acting muscarinic agonists have reported improvements in both cognitive function and behavioral disturbances (i.e., hallucinations, delusions, outbursts, and paranoia) observed in AD patients (Bodick et al., 1997; Cummings et al., 2001). If M1 receptor activation is responsible for, or plays an important role in, these effects of non-selective cholinergic agents, M1 receptor PAMs could provide a viable approach to symptomatic treatment of AD. Furthermore, in addition to potential efficacy in reducing symptoms in AD patients, recent studies suggest that mAChR activation could reduce accumulation of toxic Aβ protein, thereby also providing disease modifying effects. For instance, the muscarinic agonist AF102B was shown to decrease production of the amyloidogenic peptide Aβ42 in the cerebral spinal fluid of AD patients (Nitsch et al., 2000). Furthermore, preclinical studies with a related mAChR agonist, AF267B suggest that mAChR activation increases non-amyloidogenic processing and prevents Aβ formation (Caccamo et al., 2006). While these earlier mAChR agonists are not selective for the M1 receptor relative to other mAChR subtypes, more recent studies revealed that the M1 receptor-selective agonist, TBPB [1-[1′-(2-tolyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one], has similar effects in PC12 cells (Jones et al., 2008).
The present finding that BQCA reverses deficits in compound discrimination reversal learning in a transgenic mouse model of AD provides exciting support for the hypothesis that highly selective M1 receptor PAMs may provide efficacy in treatment of at least some domains of cognitive function in AD. Furthermore, the finding that BQCA promotes non-amyloidogenic APP processing suggests that these agents could also reduce amyloid burden. In future studies, it will be important to fully explore the effects of BQCA in animal models that reflect other domains of cognitive function that are impaired in AD patients. For instance it is possible that M1 receptor-selective PAMs will have robust efficacy in improving PFC-dependent learning, but have less efficacy in hippocampal-dependent learning. Also, other domains of cognitive function may involve different mAChR subtypes and be differentially affected by selective activators of the M1 receptor versus selective PAMs of other mAChR subtypes, such as the recently reported M4- and M5 receptor-selective PAMs (Brady et al., 2008; Chan et al., 2008; Shirey et al., 2008; Bridges et al., 2009). In addition, it will be important to expand APP processing studies to include effects of chronic dosing in vivo. Interestingly, the high subtype-selectivity of BQCA may prove to be important for achieving maximal effects in increasing non-amyloidogenic APP processing. Previous studies suggest that activation of M2 and/or M4 mAChR subtypes may have an antagonistic effect on the non-amyloidogenic APP processing shown to be promoted by M1 receptor activation (Farber et al., 1995). Thus, in addition to reducing the adverse effect profile, it is possible that selective activation of the M1 receptor may provide greater efficacy in regulating APP processing.
In addition to implications for AD, the electrophysiology studies reveal interesting findings that may provide important insights related to the potential roles of mAChRs in regulating PFC function. For instance, when added alone, BQCA induced a slight inward current and a slight increase in sEPSC frequency. This suggests that there may be a low tonic level of M1 receptor activity that can be potentiated by BQCA. Furthermore, it was interesting to find that CCh induced a small reduction in sEPSC frequency when added in the presence of a saturating concentration of VU0255035, the M1 receptor-selective antagonist. This may suggest that activation of another mAChR subtype can reduce sEPSC frequency and that this is unmasked when the M1 receptor is selectively blocked. Interestingly, while effects of CCh on sEPSC frequency were dramatically reduced in M1 receptor KO mice, CCh did induce a small effect in slices from these animals. This suggests that another mAChR subtype may be capable of eliciting this response and could partially compensate for genetic deletion of the M1 receptor. Importantly, the effect of the highly selective M1 receptor PAM, BQCA, was eliminated in M1 receptor KO mice, suggesting that the effects of this compound are fully dependent on activation of the M1 receptor. Discovery of new mAChR subtype-selective ligands for multiple mAChR subtypes over the last year will allow for a better understanding of the roles of multiple mAChR subtypes in regulating function.
Finally, it is important to note that recent clinical and animal studies raise the possibility that mAChR agonists may also provide a novel approach for treatment of schizophrenia (for review, see Felder et al., 2001; Langmead et al., 2008a; Conn et al., 2009b). For instance, Shekhar et al. (2008) recently reported that the M1/M4 receptor-preferring agonist xanomeline induced a robust improvement in positive and negative symptoms, as well as some measures of cognitive function, in schizophrenic patients. Based on animal studies, it is likely that both M1 and M4 receptors may be important for clinical efficacy in this patient population (Felder et al., 2001; Langmead et al., 2008b; Brady et al., 2008; Chan et al., 2008; Jones et al., 2008; Conn et al., 2009b). Availability of BQCA, along with the new systemically active M4 receptor-selective PAM, VU0152100 (Brady et al., 2008), should make it possible to evaluate the effects of selective activation of each of these mAChR subtypes as well as coadministration of both BQCA and VU0152100 in a range of animal models that may be relevant to the antipsychotic effects of xanomeline.
Note added in proof.
After submission of this manuscript, studies reporting the initial discovery of BQCA were reported by Ma et al. (2009).
Footnotes
This work was supported by grants from the National Institute of Mental Health (NIMH) and the National Institute of Neurological Disorders and Stroke. A.E.B. is supported by NIMH Grant 1F32 MH079678-01. T.M.B. is supported by the Integrative Training in Therapeutic Discovery (ITTD) Grant from the Vanderbilt Institute of Chemical Biology (T90-DA22873) and J.K.S. is supported by NIMH Grant 1 F31 MH80559-01. A.A.D. is supported by a predoctoral fellowship from the National Institute on Aging and the PhRMA Foundation. A.I.L. is supported by National Institutes of Health Grant NS30454. Vanderbilt is a site in the National Institutes of Health-Supported Molecular Libraries Probe Center Network. We thank Drs. T. I. Bonner (NIMH, Bethesda, MD) for the rM4 cDNA construct, B. Conklin (Gladstone Institute, University of California, San Francisco, San Francisco, CA) for the chimeric Gqi5 construct, and J. Wess for M1 KO mice used in the electrophysiological studies. We also graciously thank Drs. D. Sheffler and M. Noetzel for technical support and Dr. A. Liguori for statistical assistance.
References
- Aigner TG, Walker DL, Mishkin M. Comparison of the effects of scopolamine administered before and after acquisition in a test of visual recognition memory in monkeys. Behav Neural Biol. 1991;55:61–67. doi: 10.1016/0163-1047(91)80127-z. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev CD005593. 2006 doi: 10.1002/14651858.CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, Lindsley CW. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy n-benzyl isatins. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Fisher A, LaFerla FM. M1 agonists as a potential disease-modifying therapy for Alzheimer's disease. Curr Alzheimer Res. 2009;6:112–117. doi: 10.2174/156720509787602915. [DOI] [PubMed] [Google Scholar]
- Carr DB, Surmeier DJ. M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J Neurophysiol. 2007;97:3432–3438. doi: 10.1152/jn.00828.2006. [DOI] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, Felder CC. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci U S A. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009a;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009b;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Nadel A, Masterman D, Cyrus PA. Efficacy of metrifonate in improving the psychiatric and behavioral disturbances of patients with Alzheimer's disease. J Geriatr Psychiatry Neurol. 2001;14:101–108. doi: 10.1177/089198870101400211. [DOI] [PubMed] [Google Scholar]
- Farber SA, Nitsch RM, Schulz JG, Wurtman RJ. Regulated secretion of β-amyloid precursor protein in rat brain. J Neurosci. 1995;15:7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- Fibiger HC, Damsma G, Day JC. Behavioral pharmacology and biochemistry of central cholinergic neurotransmission. Adv Exp Med Biol. 1991;295:399–414. doi: 10.1007/978-1-4757-0145-6_23. [DOI] [PubMed] [Google Scholar]
- Fisher A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer's disease. Neurotherapeutics. 2008;5:433–442. doi: 10.1016/j.nurt.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Zhong P, Yan Z. Activation of muscarinic receptors inhibits beta-amyloid peptide-induced signaling in cortical slices. J Biol Chem. 2003;278:17546–17556. doi: 10.1074/jbc.M209892200. [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MS, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28:10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjeviæ K. Synaptic mechanisms modulated by acetylcholine in cerebral cortex. Prog Brain Res. 2004;145:81–93. [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008a;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Austin NE, Branch CL, Brown JT, Buchanan KA, Davies CH, Forbes IT, Fry VA, Hagan JJ, Herdon HJ, Jones GA, Jeggo R, Kew JN, Mazzali A, Melarange R, Patel N, Pardoe J, Randall AD, Roberts C, Roopun A, Starr KR, Teriakidis A, Wood MD, Whittington M, Wu Z, Watson J. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br J Pharmacol. 2008b;154:1104–1115. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Jacobson MA, Kreatsoulas C, Getty KL, Seabrook GR, Ray WJ. Exploring the pharmacology of BQCA, a highly selective allosteric M1 potentiator. Alzheimers Dement. 2008;4:T767. [Google Scholar]
- Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, Austin CA, Williams MB, Kim K, Williams R, Orton D, Brown HA, Lindsley CW, Weaver CD, Conn PJ. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Miller EK, Desimone R. Scopolamine affects short-term memory but not inferior temporal neurons. Neuroreport. 1993;4:81–84. doi: 10.1097/00001756-199301000-00021. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–5250. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- Morisset V, Urban L. Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J Neurophysiol. 2001;86:40–48. doi: 10.1152/jn.2001.86.1.40. [DOI] [PubMed] [Google Scholar]
- Muñoz-Torrero D. Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer's disease. Curr Med Chem. 2008;15:2433–2455. doi: 10.2174/092986708785909067. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Abeta in cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 2000;48:913–918. [PubMed] [Google Scholar]
- Rouse ST, Hamilton SE, Potter LT, Nathanson NM, Conn PJ. Muscarinic-induced modulation of potassium conductances is unchanged in mouse hippocampal pyramidal cells that lack functional M1 receptors. Neurosci Lett. 2000;278:61–64. doi: 10.1016/s0304-3940(99)00914-3. [DOI] [PubMed] [Google Scholar]
- Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Lindsley CW, Conn PJ. A novel selective muscarinic acetylcholine receptor subtype 1 (M1 mAChR) antagonist reduces seizures without impairing hippocampal-dependent learning. Mol Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Spalding TA, Trotter C, Skjaerbaek N, Messier TL, Currier EA, Burstein ES, Li D, Hacksell U, Brann MR. Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol Pharmacol. 2002;61:1297–1302. doi: 10.1124/mol.61.6.1297. [DOI] [PubMed] [Google Scholar]
- Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc Natl Acad Sci U S A. 2003;100:13674–13679. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Zhuo JM, Prescott SL, Murray ME, Zhang HY, Baxter MG, Nicolle MM. Early discrimination reversal learning impairment and preserved spatial learning in a longitudinal study of Tg2576 APPsw mice. Neurobiol Aging. 2007;28:1248–1257. doi: 10.1016/j.neurobiolaging.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Zhuo JM, Prakasam A, Murray ME, Zhang HY, Baxter MG, Sambamurti K, Nicolle MM. An increase in Abeta42 in the prefrontal cortex is associated with a reversal-learning impairment in Alzheimer's disease model Tg2576 APPsw mice. Curr Alzheimer Res. 2008;5:385–391. doi: 10.2174/156720508785132280. [DOI] [PubMed] [Google Scholar]