

Abstract
The engineering of proteins can illuminate their biological function and improve their performance in a variety of applications. Within the past decade, methods have been developed that facilitate the ability of chemists to manipulate proteins in a controlled manner. Here, we present the traceless Staudinger ligation as a strategy for the convergent chemical synthesis of proteins. This reaction unites a phosphinothioester and an azide to form an amide bond with no residual atoms. An important feature of this reaction is its ability to ligate peptides at non-cysteine residues, thereby overcoming a limitation of alternative strategies. Attributes of the traceless Staudinger ligation are discussed, and an overall comparison of known reagents for effecting the reaction is presented. General methods are elaborated for the synthesis of the most efficacious phosphinothiol for mediating the traceless Staudinger ligation, as well as for the preparation of phosphinothioester and azide fragments and the ligation of peptides immobilized on a solid support. Together, this information facilitates the use of this emerging method to engineer proteins.
Introduction
The advent of recombinant DNA technology and site-directed mutagenesis has made facile the substitution of one amino acid for another at any site within a protein (Smith, 1994). For protein chemists, however, there remains a major barrier—the genetic code, which only tolerates the introduction of twenty amino acids. Methods that overcome this limitation but still rely on the ribosome are limited to the substitution of a subset of α-amino acids and α-hydroxyacids.
Driven by the desire to achieve complete flexibility in the manipulation of primary structure, protein chemists are developing methods that enable nonnatural amino acids and artificial modules to be incorporated into proteins. The most popular such method is “native chemical ligation”, which was developed by Kent and coworkers as a means to join large peptide fragments (Kent, 2003). In native chemical ligation, the thiolate of an N-terminal cysteine residue of one peptide reacts with a C-terminal thioester of a second peptide, forming an amide bond after rapid S→N acyl group transfer. The ligation also works with selenocysteine—the rare “21st” amino acid—in the place of cysteine (Hondal et al., 2001; Hondal & Raines, 2002). An extension of native chemical ligation, “expressed protein ligation”, employs an engineered intein to access a polypeptide containing the C-terminal thioester (Muir, 2003). Although these methods have produced landmark results, both require a cysteine residue at the ligation juncture. Cysteine is uncommon, comprising <2% of all protein residues. The introduction of a new cysteine residue can be detrimental, as its high nucleophilicity and propensity to oxidize leads to undesirable side reactions. Accordingly, many natural proteins can be neither synthesized nor modified by a ligation method that relies on cysteine residues.
Traceless Staudinger Ligation
Emerging strategies for the unconstrained engineering of proteins avoid the requisite cysteine residues (Nilsson et al., 2005). Here, we describe one such strategy—the Staudinger ligation, which is based on the Staudinger reaction (Staudinger & Meyer, 1919). In the Staudinger reaction, a phosphine is used to reduce an azide to an amine: PR3 + N3R′ + H2O → O=PR3 + H2NR′ + N2(g). This reaction occurs via a stable intermediate, an iminophosphorane (R3P+−−NR′, also known less precisely as an “aza-ylide”), which has a nucleophilic nitrogen. Vilarrasa and others showed that this nitrogen can be acylated, both in intermolecular (i.e., three-component) and intramolecular (i.e., two-component) ligations (Bosch et al., 1995; Velasco et al., 2000). Hydrolysis of the resulting amidophosphonium salt gives an amide and a phosphine oxide. Bertozzi and coworkers showed that the phosphine itself can serve as the acyl group donor in a two-component ligation (Saxon & Bertozzi, 2000).
To apply the Staudinger reaction to peptide synthesis, we developed the use of a phosphinothiol to unite a thioester and azide, as shown in Fig. 1 (Nilsson et al., 2000; Nilsson et al., 2001). This phosphinothiol is bifunctional, having a thiol group that can be tethered to the C-terminus of a peptide fragment, and a phosphino group that can react with a peptide fragment that has an azido group at its N-terminus to form an iminophosphorane intermediate. Attack of the iminophosphorane nitrogen on the conjoined thioester carbon leads first to a tetrahedral intermediate, and then to an amidophosphonium salt (Soellner et al., 2006a). Hydrolysis of the amidophosphonium salt releases a phosphine oxide and produces a native amide bond between the two peptides. Significantly, no extraneous atoms remain in the amide product—the reaction is “traceless” (Nilsson et al., 2000). This attribute is a strict requirement for the use of the Staudinger ligation in the chemical synthesis of proteins or other molecules. It is noteworthy that the traceless Staudinger ligation mediated by a phosphinothiol couples the energetics of native chemical ligation with that of the Staudinger reaction (which is highly exergonic), resulting in an enormous thermodynamic driving force for the overall transformation (Nilsson et al., 2005).
FIG. 1.

Putative mechanism for the traceless Staudinger ligation of two peptides.
The kinetics of the traceless Staudinger ligation have been characterized by using a sensitive and continuous assay based on 13C NMR spectroscopy (Soellner et al., 2006a). In this assay, a phosphinothioester is allowed to react with a 13Cα-labeled azide in a deuterated solvent, and the course of the reaction is monitored over time. Significantly, intermediates do not accumulate, indicating that the rate-limiting step is the association of the phosphinothioester and the azide. For the reaction of AcGlySCH2PPh2 and N313CH2C(O)NHBn at room temperature, t1/2 = 7 min. The traceless Staudinger ligation proceeds without detectable (<0.5%) epimerization of the α-carbon of the azido acid (Soellner et al., 2002). This attribute is crucial for its application in protein chemistry, as all twenty proteinogenic amino acids except glycine have a stereogenic center at their α-carbon. The reaction of phosphinothioesters (but not phosphinoesters) with azides is also chemoselective in the presence of the functional groups in native proteins, and unprotected peptide fragments can be ligated with no undesirable side reactions (Soellner et al., 2006a). These attributes endow the Staudinger ligation with broad utility.
The traceless Staudinger ligation has been applied to the assembly of a protein from constituent peptides (Nilsson et al., 2003a), as well as the site-specific immobilization of peptides and proteins to a surface (Soellner et al., 2003; Gauchet et al., 2006). Variations of the Staudinger ligation have also been used in the synthesis of glycopeptides (He et al., 2004; Bianchi et al., 2005; Liu et al., 2006; Bianchi & Bernardi, 2006) and biomolecular labeling experiments in vitro (Tsao et al., 2005; Grandjean et al., 2005) and in vivo (Dube et al., 2006), and for drug delivery (Azoulay et al., 2006). As with auxiliary-mediated ligations (Nilsson et al., 2005), steric hindrance at the ligation junction (as in non-glycyl couplings) diminishes the ligation yield. Phosphinothiols that mediate the efficient coupling of non-glycyl amino acids are, however, now known (Soellner et al., 2006b; Tam et al., 2008).
Choice of Coupling Reagent
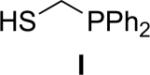
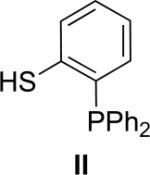
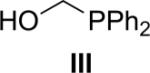
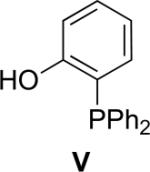
Several coupling reagents have been used in the traceless Staudinger ligation, with varied success. These compounds include phosphinomethanethiol I (Nilsson et al., 2001), phosphinothiophenol II (Nilsson et al., 2000), phosphinomethanol III (Saxon et al., 2000), phosphinoethanethiol IV (Han & Viola, 2004), and phosphinophenol V (Saxon et al., 2000). The efficacy of these coupling reagents in a model reaction between its AcGly(thio)ester and 13Cα-labeled N3GlyNHBn in a wet organic solvent has been compared directly (Soellner et al., 2006b), and the key results are listed in Table I. Traceless Staudinger ligations mediated by reagents II, III, and IV are sluggish compared to those by reagents I and V. Furthermore, coupling reagents II, III, and IV also display low ligation yields. The low rate and yield with II and IV could be due to the increased size of the ring that is formed during the nucleophilic attack of the iminophosphorane nitrogen on the thioester (e.g., to produce the tetrahedral intermediate in Fig. 1). Reagent III enabled a direct comparison of an ester and thioester reagent, and highlights the advantage of a good leaving group (thiolate versus alkoxide) in mediating the traceless Staudinger ligation. Finally, phosphinophenol V gave amide yields and reaction rates nearly indistinguishable from phosphinomethanethiol I. Although Staudinger ligation with V requires the formation of a six- rather than a five-membered ring during S→N acyl group transfer (Fig. 1), the conjugate base of V is a somewhat better leaving group than is that of I. Upon further investigation, ligations mediated by V were found to suffer a decrease in amide yield in the presence of the functional groups found in proteinogenic amino acids. This result is presumably due to the aryl ester of V being more electrophilic than the thioester of I, increasing its susceptibility to nonspecific acyl transfer reactions (e.g., with the ε-amino group of a lysine residue). On the contrary, Staudinger ligations performed with I can be performed on unprotected peptide fragments (Soellner et al., 2003; Liu et al., 2006; Gauchet et al., 2006).
Table I.
Effect of Coupling Reagent on the Rate and Product Distribution of the Staudinger Ligation

| Coupling reagent (HR) | k2 (10−3 M−1s−1) | Yield (%) |
|---|---|---|
 |
7.7 ± 0.3 | 95 |
 |
1.04 ± 0.05 | 38 |
 |
0.12 ± 0.01 | 11 |
 |
0.65 ± 0.01 | 39 |
 |
7.43 ± 0.03 | 99 |
Because of its high reaction rate, high ligation yields, and chemoselectivity, (diphenylphosphino)methanethiol (I) is the most efficacious of known reagents for mediating the traceless Staudinger ligation (Soellner et al., 2006b). Thiol-based reagents, (e.g., I) have another intrinsic advantage over hydroxyl-based reagents (e.g., V). The thiol-based reagents react readily with thioester fragments generated by expressed protein ligation or other methods to form phosphinothioesters poised for a traceless Staudinger ligation.
Alternatively, phosphinothiol I can be prepared from diphenylphosphine–borane complex and other commercial materials by two routes, designated as a (Soellner et al., 2002) and b (He et al., 2004) in Fig. 2, both with overall yields of 55%. A precursor that is common to both routes, phosphine–borane complex X, is stable to air and moisture and can be stored on the shelf at room temperature for months without any sign of oxidation or decomposition. Phosphine–borane complex X is also available from a commercial vendor (Sigma–Aldrich product #670359). Although fully deprotected phosphinothiol I is stable under Ar(g) for several days, it is best when prepared freshly from phosphine–borane complex X.
FIG. 2.

Routes for the synthesis of (diphenylphosphino)methanethiol (I).
Experimental Procedure: Synthesis of Phosphinothiol I
Route a
In route a of Fig. 2 (Soellner et al., 2002), a P–C bond is made by alkylation of a diphenylphosphine–borane complex with agent VII, which was known previously (Farrington et al., 1989). Thioacetic acid (50 g, 0.65 mol) and paraformaldehyde (20 g) are mixed and heated at 100° for 2 hr under Ar(g). The reaction mixture becomes clear and light yellow, which indicates that the reaction is complete. Distillation under a high vacuum (bp 36° at 0.1 mm Hg) gives the AcSCH2OH (VI) as a colorless oil (typical yield: 59 g, 0.65 mmol, 86%). AcSCH2OH (VI, 59 g, 0.56 mol) is cooled under Ar(g) in an ice bath, and PBr3 (50.5 g, 0.19 mol) is added dropwise slowly such that the reaction temperature does not exceed 8°. After the complete addition of PBr3, the reaction mixture is stirred for an additional 30 min in an ice bath, and then allowed to warm to room temperature. The reaction mixture is poured over an ice/water mixture (100 ml), and extracted with ether (3 × 100 ml). The organic extracts are dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure. The residue is distilled under a high vacuum (bp 53° at 0.1 mm Hg) to give alkylating agent VII as a colorless oil (typical yield: 0.80 g, 0.47 mmol, 84%). Spectral data should be as reported previously (Farrington et al., 1989).
Diphenylphosphine–borane complex (10.33 g, 51.6 mmol) is dissolved in dry DMF under Ar(g) and cooled to 0°. NaH (1.24 g, 51.6 mmol) is added slowly, and the mixture is stirred at 0° until bubbling ceases. Alkylating agent VII (8.73 g, 51.6 mmol) is then added, and the mixture is allowed to warm to room temperature and stirred for 12 hr. The product is concentrated under reduced pressure, and the residue is purified by flash chromatography (silica gel, 10% v/v EtOAc in hexanes). Phosphine–borane complex X is isolated as a colorless oil (typical yield: 12.8 g, 44.4 mmol, 86%), and can be stored under air in a flask or bottle for extended periods in this form. 1H NMR (300 MHz, CDCl3) δ 7.74–7.67 (m, 4 H), 7.54–7.41 (m, 6 H), 3.72 (d, J = 6 Hz, 2 H), 2.23 (s, 3 H), 1.51–0.53 (broad m, 3 H) ppm; 13C NMR (75 MHz, CDCl3) δ 192.94, 132.26 (d, J = 9.2 Hz), 131.61 (d, J = 2.3 Hz), 128.71 (d, J = 10.2 Hz), 127.43 (d, J = 55.4 Hz), 29.87, 23.59 (d, J = 35.5 Hz) ppm; 31P NMR (121 MHz, CDCl3) δ 19.40 (d, J = 59.3 Hz) ppm; typical MS (ESI) m/z 311.0806 (MNa+ = 311.0807).
Route b
In Route b of Fig. 2 (He et al., 2004), a P–C bond is made by addition of a diphenylphosphine–borane complex to formaldehyde. Diphenylphosphine–borane complex (2.45 g, 12.2 mmol) is dissolved in THF (7 mL). Formaldehyde (37% v/v in H2O, 7.16 mL) is added to the solution, followed by potassium hydroxide (825 mg, 14.7 mmol). The resulting bilayered solution is stirred overnight, and then concentrated under reduced pressure. The residue is dissolved in ethyl acetate (10 mL), and the layers are separated. The organic extracts are washed with brine, dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure. The crude oil is purified by flash chromatography (silica gel, 50% v/v CH2Cl2 in hexanes) to give phosphine–borane complex VIII as a colorless oil (typical yield: 2.81 g, 12.2 mmol, 99%). 1H NMR (CDCl3, 300 MHz) δ 7.73–7.68 (m, 4 H), 7.52–7.41 (m, 6 H), 4.40 (broad s, 2 H), 2.38 (broad s, 1 H), 1.50–0.50 (broad m, 3 H) ppm; 13C NMR (CDCl3, 75 MHz) δ 132.89 (d, J = 8.9 Hz), 131.79, 129.10 (d, J = 10.8 Hz), 126.88 (d, J = 54.5 Hz), 60.47 (d, J = 41.4 Hz) ppm; 31P NMR (CDCl3, 121 MHz) δ 17.61 (d, J = 58.9 Hz) ppm; typical MS (ESI) m/z 253.0927 (MNa+ = 253.0930).
Triethylamine (2.56 mL, 18.35 mmol) is added to a solution of phosphine–borane complex VIII (2.81 g, 12.2 mmol) in CH2Cl2 (36 mL), and the reaction mixture is cooled to 0° with an ice bath. Methanesulfonyl chloride (1.33 mL, 17.1 mmol) is added dropwise, and the resulting solution is allowed to warm to room temperature slowly (e.g., overnight). The solution is washed with 0.1 N HCl and brine, and the combined organic extracts are dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure. The residue is purified by flash chromatography (silica gel, 30% v/v ethyl acetate in hexanes) to give phosphine–borane complex IX as a pale yellow oil (typical yield: 3.14 g, 10.16 mmol, 83% yield). 1H NMR (CDCl3, 300 MHz) δ 7.76–7.71 (m, 4 H), 7.59–7.48 (m, 6 H), 4.90 (d, J = 1.90 Hz, 2 H), 2.87 (s, 3 H), 1.50–0.50 (broad m, 3 H) ppm; 13C NMR (CDCl3, 75 MHz) δ 133.09 (d, J = 9.50 Hz), 132.48, 129.343 (d, J = 10.0 Hz), 125.32 (d, J = 58.3 Hz), 64.68 (d, J = 37.8 Hz), 37.65 ppm; 31P NMR (CDCl3, 121 MHz) δ 18.87 (d, J = 57.8 Hz) ppm; typical MS (ESI) m/z 331.0719 (MNa+ = 331.0705).
Potassium thioacetate (1.4 g, 12.2 mmol) is added to a solution of phosphine–borane complex IX (3.14 g, 10.16 mmol) in anhydrous DMF (50 mL) under Ar(g). The resulting solution is stirred overnight, and then concentrated under reduced pressure. The residue is dissolved in ethyl acetate (25 mL), and the resulting solution is washed with water and brine. The combined organic extracts are dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure. The residue is purified by flash chromatography (silica gel, 30% v/v CH2Cl2 in hexanes) to give phosphine–borane complex X as a colorless oil (typical yield: 2.22 g, 7.7 mmol, 76%). Spectral data should be as reported for route a.
Phosphine–borane complex X (4.00 g, 13.9 mmol) is dissolved in toluene (140 ml) under Ar(g). 1,4-Diazabicylo[2.2.2]octane (DABCO) (1.56 g, 13.9) is added, and the mixture is heated at 40° for 4 hr. The product is concentrated under reduced pressure, dissolved in CH2Cl2 (50 ml), and washed with both 1 N HCl (20 ml) and saturated brine (20 ml). The organic layer is dried over MgSO4(s), and concentrated under reduced pressure. Phosphine XI is isolated as a colorless oil (typical yield: 3.62 g, 13.2 mmol, 95%) and is used without further purification. 1H NMR (CDCl3, 500 MHz) δ 7.43–7.40 (m, 4 H), 7.33–7.30 (m, 6 H), 3.50 (d, J = 4 Hz, 2 H), 2.23 (s, 3 H) ppm; 13C NMR (CDCl3, 125 MHz) δ 194.01, 136.42 (d, J = 13.6 Hz), 132.28 (d, J = 19.4 Hz), 128.69, 128.11 (d, J = 6.8 Hz), 29.83, 25.41 (d, J = 23.4 Hz) ppm; 31P NMR (CDCl3, 202 MHz) δ –15.11 ppm; typical MS (ESI) m/z 274.06 (MH+ = 275.0, fragments at 233.0, 199.2, 121.2).
Phosphine XI (17.27 g, 63.0 mmol) is dissolved in anhydrous methanol (0.40 L), and Ar(g) is bubbled through the solution for 1 hr. Sodium hydroxide (5.04 g, 126 mmol) is then added, and the mixture is stirred under argon for 2 hr. The product is concentrated under reduced pressure, and then dissolved in methylene chloride (0.30 L). The resulting solution is washed with 2 N HCl (2 × 0.10 L) and brine (0.10 L). The organic layer is dried over MgSO4(s), filtered, and concentrated under reduced pressure. The residue is purified by flash chromatography (alumina, 25% ethyl acetate in hexanes). (Diphenylphosphino)methanethiol (I) is isolated as a colorless oil (typical yield: 10.8 g, 46.6 mmol, 74%). 1H NMR (CDCl3, 300 MHz) δ 7.41–7.38 (m, 4 H), 7.33–7.26 (m, 6 H), 3.02 (d, J = 7.8 Hz, 2 H), 1.38 (t, J = 7.5 Hz, 1 H) ppm; 13C NMR (CDCl3, 75 MHz) δ 132.54 (d, J = 17.1 Hz), 128.86, 128.36, 128.14, 20.60 (d, J = 21.7 Hz) ppm; 31P NMR (CDCl3, 121 MHz) δ –7.94 ppm; typical MS (ESI) m/z 232.05 (MH+ = 233.0, fragments at 183.0, 155.0, 139.0, 91.2).
Preparation of the Azido Fragment
Three methods have been described for preparing a peptide with an N-terminal azido group. These methods are listed in Table II. Two of these methods involve a protected peptide on a solid support; the third involves an unprotected peptide in solution.
-
N1.
A synthetic azido acid (or peptide) is coupled to the N-terminus of a synthetic peptide on a solid support (Nilsson et al., 2003a; Soellner et al., 2003). Side-chain functional groups are protected from side reactions (as indicated by the triangles).
-
N2.
The N-terminal amino group of a peptide on solid support is converted into an azide by diazo transfer from (for example) triflyl azide in the presence of divalent copper ions (Rijkers et al., 2002). Side-chain functional groups are protected from reaction.
-
N3.
A protease-catalyzed peptide condensation reaction is used to introduce azido-dipeptides to the N-terminus of an unprotected peptide fragment (Liu et al., 2006). A large excess (10 equiv) of synthetic azido-dipeptides is needed, along with the protease subtilisin.
Table II.
Strategies for Preparation of the Azide Fragment
| Strategy | Route |
|---|---|
| N1 | 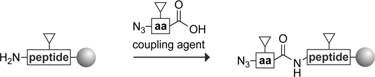 |
| N2 | |
| N3 | 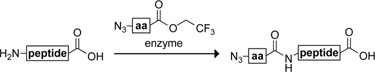 |
 = side-chain protecting group
= side-chain protecting group
Experimental Procedure: Strategy N1
The azido derivatives of amino acids can be prepared by a method described previously (Lundquist & Pelletier, 2001). In our example, azido-glycine is prepared by partially dissolving sodium azide (20.56 g, 317 mmol) by stirring in DMSO (880 ml) for 1.5 hr. Bromoacetic acid (20.96 g, 151 mmol) is added to this slurry, and the remaining NaN3 dissolves within minutes. The reaction mixture is stirred overnight at room temperature, before diluting with H2O (1.0 L) and adjusting the pH to 2.5 with concentrated HCl. The desired azido-glycine is extracted with EtOAc (2 × 1 L). The organic extracts are dried over anhydrous MgSO4(s), and then concentrated under reduced pressure to yield azido-glycine as a pale oil (typical yield: 11.1 g, 110 mmol, 73%). Spectral data should be as reported previously (Lundquist & Pelletier, 2001).
A desired (n – 1) peptide fragment (in our example, RNase A fragment 113–124: NPYVPVHFDASV (Nilsson et al., 2003a)) is synthesized by solid-phase peptide synthesis on a Novasyn® TGA resin loaded with FmocValOH (110 mg, 22 μmol) by using standard methods on an automated synthesizer. The resin containing the (n – 1) peptide fragment is swollen in DMF (1 ml) for 1 hr. Azido-glycine (10.1 mg, 100 μmol), PyBOP (52 mg, 100 μmol), HOBT (14 mg, 100 μmol), and diisopropylethylamine (DIEA, 35 μL, 200 μmol) are dissolved in DMF (4 ml), and this mixture is added to the resin. The resin is agitated for 2 hr by bubbling Ar(g) through the slurry. The resin is filtered, and this coupling protocol is repeated to ensure maximal coupling. After the second coupling, the resin containing the azido peptide is rinsed and dried under Ar(g).
Preparation of the Phosphinothioester Fragment
Many methods are known for installing a phosphinothioester at the C-terminus of a synthetic peptide (or module). Five of these methods are listed in Table III. All peptide fragments synthesized via solid-phase peptide synthesis have the potential of incorporating unnatural amino acids and synthetic modules anywhere within the peptide fragment.
-
C1.
A peptide fragment is synthesized on a sulfonamide-linker (“safety-catch”) resin (Backes & Ellman, 1999). After activation of the fully loaded resin with iodoacetonitrile, treatment with an excess of phosphinothiol I liberates the thioester fragment.
-
C2.
A peptide fragment is synthesized on an acid-sensitive resin (e.g., NovaSyn TGA resin or 2-chlorotrityl resin) and liberated with 1% (v/v) TFA, which leaves intact the amino-acid protecting groups. The C-terminus is then activated (e.g., with DCC, PyBOP, or NHS) and coupled with phosphinothiol I.
-
C3.
A peptide fragment is assembled by standard Fmoc chemistry on a 4-hydroxymethylphenylacetamidomethyl (PAM) or 4-hydroxymethylbenzoic acid (HMBA) resin (Sewing & Hilvert, 2001). The ester linkage is activated for cleavage by AlMe3 in the presence of an excess of phosphinothiol I. Epimerization at the C-terminal residue can occur, limiting this strategy to peptide fragments with a C-terminal glycine.
-
C4.
A peptide fragment is assembled by standard Fmoc chemistry on an ester-linked (acid-stable) resin, which is loaded with N-4,5-dimethoxy-2-mercaptobenzyl (Dmmb)–Ala. The Dmmb group undergoes an N→S acyl group shift under acidic conditions, and the resulting thioester can undergo transthioesterification (Kawakami et al., 2005).
-
C5.
A polypeptide with a C-terminal intein and resin-binding domain is produced by recombinant DNA technology. Transthioesterification with water-soluble phosphinothiol XII liberates the peptide from the resin, simultaneously forming the C-terminal phosphinothioester (Tam et al., 2007; Tam & Raines, 2009).
Table III.
Strategies for Preparation of the Phosphinothioester Fragment
| Strategy | Route |
|---|---|
| C1 | |
| C2 | |
| C3 | |
| C4 |  |
| C5 |  |
 = side-chain protecting group
= side-chain protecting group
Experimental Procedure: Strategy C1
First, a peptide is synthesized on resin. In our example (Nilsson et al., 2003a), FmocGlu(OtBu)OH is loaded onto 4-sulfamybutyryl resin as described previously (Backes & Ellman, 1999). 4-Sulfamylbutyryl resin (1 g, 1.12 mmol) is swollen in CHCl3 (25 ml) for 1 hr. DIEA (1.56 ml, 8.96 mmol) and FmocGlu(OtBu)OH (1.91 g, 4.48 mmol) is added to the resin. The reaction mixture is cooled to −20° under a flow of Ar(g). After 20 min, PyBOP (2.33 g, 4.48 mmol) is added to the solution and the resulting mixture is stirred, allowing the temperature to warm slowly to room temperature over a period of 8 hr. The resin is filtered immediately and rinsed with CHCl3. It is important to terminate the reaction after 8 hr so as to minimize epimerization (Backes & Ellman, 1999). The coupling protocol is repeated to ensure maximal loading. After the second coupling is complete, the resin is filtered, rinsed with CHCl3, and dried under Ar(g).
Fmoc-deprotection is achieved by swelling the resin in DMF. A solution of piperidine in DMF (30% v/v, 10 ml) is then added to the resin, and agitated for 2 hr. The resin is filtered, and rinsed with DMF (10 × 5 ml) and CH2Cl2 (10 × 5 ml).
To couple the subsequent amino acid, FmocCys(Trt)OH (2.62 g, 4.48 mmol), PyBOP (2.33 g, 4.48 mmol), and HOBT (0.605 g, 4.48 mmol) are dissolved in DMF (10 ml). DIEA (1.56 ml, 8.96 mmol) is added to the mixture, and the resulting solution is added to the resin described above. After agitating for 3 h, the resin is filtered and rinsed with DMF (5 × 10 ml) and CH2Cl2 (5 × 10 ml).
The linker between a resin and its pendant synthetic peptide (in our example, RNase A fragment 110–111) is then activated with iodoacetonitrile as follows (Nilsson et al., 2003a). The resin is swollen in CH2Cl2. A solution of iodoacetonitrile (3.4 ml, 46.8 mmol), DIEA (3.2 ml, 18.7 mmol), and NMP (75 ml) is filtered through a plug of basic alumina, and added to the resin. The resin is agitated for 18 hr, filtered, and washed with NMP (5 × 10 ml) and CH2Cl2 (5 × 10 ml).
The phosphinothioester is liberated by incubating the above resin (1.0 g, 1.12 mmol peptide loading) with a solution of phosphinothiol I (2.1 g, 9.0 mmol) in DMF (15 ml) for 12 hr under Ar(g). The resin is filtered, and rinsed with DMF (5 × 10 ml) and CH2Cl2 (5 × 10 ml), and the filtrate is concentrated under reduced pressure. The residue is purified by flash chromatography (silica gel, 30% v/v EtOAc in hexanes) to yield FmocCys(Trt)Glu(OtBu)SCH2PPh2 (typical yield: 0.71 g, 0.72 mmol, 64% based on a 1.12-mmol resin loading). 1H NMR (CDCl3, 300 MHz) δ 7.75–7.70 (m, 2 H), 7.57–7.55 (m, 2 H), 7.42–7.14 (m, 29 H), 6.68 (d, J = 6.6 Hz, 1 H), 5.13 (d, J = 8.1 Hz, 1 H), 4.56–4.50 (m, 1 H), 4.36–4.34 (m, 2 H), 4.19–4.17 (m, 1 H), 3.81–3.80 (m, 1 H), 3.44–3.38 (m, 2 H), 2.78–2.68 (m, 1 H), 2.61–2.57 (m, 1 H), 2.27–2.23 (m, 2 H), 2.11–1.95 (m, 1 H), 1.83–1.70 (m, 1 H), 1.37 (s, 9 H) ppm; 13C NMR (CDCl3, 75 MHz) δ 198.14, 171.89, 170.19, 155.81, 144.17, 143.59, 143.46, 141.10, 136.49 (d, J = 14 Hz), 132.69 (d, J = 4.2 Hz), 132.44 (d, J = 4.1 Hz), 129.41, 128.98, 128.38 (d, J = 6.6 Hz), 127.93, 127.59, 126.94, 126.74, 124.93, 119.80, 80.74, 67.21, 67.00, 58.50, 53.83, 46.89, 31.00, 27.85, 27.23, 25.45 (d, J = 24.8 Hz) ppm; 31P NMR (CDCl3, 121 MHz) δ −14.51 ppm; typical MS (ESI) m/z 1007.3340 (MNa+ = 1007.3371).
Experimental Procedure: Strategy C5
A water-soluble phosphinothiol can effect the traceless Staudinger ligation in purely aqueous medium in moderate yields, thereby integrating the traceless Staudinger ligation with expressed protein ligation (Tam et al., 2007). Incubation of the phosphinothiol and the chitin-bound peptide expressed via rDNA technology, and direct elution from the chitin resin yields the C-terminal phosphinothioester, which can then be used in Staudinger ligation with an azido-peptide fragment.
Proteins and peptide fragments can be produced with rDNA methods in which the fragment is fused with the Mxe intein and a chitin-binding domain (CBD) (Arnold et al., 2002). In our example (Tam et al., 2007), this method is performed on Met(−1)RNase A–Gly–intein–CBD fusion protein to generate its C-terminal phosphinothioester. The desired plasmid is transformed into E. coli BL21(DE3) cells. Luria–Bertani (LB) medium (5 ml) containing ampicillin (0.10 mg/ml) is inoculated with a single colony and grown for 16 hr at 37°. The cells are collected by centrifugation (2000g for 2 min), and resuspended in LB medium (4 ml). Four 4-liter flasks each containing 1 liter of LB medium with ampicillin (0.10 mg/ml) are then inoculated with the resuspended cells (1 ml to each flask) from the 16 hr culture. Cultures are grown with shaking at 37° until OD = 0.5 at 600 nm. Gene expression is then induced by the addition of isopropyl β-D-thiogalactopyranoside (IPTG; to 0.5 mM), and the cultures are grown for an additional 3–4 hr at 25°. The lower temperature prevents the formation of inclusion bodies. Cells are harvested by centrifugation, and the cell pellet is stored at −20°.
Frozen cells are thawed and suspended in lysis and column buffer (LCB), which is 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS)-NaOH buffer (pH 6.8) containing NaCl (0.5 M), ethylenediaminetetraacetic acid (EDTA; 0.1 mM), Triton X-100 (0.1% w/w). Cells are lysed by sonication, and the lysate is subjected to centrifugation at 15,000g for 30 min. The supernatant is applied slowly to an LCB-equilibrated column of chitin resin (New England Biolabs, Ipswich, MA). Approximately 6 ml of chitin resin is needed for 1 g of cells. The loaded resin is washed thoroughly with LCB (8 column-volumes), and LCB containing 0.5 M NaCl (2 column-volumes).
Intein-mediated cleavage is induced by incubating the resin with degassed cleavage buffer, which is 50 mM MOPS–NaOH buffer (pH 6.8) containing NaCl (0.5 M), EDTA (0.1 mM), and a water-soluble thiol such as 2-mercaptoethanesulfonic acid (MESNA) for 14 hr under Ar(g). The thiol effects the transthioesterification of the fusion protein to form a C-terminal thioester of the protein, which is eluted from the resin with 0.5 M NaCl (2 ml). The peptide thioester is precipitated by the addition to 1% (v/v) of an aqueous solution of sodium deoxycholate (NaDOC) (1% v/v) and by the addition to 2% (v/v) of an aqueous solution of trichloroacetic acid (TCA, 50% w/v). After mixing, the precipitate is collected by centrifugation (5,000g for 5 min), decanted, and resuspended in acetone to remove small-molecule additives. MALDI mass spectrometry can be used to confirm the identity of the peptide thioester. After dissolving the peptide thioester in the appropriate solvent/buffer, transthioesterification can be performed with a phosphinothiol to generate the C-terminal phosphinothioester. The resulting peptide phosphinothioester can be isolated by the above precipitation procedure using NaDOC and TCA.
Protein Assembly by Orthogonal Chemical Ligations
Perhaps the most well-characterized protein, bovine pancreatic ribonuclease (RNase A (Raines, 1998)), has been used to evaluate the efficacy of some of the strategies above. The 124 amino acids of RNase A were assembled by using a variety of sequential and convergent amide-bond forming reactions, including the traceless Staudinger ligation, as depicted in Fig. 3 (Nilsson et al., 2003a). The enzyme thus created is remarkable in that its peptide bonds were synthesized by four distinct processes, two of which are sequential (mRNA translation by a ribosome and solid-phase peptide synthesis) and two of which are convergent (native chemical ligation and traceless Staudinger ligation).
FIG. 3.

Route for the assembly of RNase A with solid-phase peptide synthesis, Staudinger ligation, and expressed protein ligation.
Experimental Procedure: Traceless Staudinger Ligation on a Solid Phase
The resin-bound azido-peptide (RNase A fragment 112–124: GNPYVPVHFDASV, 180 mg, 25 μmol) as synthesized with Strategy N1 is swollen in DMF for 1 hr. The C-terminal phosphinothioester of RNase A fragment 110–111 is synthesized with Strategy C1 as FmocCys(Trt)Glu(OtBu)SCH2PPh2 (99 mg, 100 μmol), dissolved in 10:1 DMF/H2O (1.5 ml), and added to the swollen resin. The slurry is agitated gently for 12 h, after which the solvent is removed by filtration, and the resin is rinsed with DMF (5 × 10 ml) and CH2Cl2 (5 × 10 ml). The resin is dried under high vacuum and then treated with a cleavage cocktail (38:1:1 TFA/H2O/ethanedithiol, 2 ml) for 2 hr. The resin is filtered, and added to ice-cold diethyl ether (20 ml) to precipitate the deprotected peptide, RNase A fragment 110–124. The peptide is purified by reverse-phase HPLC and can be analyzed by MALDI mass spectrometry. The ligated peptide can be elaborated further with orthogonal ligation methods. In our example, expressed protein ligation with the C-terminal thioester of RNase A fragment 1–109 gives full-length RNase A, as shown.
Prospectus
The traceless Staudinger ligation has joined the repertoire of ligation methods for the convergent synthesis of proteins. This method has been used along with others to assemble an entire protein. A putative strategy for the assembly of proteins is depicted in Fig. 4 (Nilsson et al., 2003b). Here, a target protein is divided into shorter fragments, and the ultimate C-terminal fragment is attached to a solid support. This immobilized fragment is capped with an α-azido acid and then reacted with a protected C-terminal phosphinothioester peptide fragment. The cycle is repeated until all fragments have been added. Deprotection and folding of the nascent polypeptide while still attached to the solid support (to avoid aggregation) yields a functional protein. The protein can be left attached to the resin for high-throughput assays or liberated for structure-function analyses in solution. The entire process is amenable to automation. Most notably, nonnatural amino acids or synthetic modules can be substituted for native ones, affording otherwise inaccessible proteins for otherwise unattainable goals.
FIG. 4.

Strategy for the chemical synthesis of proteins by iterative cycles of solid-phase peptide synthesis and solid-phase Staudinger ligation.
Experimental Procedure: General
All chemicals and reagents are available from Aldrich Chemical (Milwaukee, WI), with the exception of Fmoc-protected amino acids and alkanesulfonamide safety-catch resins, which are available from Novabiochem (San Diego, CA). Solution-phase reactions are monitored by thin-layer chromatography and visualized by UV light or staining with I2. Flash chromatography is performed with columns of silica gel 60, 230–400 mesh (Silicycle, Québec City, Québec, Canada). HPLC purification is performed on a C18 reverse-phase column.
The term “concentrated under reduced pressure” refers to the removal of solvents and other volatile materials using a rotary evaporator at water-aspirator pressure (<20 mm Hg) while maintaining the water-bath temperature below 40°. The term “high vacuum” refers to a vacuum (≤0.1 mm Hg) achieved by a mechanical belt-drive oil pump.
Peptide synthesis is performed by standard Fmoc-protection strategies using an automated synthesizer with HATU activation. Phosphorus-31 NMR spectra are proton-decoupled and referenced against an external standard of deuterated phosphoric acid. Mass spectra are obtained with electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI) techniques.
Acknowledgement
We are grateful to L. L. Kiessling for contributive discussions. Research on protein chemistry in the authors' laboratory is supported by Grant GM44783 (NIH).
References
- Arnold U, Hinderaker MP, Nilsson BL, Huck BR, Gellman SH, Raines RT. Protein prosthesis: A semisynthetic enzyme with a β-peptide reverse turn. J. Am. Chem. Soc. 2002;124:368–369. doi: 10.1021/ja026114n. [DOI] [PubMed] [Google Scholar]
- Azoulay M, Tuffin G, Sallem W, Florent JC. A new drug-release method using the Staudinger ligation. Bioorg. Med. Chem. Lett. 2006;16:3147–3149. doi: 10.1016/j.bmcl.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Backes BJ, Ellman JA. An alkanesulfonamide “safety-catch” linker for solid-phase synthesis. J. Org. Chem. 1999;64:2322–2330. [Google Scholar]
- Bianchi A, Russo A, Bernardi A. Neo-glycoconjugates: Stereoselective synthesis of α-glycosyl amides via Staudinger ligation reactions. Tetrahedron–Asymmetry. 2005;16:381–386. [Google Scholar]
- Bianchi A, Bernardi A. Traceless Staudinger ligation of glycosyl azides with triaryl phosphines: Stereoselective synthesis of glycosyl amides. J. Org. Chem. 2006;71:4565–4577. doi: 10.1021/jo060409s. [DOI] [PubMed] [Google Scholar]
- Bosch I, Urpí F, Vilarrasa J. Epimerization-free peptide formation from carboxylic-acid anhydrides and azido derivatives. Chem. Commun. 1995:91–92. [Google Scholar]
- Dube DH, Prescher JA, Quang CN, Bertozzi CR. Probing mucin-type O-linked glycosylation in living animals. Proc. Natl. Acad. Sci. USA. 2006;103:4819–4824. doi: 10.1073/pnas.0506855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrington GK, Kumar A, Wedler FC. A convenient synthesis of diethyl (mercaptomethyl)phosphonate. Org. Prep. Proced. Int. 1989;21:390–392. [Google Scholar]
- Gauchet C, Labadie GR, Poulter CD. Regio- and chemoselective covalent immobilization of proteins through unnatural amino acids. J. Am. Chem. Soc. 2006;128:9274–9275. doi: 10.1021/ja061131o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. On the preparation of carbohydrate-protein conjugates using the traceless Staudinger ligation. J. Org. Chem. 2005;70:7123–7132. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- Han S, Viola RE. Splicing of unnatural amino acids into proteins: A peptide model study. Prot. Pept. Lett. 2004;11:107–114. doi: 10.2174/0929866043478301. [DOI] [PubMed] [Google Scholar]
- He Y, Hinklin RJ, Chang JY, Kiessling LL. Stereoselective N-glycosylation by Staudinger ligation. Org. Lett. 2004;6:4479–4482. doi: 10.1021/ol048271s. [DOI] [PubMed] [Google Scholar]
- Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J. Am. Chem. Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002;347:70–83. doi: 10.1016/s0076-6879(02)47009-7. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Sumida M, Nakamura K, Vorherr T, Aimoto S. Peptide thioester preparation based on an N-S acyl shift reaction mediated by a thiol ligation auxiliary. Tetrahedron Lett. 2005;46:8805–8807. [Google Scholar]
- Kent S. Total chemical synthesis of enzymes. J. Pep. Sci. 2003;9:574–593. doi: 10.1002/psc.475. [DOI] [PubMed] [Google Scholar]
- Liu L, Hong ZY, Wong CH. Convergent glycopeptide synthesis by traceless Staudinger ligation and enzymatic coupling. ChemBioChem. 2006;7:429–432. doi: 10.1002/cbic.200500437. [DOI] [PubMed] [Google Scholar]
- Lundquist JT, Pelletier JC. Improved solid-phase peptide synthesis method utilizing α-azide-protected amino acids. Org. Lett. 2001;3:781–783. doi: 10.1021/ol0155485. [DOI] [PubMed] [Google Scholar]
- Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- Nilsson BL, Kiessling LL, Raines RT. Staudinger ligation: A peptide from a thioester and azide. Org. Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- Nilsson BL, Kiessling LL, Raines RT. High-yielding Staudinger ligation of a phosphinothioester and azide to form a peptide. Org. Lett. 2001;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]
- Nilsson BL, Hondal RJ, Soellner MB, Raines RT. Protein assembly by orthogonal chemical ligation methods. J. Am. Chem. Soc. 2003a;125:5268–5269. doi: 10.1021/ja029752e. [DOI] [PubMed] [Google Scholar]
- Nilsson BL, Soellner MB, Raines RT. Protein assembly to mine the human genome. In: Schneider MP, editor. Chemical Probes in Biology (NATO ASI Series) Kluwer Academic; Boston, MA: 2003b. pp. 359–369. [Google Scholar]
- Nilsson BL, Soellner MB, Raines RT. Chemical synthesis of proteins. Annu. Rev. Biophys. Biomol. Struct. 2005;34:91–118. doi: 10.1146/annurev.biophys.34.040204.144700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines RT. Ribonuclease A. Chem. Rev. 1998;98:1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- Rijkers DTS, van Vugt HHR, Jacobs HJF, Liskamp RMJ. A convenient synthesis of azido peptides by post-assembly diazo transfer on the solid phase applicable to large peptides. Tetrahedron Lett. 2002;43:3657–3660. [Google Scholar]
- Saxon E, Armstrong JI, Bertozzi CR. A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Sewing A, Hilvert D. Fmoc-compatible solid-phase peptide synthesis of long C-terminal peptide thioesters. Angew. Chem. Int. Ed. 2001;40:3395–3396. doi: 10.1002/1521-3773(20010917)40:18<3395::aid-anie3395>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Smith M. Nobel lecture. Synthetic DNA and biology. Biosci. Rep. 1994;14:51–66. doi: 10.1007/BF01210301. [DOI] [PubMed] [Google Scholar]
- Soellner MB, Nilsson BL, Raines RT. Staudinger ligation of α-azido acids retains stereochemistry. J. Org. Chem. 2002;67:4993–4996. doi: 10.1021/jo025631l. [DOI] [PubMed] [Google Scholar]
- Soellner MB, Dickson KA, Nilsson BL, Raines RT. Site-specific protein immobilization by Staudinger ligation. J. Am. Chem. Soc. 2003;125:11790–11791. doi: 10.1021/ja036712h. [DOI] [PubMed] [Google Scholar]
- Soellner MB, Nilsson BL, Raines RT. Reaction mechanism and kinetics of the traceless Staudinger ligation. J. Am. Chem. Soc. 2006a;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]
- Soellner MB, Tam A, Raines RT. Staudinger ligation of peptides at non-glycyl residues. J. Org. Chem. 2006b;71:9824–9830. doi: 10.1021/jo0620056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger H, Meyer J. New organic compounds of phosphorus. III. Phosphinemethylene derivatives and phosphinimines. Helv. Chim. Acta. 1919;2:635–646. [Google Scholar]
- Tam A, Soellner MB, Raines RT. Water-soluble phosphinothiols for traceless Staudinger ligation and integration with expressed protein ligation. J. Am. Chem. Soc. 2007;129:11421–11430. doi: 10.1021/ja073204p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, Raines RT. Coulombic effects on the traceless Staudinger ligation in water. Bioorg. Med. Chem. 2009;17:1055–1063. doi: 10.1016/j.bmc.2008.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, Soellner MB, Raines RT. Electronic and steric effects on the rate of the traceless Staudinger ligation. Org. Biomol. Chem. 2008;6:1173–1175. doi: 10.1039/b802336k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao ML, Tian F, Schultz PG. Selective Staudinger modification of proteins containing p-azidophenylalanine. ChemBioChem. 2005;6:2147–2149. doi: 10.1002/cbic.200500314. [DOI] [PubMed] [Google Scholar]
- Velasco MD, Molina P, Fresneda PM, Sanz MA. Isolation, reactivity and intramolecular trapping of phosphazide intermediates in the Staudinger reaction of tertiary phosphines with azides. Tetrahedron. 2000;56:4079–4084. [Google Scholar]