

Abstract
We conducted an association study to identify risk variants for familial prostate cancer within the HPCX locus at Xq27 among Americans of Northern European descent. We investigated a total of 507 familial prostate cancer probands and 507 age-matched controls without a personal or family history of prostate cancer. The study population was subdivided into a set of training subjects to explore genetic variation of the locus potentially impacting risk of prostate cancer, and an independent set of test subjects to confirm the association and to assign significance, addressing multiple comparisons. We identified a 22.9 kb haplotype nominally associated with prostate cancer among training subjects (292 cases, 292 controls; χ2 = 5.08, P = 0.020), that was confirmed among test subjects (215 cases, 215 controls; χ 2 = 3.73, P = 0.040). The haplotype predisposed to prostate cancer with an odds ratio of 3.41 (95% CI 1.04–11.17, P = 0.034) among test subjects. The haplotype extending from rs5907859 to rs1493189 is concordant with a prior study of the region within the Finnish founder population, and warrants further independent investigation.
BACKGROUND
Linkage and genetic epidemiological data support the existence of genetic variants on the X chromosome that predispose to prostate cancer (Woolf 1960). Prostate cancer loci on both arms of the X chromosome have been identified, including the HPCX locus at Xq27-28 (Bochum et al. 2002; Brown et al. 2004; Chang et al. 2005; Cunningham et al. 2003; Farnham et al. 2005; Gillanders et al. 2004; Gudmundsson et al. 2008; Lange et al. 1999; Schleutker et al. 2000; Xu et al. 1998). The ~14 Mb linkage interval of HPCX was originally delineated within US, Swedish, and Finnish hereditary prostate cancer pedigrees (Xu et al. 1998). Further shared haplotype analysis among Finnish probands refined the locus to a candidate interval flanked on either side by a notable 113 kb inverted repeat (Baffoe-Bonnie et al. 2005a, b). The 352 kb area between these inverted repeats was the candidate interval for the present study (Fig. 1), which sought evidence of association with familial prostate cancer among Americans of Northern European descent. Our study population was uniquely comprised of independent familial prostate cancer probands, matched to controls with no personal or family history of prostate cancer. These two groups represent extremes of potential genetic load for prostate cancer. Our study included a training set of 292 case-control pairs to identify nominal associations, and a test set of 215 case-control pairs to confirm or to refute observations within the training set. We conducted extensive allele discovery and validation within the study population, characterized linkage disequilibrium (LD) patterns, and selected tagging SNPs for tests of association by haplotype-based methods. Our investigation comprehensively tested association of the candidate interval with prostate cancer, and included non-unique genomic regions that are not amenable to current high-throughput techniques.
Fig. 1. HPCX Candidate Interval.

3 MB of Xq27.1-27.3 depicting previously genotyped STR markers, annotated genes, and a complex HPCX specific repeat is shown at top (red bounding box, NCBI build 36.1 ChrX: 139005624-142009903 bp). The candidate interval for study is zoomed at bottom (blue bounding box, ChrX:140046709-140391709 bp). The interval contains SPANXC and LDOC1, as well as a predicted RPL44 homolog and a pseudogene of RBMX2. As members of larger gene families, unique long range amplimers (denoted) were required to ensure site-specific assays. Polymorphic SNPs (N=246) are positioned on the map (tagging SNPs in pink). At bottom is a pairwise LD matrix for 141 controls across the subset of 220 SNPs with MAF ≥ 0.05. Red, D' = 1 (lod ≥ 2); blue, D' = 1 (lod < 2); pink, D' < 1 (lod ≥ 2); white, D' < 1 (lod < 2). Blocks of LD are denoted A, B, C and D. Block A contains all four candidate gene regions.
MATERIALS AND METHODS
Samples
Study subjects were Americans of Northern European descent, ascertained with informed consent between 2002 and 2007 from Vanderbilt University Medical Center and from the VA Tennessee Valley Healthcare System (adjacent hospitals) with institutional review board oversight. Subjects were residents of Tennessee (75%), Kentucky (15%), Georgia (2%), Alabama (1%), Mississippi (1%), Virginia (1%), and other states (4%). Familial prostate cancer cases were ascertained at the time of treatment for the principal diagnosis of prostate cancer, and controls were ascertained at the time of routine preventative screening for prostate cancer. All prostate cancer probands included in the study are from pedigrees with a family history of prostate cancer, and all control probands are from pedigrees without a family history of prostate cancer. Family history included 1st and 2nd degree relatives. Controls had a screening prostate specific antigen (PSA) test < 4 ng/ml at the time of ascertainment, and had no record of a PSA test ≥ 4 ng/ml or record of abnormal digital rectal examination. Controls were matched to cases on age (± 2.5 years; age at screen for controls, age at diagnosis for cases). Case and control pedigrees were of comparable size. The mean number of at-risk male siblings was 1.7 for cases, and 1.8 for controls. Initial accruals included 292 unrelated, independent familial prostate cancer probands and 292 age-matched controls, comprising a training study group. Subsequent accruals included 215 additional unrelated, independent prostate cancer probands and 215 additional age-matched controls, comprising a separate test study group. The geographical distribution (by state of residence) of training and test subjects was not significantly different. Analyses preferentially employed prostatectomy specimen over biopsy Gleason score (available for 87% of cases). Table 1 provides characteristics of the study population.
Table 1.
Study population characteristics
| Training | Test | Combined | |||||
|---|---|---|---|---|---|---|---|
| Controls | Cases | Controls | Cases | Controls | Cases | ||
| No. | 292 | 292 | 215 | 215 | 507 | 507 | |
| Mean Agea, y | 63.4 | 61.3 | 60.7 | 60.6 | 62.3 | 61.0 | |
| Median PSAa | 0.95 | 5.7 | 0.92 | 5.6 | 0.92 | 5.7 | |
| Median Gleason Sum | - | 6 | - | 6 | - | 6 | |
| Gleason Sum ≤ 6, No. | - | 145 | - | 114 | - | 259 | |
| Gleason Sum ≥ 7, No. | - | 130 | - | 96 | - | 226 | |
| Affected in Pedigree, No.b | 0 | 292 | - | 215 | - | 507 | - |
| 2 | - | 184 | - | 142 | - | 326 | |
| ≥3 | - | 108 | - | 73 | - | 181 | |
At diagnosis for cases, at screen for controls.
Proband plus 1st and 2nd degree affected relatives
Genotyping Methods
DNA was extracted from whole blood using the Puregene DNA Purification System Standard Protocol (Qiagen, Valencia, CA). DNA was quantified using the PicoGreen dsDNA Quantitation Kit (Invitrogen, Carlsbad, CA), imaged with a Molecular Devices / LJL Analyst HT (Molecular Devices, Union City, CA). We genotyped SNPs by single nucleotide primer extension and fluorescence polarization, as previously described (Yaspan et al. 2007). Both forward and reverse strand extension primers were tested to select the most robust assay. Amplimer and extension primer sequences for tagging SNPs are provided in Supplementary Table 1.
Candidate Polymorphisms
To capture genetic diversity across the candidate interval, database SNPs annotated in dbSNP were screened for common polymorphism in a set of 40 independent familial prostate cancer probands from the training study group. This included 415 annotated SNPs on chromosome X between positions 140,036,557 and 140,388,361 (NCBI Build 36.1). The screening set was estimated to provide 98% power to detect a polymorphism with a minor variant frequency of 0.10, and 87% power with a frequency of 0.05. These 40 prostate cancer cases were also used for de novo SNP discovery at known and predicted genes within the candidate interval: 4.6 kb 5’ to 0.2 kb 3’ of LDOC1; 1.6 kb 5’ to 4.4 kb 3’ of SPANXC; 3.0 kb 5’ to 1.4 kb 3’ of a predicted coding region containing homology to ribosomal protein L44 (“hRPL44”); and 2.3 kb 5’ to 0.8 kb 3’ of a predicted pseudogene containing homology to RBMX2 (“RBMX2P1”). The latter two annotations were identified by aligning transcript evidence on the genomic map. We employed two single-stranded conformation polymorphism methods (redundant) and re-sequencing for SNP discovery, as previously described (Yaspan et al. 2007). Exons of the four genes were also re-sequenced for all 40 prostate cancer cases in the screening set.
Nested Amplification of Non-unique Regions
Non-unique regions of SPANXC, hRPL44 and RBMX2P1 were assayed using a nested reaction strategy. Unique priming sites were identified flanking non-unique regions and amplified using the Expand Long Template PCR System (Roche Diagnostics, Indianapolis, IN). Amplimer primers for long range PCR are provided in Supplementary Table 1. Long-range PCR product was then diluted 1:5,000,000 to dilute carry-over genomic template to non-amplifiable levels while retaining the ability to amplify from the long-range PCR product. This was verified by successful test of amplimers nested within the template long-range PCR product, but failure of amplimers elsewhere in the genome. Nested amplimers were designed in overlapping fashion along a given long range amplimer, enabling PCR-based assays. These primers are provided in Supplementary Table 2. High pairwise LD between SNPs within a given non-unique region and flanking unique SNPs supported correct non-unique copy assay (visible in Fig. 1). All nested assays within long-range amplimers yielded one allele per subject, concordant with a unique X-chromosomal region for a male.
Tag SNP Determination
Tagging SNP determination was conducted in a subset of 141 training set control subjects that were genotyped at 246 SNPs (including 194 validated from dbSNP and 52 identified by de novo discovery efforts). Pairwise LD was visualized using Haploview v4 (Barrett et al. 2005). LDSelect was used for tagging SNP selection, specifying a MAF threshold of 0.05 and an r2 threshold of 0.9 (Carlson et al. 2004). A total of 128 tagging SNPs were selected for assay in the remaining subjects of the training set (totaling 292 independent familial case probands and their 292 age-matched controls). Data was obtained for 96.2% of the 74,752 tagging genotypes sought in the training subjects, with a per marker range from 88.4% to 100%. SNPs in this tagging set and their assay primers are listed in Supplementary Table 1.
Statistical Analyses
A sliding window approach tested a haplotype window of N markers, sliding the window along the map in single marker increments (Fallin et al. 2001; Mathias et al. 2006; Yaspan et al. 2007). Each N-marker haplotype was compared to the remaining haplotypes as a group among the training group of 292 cases and 292 matched controls. The resulting 2 × 2 contingency table was evaluated by the χ2 test statistic. Haplotype windows of 1–10 markers were evaluated in the exploratory analyses of training subjects. In a given map region that was nominally associated with prostate cancer within the training subjects (P ≤ 0.05), haplotype tagging SNPs (htSNPs) were selected that most efficiently distinguished the risk haplotype from others in the region. Nominally significant tagged haplotypes (two observed) were genotyped in a subsequently ascertained independent test group of 215 cases and their 215 matched controls to address multiple comparisons. Significance for a given tagged haplotype candidate was adjusted for the two comparisons among test subjects through permutation testing. We generated 5,000 copies of the data set in which pseudo case status was permuted among cases and controls of the test group. A χ2 value for each tagged haplotype was calculated for each simulated data set, as it was for the real data. Since the null hypothesis is true for each randomized subject set, the proportion of simulated χ2 values greater than the real χ2 value was used as a P value for the association, adjusted for multiple comparisons. Unless specifically noted, P values are unadjusted for multiple comparisons.
A risk haplotype that was significant after adjustment for multiple comparisons among test subjects was subsequently modeled by conditional logistic regression to obtain an estimate of effect size (Intercooled Stata 9, Stata Corporation, College Station, TX). The regression model was adjusted for the matching variable: age (age at diagnosis for cases, age at negative screen for controls). Permutation testing was employed to assign significance.
RESULTS AND DISCUSSION
Our allele discovery and characterization was done within a screening set of 40 familial prostate cancer probands from the training group. We evaluated 415 SNPs annotated in dbSNP, 194 of which were polymorphic in these subjects. We also undertook de novo SNP discovery efforts across LDOC1 and SPANXC, as well as across an RPL44 homolog and RBMX2P1 pseudogene. Among these, only LDOC1 resides within a region of unique genomic sequence. We devised a nested amplification system to allow assay of non-unique genomic sequence flanked by unique sequence. Collectively, we discovered 52 common SNPs amenable to assay. The 246 polymorphic SNPs (194 from dbSNP, 52 newly discovered) of the screening subjects were genotyped in a subset of the training study population (141 cases and 141 controls) for assessment of allele frequency and for tagging SNP selection based upon LD patterns. Within this data, 220 SNPs had a minor allele frequency ≥ 0.05 for inclusion in subsequent analyses. Pairwise LD across the candidate interval for these SNPs highlights four LD blocks (denoted A, B, C, and D in Fig. 1).
Among the 220 informative SNPs, we selected 128 tagging SNPs for genotyping in the full group of training subjects (292 familial case probands and 292 age-matched controls). We explored evidence of association with prostate cancer using a haplotype-based sliding window approach. This entailed evaluation of 1,235 haplotype windows across the candidate interval. All haplotype windows of statistical significance were from four distinct regions. At each of the four regions, multiple overlapping windows were consistent with the redundant identification of one haplotype associated with prostate cancer risk. These four candidate risk haplotypes are numbered 1 to 4 in Table 2.
Table 2.
Sliding window risk haplotypes at Xq27 - Training subjects
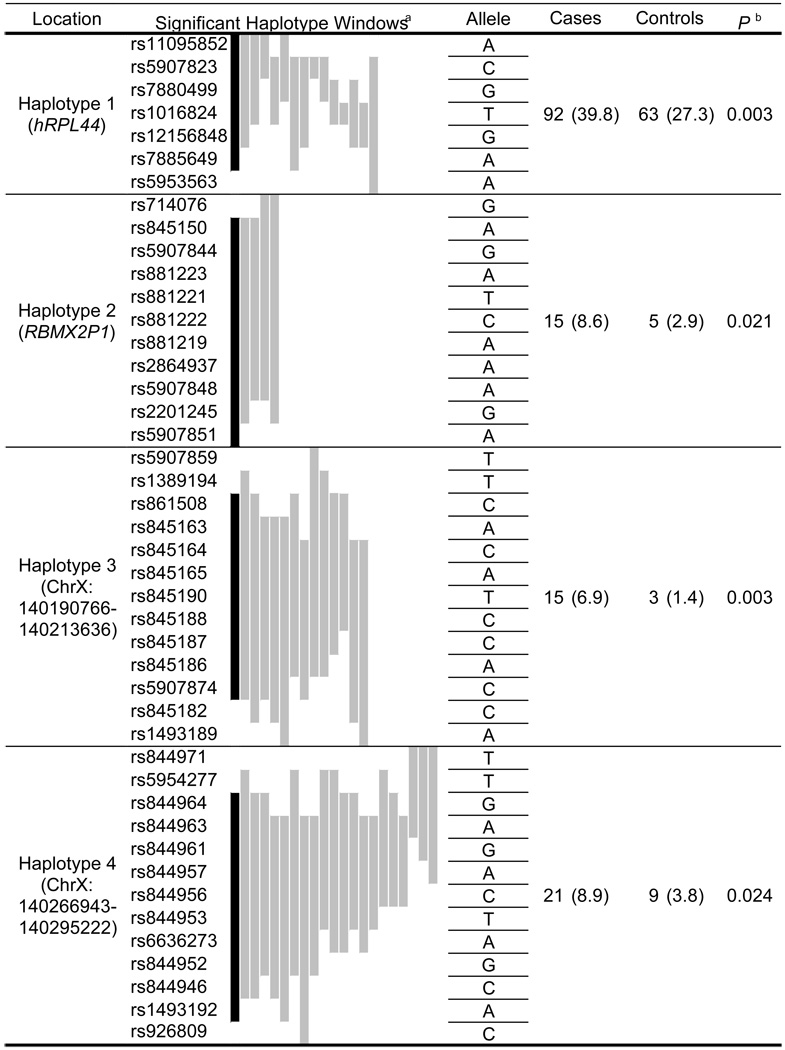 |
Sliding haplotype windows of P ≤ 0.05, graphically ordered as most (black) to least significant (left to right).
P for haplotype designated in black, with corresponding numbers of cases and controls, and haplotype frequencies (%).
Only a subset of SNPs in each of the four regions was required to distinguish the candidate risk haplotype from remaining haplotypes. We identified haplotype-tagging SNPs (htSNPs) efficiently capturing the four candidate risk haplotypes (full span of windows P ≤ 0.05) of Table 2. As our analysis required complete data for each subject across the multiple SNPs of the haplotype, the restricted set of htSNPs provided a better estimate of haplotype frequency. Only two of the four haplotypes were nominally significant when assessed by htSNPs among training subjects (Table 3, haplotype 1 (χ2 = 5.24, P = 0.023) and haplotype 3 (χ2 = 5.08, P = 0.020)). We evaluated evidence of association between these two htSNP haplotypes and prostate cancer in a second, independent study group of 215 familial prostate cancer probands and 215 age-matched controls. These subjects were accrued after the training subjects over the course of the ongoing study. Numerous exploratory tests were conducted among training subjects, but only two tests were conducted among test subjects, a greatly restricted number of comparisons. Only haplotype 3 was significant among test subjects (Table 3, χ2 = 3.73, P = 0.040). Permutation testing was used to correct this value for the two comparisons conducted in test subjects, yielding P = 0.048. Our study identifies haplotype 3 as the most likely genetic variant of the interval to be associated with familial prostate cancer, with a nominal significance of P = 0.003 in the combined training and test subjects.
Table 3.
Tagged risk haplotypes at Xq27 -Training and test subjects
| Location | htSNP | Allele | Training | Test | ||||
|---|---|---|---|---|---|---|---|---|
| Cases | Controls | P | Cases | Controls | P | |||
|
Haplotype 1 (hRPL44) |
rs5907823 | C | 95 (38.6) |
71 (28.9) |
0.023 | 73 (39.3) |
79 (42.5) |
0.536 |
| rs7880499 | G | |||||||
| rs1016824 | T | |||||||
| rs12156848 | G | |||||||
| rs7885649 | A | |||||||
| Haplotype 2 (RBMX2P1) |
rs845150 | A | 19 (6.6) |
9 (3.1) |
0.062 | |||
| Haplotype 3 (ChrX: 140190766- 140213636) |
rs861508 | C | 18 (6.8) |
7 (2.6) |
0.020 | 13 (6.6) |
5 (2.5) |
0.040 |
| rs845165 | A | |||||||
| rs845190 | T | |||||||
| rs845187 | C | |||||||
| rs845186 | A | |||||||
| rs1493189 | A | |||||||
| Haplotype 4 (ChrX: 140266943- 140295222) |
rs844963 rs844956 |
A C |
22 (7.7) |
14 (4.9) |
0.168 | |||
Numbers of subjects and tagged haplotype frequencies (%) are indicated.
Under logistic regression modeling to assess effect size, haplotype 3 was associated with prostate cancer with an odds ratio of 3.41 (95% CI 1.04–11.17, P = 0.034) among test subjects, and an odds ratio of 2.52 (95% CI 1.25 – 5.10, P = 0.006) among combined training and test subjects. The number of cases with aggressive prostate cancer among the test subjects was too few to provide a significant estimate of effect size. However, the 226 cases with aggressive prostate cancer (Gleason score ≥ 7) among the full study population yielded a more marked odds ratio of 4.06 (95% CI 1.15 – 14.31, P = 0.021). Gleason score is among the most important criteria in defining clinically significant disease. Our results are consistent with linkage data at the locus under stratification for clinically significant disease (Chang et al. 2005).
The location of the haplotype found to be significantly associated with prostate cancer in this study coincides with that described through prior high-density simple tandem repeat mapping within a Finnish study population (Baffoe-Bonnie et al. 2005a, b). Among the simple tandem repeats, bG82i1.1 was most significantly associated with prostate cancer in the prior study. The peak associated haplotype was comprised of alleles at bG82i1.1 (centromeric) and bG82i1.0 (telomeric), P = 0.0014. Haplotype 3 of our study directly overlays the recombination hotspot between LD blocks A and B of Fig. 1. The most centromeric SNP of the associated haplotype (rs5907859) is 4.0 kb downstream from bG82i1.1. The most telomeric SNP of the associated haplotype (rs1493189) is 1.9 kb downstream from bG82i1.0. The same genomic region of interest is highlighted by our present study of Americans of Northern European descent and the prior study of Finns. We further note that among SNPs evaluated in the genome wide association study of prostate cancer recently published by Thomas et al., rs845189 has a Whole Genome Rank of 1135 out of 527,869 SNPs assessed, with a significance of P = 0.002 (Thomas et al. 2008). This SNP resides at the LD break centered within the disease-associated haplotype of our study.
The associated haplotype region does not harbor known genes. All missense variants of potential interest in the entire candidate interval of 352 kb were within SPANXC, 30 kb from the associated haplotype. These missense variants clustered into two LD groups. The first group (all in exon 1) included D17E, A21V, and M24T. The second group (all in exon 2) included P29S, T30S, D32Y, and M42L. Within a group, a male subject had either each first or each second allele as listed. Additional SPANXC missense variants, E23K, V59F and L68V, did not appear to be in these two LD groups. This allele structure in SPANXC is also evident in data of an independent study (Kouprina et al. 2007). That study also found no evidence to support an association between SPANXC alleles and risk of prostate cancer. The coding regions of hRPL44 and LDOC1 were without missense variants. We denote RBMX2P1 as a pseudogene, having a mutated initiator methionine, multiple frameshift mutations, and an internal Alu insertion. Thus, the missense variants at SPANXC were among the best potential candidates for association with prostate cancer at Xq27.
The haplotype significantly associated with prostate cancer in this study straddles an LD break, potentially detecting a pair of contributing components located within each of the two bounding LD blocks (e.g. a gene and a long-range regulatory element). In a sliding window haplotype analysis, a haplotype overlapping the two blocks would be particularly suited to detect such a combination. We considered the possibility that causal variants are a pair of non-contiguous SNPs within each LD block. We divided haplotype 3 so that those SNPs in LD block A comprised sub-haplotype 3A, and those in block B comprised sub-haplotype 3B. Eighteen of the SNPs within block A and only one SNP within block B had an r2 > 0.8 with the respective sub-haplotypes. A matrix depiction of pairwise r2 values between these is illustrated in Supplementary Fig. 1. The T30S (ss78456788) variant of SPANXC (in LD Block A) also demonstrated modest LD with sub-haplotype 3A (r2 = 0.73). Another variant altering an open reading frame within RBMX2P1 (rs1968987) directly marked sub-haplotype 3A (r2 = 1).
Only a subset of the SNPs demonstrating LD with the two sub-haplotypes had been genotyped as tagging SNPs in the training study group to enable an assessment of disease association. These included rs1012777, ss78456788 (T30S), rs12394263, ss78456800, rs5953578, rs845144, rs714076, rs881223, and ss78456818 in LD block A, and rs5907874 in LD block B. With only one exception, χ2 tests of association for these block A SNP--block B SNP pair haplotypes were each associated with prostate cancer in our training group (P range 0.0008 to 0.030). These SNP pairs and the original sliding window haplotype spanning the LD break each detect the association with prostate cancer with varying efficiencies.
Our study sought to identify the genetic variant predisposing to familial prostate cancer at Xq27, a locus initially identified by linkage study of American, Swedish, and Finnish hereditary prostate cancer pedigrees, and subsequently refined by linkage disequilibrium analysis of the Finnish familial prostate cancer cases. After a comprehensive effort in the present study, we identified a single candidate haplotype that was associated with familial prostate cancer within independent training and test study subjects. Although the replication was encouraging, the sample size of our test group was sufficiently small that an independent assessment of significance is warranted. Population structure is unlikely to represent a confounding factor within our study, as self-described ethnicity has recently been shown to accurately represent genetic ancestry among Americans of Northern European descent (Hunter et al. 2007; Tang et al. 2005). We believe that this haplotype represents the best candidate within the region for further investigation within additional study populations. If confirmed, these findings begin to clarify the X-linked heritable component of prostate cancer risk.
Supplementary Material
ACKNOWLEDGMENTS
We extend particular thanks to the study participants and to Drs Joseph Smith, Michael Cookson, Sam Chang, Richard Hock, William Maynard, Jason Pereira, and William Dupont. This work was supported by an award from the V Foundation, by a MERIT grant from the US Department of Veterans Affairs, by grant W81XWH-06-1-0057 from the Department of the Army, and by General Clinical Research Center grant M01 RR-00095 from the National Center for Research Resources, National Institutes of Health.
REFERENCES
- Baffoe-Bonnie AB, Smith JR, Stephan DA, Schleutker J, Carpten JD, Kainu T, Gillanders EM, Matikainen M, Teslovich TM, Tammela T, Sood R, Balshem AM, Scarborough SD, Xu J, Isaacs WB, Trent JM, Kallioniemi OP, Bailey-Wilson JE. A major locus for hereditary prostate cancer in Finland: localization by linkage disequilibrium of a haplotype in the HPCX region. Hum Genet. 2005a;117:307–316. doi: 10.1007/s00439-005-1306-z. [DOI] [PubMed] [Google Scholar]
- Baffoe-Bonnie AB, Smith JR, Stephan DA, Schleutker J, Carpten JD, Kainu T, Gillanders EM, Matikainen M, Teslovich TM, Tammela T, Sood R, Balshem AM, Scarborough SD, Xu J, Isaacs WB, Trent JM, Kallioniemi OP, Bailey-Wilson JE. A major locus for hereditary prostate cancer in Finland: localization by linkage disequilibrium of a haplotype in the HPCX region (Erratum) Hum Genet. 2005b;118:307. doi: 10.1007/s00439-005-1306-z. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bochum S, Paiss T, Vogel W, Herkommer K, Hautmann R, Haeussler J. Confirmation of the prostate cancer susceptibility locus HPCX in a set of 104 German prostate cancer families. Prostate. 2002;52:12–19. doi: 10.1002/pros.10078. [DOI] [PubMed] [Google Scholar]
- Brown WM, Lange EM, Chen H, Zheng SL, Chang B, Wiley KE, Isaacs SD, Walsh PC, Isaacs WB, Xu J, Cooney KA. Hereditary prostate cancer in African American families: linkage analysis using markers that map to five candidate susceptibility loci. Br J Cancer. 2004;90:510–514. doi: 10.1038/sj.bjc.6601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BL, Isaacs SD, Wiley KE, Gillanders EM, Zheng SL, Meyers DA, Walsh PC, Trent JM, Xu J, Isaacs WB. Genome-wide screen for prostate cancer susceptibility genes in men with clinically significant disease. Prostate. 2005;64:356–361. doi: 10.1002/pros.20249. [DOI] [PubMed] [Google Scholar]
- Cunningham JM, McDonnell SK, Marks A, Hebbring S, Anderson SA, Peterson BJ, Slager S, French A, Blute ML, Schaid DJ, Thibodeau SN. Genome linkage screen for prostate cancer susceptibility loci: results from the Mayo Clinic Familial Prostate Cancer Study. Prostate. 2003;57:335–346. doi: 10.1002/pros.10308. [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, Schork NJ. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res. 2001;11:143–151. doi: 10.1101/gr.148401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham JM, Camp NJ, Swensen J, Tavtigian SV, Albright LA. Confirmation of the HPCX prostate cancer predisposition locus in large Utah prostate cancer pedigrees. Hum Genet. 2005;116:179–185. doi: 10.1007/s00439-004-1220-9. [DOI] [PubMed] [Google Scholar]
- Gillanders EM, Xu J, Chang BL, Lange EM, Wiklund F, Bailey-Wilson JE, Baffoe-Bonnie A, Jones M, Gildea D, Riedesel E, Albertus J, Isaacs SD, Wiley KE, Mohai CE, Matikainen MP, Tammela TL, Zheng SL, Brown WM, Rokman A, Carpten JD, Meyers DA, Walsh PC, Schleutker J, Gronberg H, Cooney KA, Isaacs WB, Trent JM. Combined genome-wide scan for prostate cancer susceptibility genes. J Natl Cancer Inst. 2004;96:1240–1247. doi: 10.1093/jnci/djh228. [DOI] [PubMed] [Google Scholar]
- Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, Gudbjartsson D, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Blondal T, Jakobsdottir M, Stacey SN, Kostic J, Kristinsson KT, Birgisdottir B, Ghosh S, Magnusdottir DN, Thorlacius S, Thorleifsson G, Zheng SL, Sun J, Chang BL, Elmore JB, Breyer JP, McReynolds KM, Bradley KM, Yaspan BL, Wiklund F, Stattin P, Lindstrom S, Adami HO, McDonnell SK, Schaid DJ, Cunningham JM, Wang L, Cerhan JR, St Sauver JL, Isaacs SD, Wiley KE, Partin AW, Walsh PC, Polo S, Ruiz-Echarri M, Navarrete S, Fuertes F, Saez B, Godino J, Weijerman PC, Swinkels DW, Aben KK, Witjes JA, Suarez BK, Helfand BT, Frigge ML, Kristjansson K, Ober C, Jonsson E, Einarsson GV, Xu J, Gronberg H, Smith JR, Thibodeau SN, Isaacs WB, Catalona WJ, Mayordomo JI, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008 doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, Noskov VN, Solomon G, Otstot J, Isaacs W, Xu J, Schleutker J, Larionov V. Mutational analysis of SPANX genes in families with X-linked prostate cancer. Prostate. 2007;67:820–828. doi: 10.1002/pros.20561. [DOI] [PubMed] [Google Scholar]
- Lange EM, Chen H, Brierley K, Perrone EE, Bock CH, Gillanders E, Ray ME, Cooney KA. Linkage analysis of 153 prostate cancer families over a 30-cM region containing the putative susceptibility locus HPCX. Clin Cancer Res. 1999;5:4013–4020. [PubMed] [Google Scholar]
- Mathias RA, Gao P, Goldstein JL, Wilson AF, Pugh EW, Furbert-Harris P, Dunston GM, Malveaux FJ, Togias A, Barnes KC, Beaty TH, Huang SK. A graphical assessment of p-values from sliding window haplotype tests of association to identify asthma susceptibility loci on chromosome 11q. BMC Genet. 2006;7:38. doi: 10.1186/1471-2156-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleutker J, Matikainen M, Smith J, Koivisto P, Baffoe-Bonnie A, Kainu T, Gillanders E, Sankila R, Pukkala E, Carpten J, Stephan D, Tammela T, Brownstein M, Bailey-Wilson J, Trent J, Kallioniemi OP. A genetic epidemiological study of hereditary prostate cancer (HPC) in Finland: frequent HPCX linkage in families with late-onset disease. Clin Cancer Res. 2000;6:4810–4815. [PubMed] [Google Scholar]
- Tang H, Quertermous T, Rodriguez B, Kardia SL, Zhu X, Brown A, Pankow JS, Province MA, Hunt SC, Boerwinkle E, Schork NJ, Risch NJ. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet. 2005;76:268–275. doi: 10.1086/427888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008 doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- Woolf CM. An investigation of the familial aspects of carcinoma of the prostate. Cancer. 1960;13:739–744. doi: 10.1002/1097-0142(196007/08)13:4<739::aid-cncr2820130414>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, Wilkens E, Bujnovszky P, Bova GS, Walsh P, Isaacs W, Schleutker J, Matikainen M, Tammela T, Visakorpi T, Kallioniemi OP, Berry R, Schaid D, French A, McDonnell S, Schroeder J, Blute M, Thibodeau S, Gronberg H, Emanuelsson M, Damber JE, Bergh A, Jonsson BA, Smith J, Bailey-Wilson J, Carpten J, Stephan D, Gillanders E, Amundson I, Kainu T, Freas-Lutz D, Baffoe-Bonnie A, Van Aucken A, Sood R, Collins F, Brownstein M, Trent J. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20:175–179. doi: 10.1038/2477. [DOI] [PubMed] [Google Scholar]
- Yaspan BL, Breyer JP, Cai Q, Dai Q, Elmore JB, Amundson I, Bradley KM, Shu XO, Gao YT, Dupont WD, Zheng W, Smith JR. Haplotype analysis of CYP11A1 identifies promoter variants associated with breast cancer risk. Cancer Res. 2007;67:5673–5682. doi: 10.1158/0008-5472.CAN-07-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.