

Abstract
Polyomavirus JC (JCV) infects oligodendrocytes and astrocytes in the brain and is the cause of the demyelinating disease progressive multifocal leukoencephalopathy (PML). In cell culture, JCV infection is characterized by severe damage to cellular DNA, which begins early in infection, and a viral cytopathic effect, which is observed late in infection. Nevertheless, these JCV-infected cells show a low level of apoptosis, at both the early and late stages of infection. This suggests that there is conflicting interplay between viral anti-apoptotic pathways that seek to optimize virus production, e.g. through T antigen (T-Ag)–p53 interaction, and cellular pro-apoptotic pathways that seek to eliminate virally infected cells. The apoptosis regulatory protein BAG3 is a member of the human Bcl-2-associated athanogene (BAG) family of proteins, which function as molecular co-chaperones through their interaction with Hsc70/Hsp70 and function in the regulation of the cellular stress response, proliferation and apoptosis. This study showed that BAG3 protein is downregulated upon JCV infection and that this effect is mediated by JCV T-Ag via repression of the BAG3 promoter. The site of action of T-Ag was mapped to an AP2 site in the BAG3 promoter, and gel shift and chromatin immunoprecipitation assays showed that T-Ag inhibited AP2 binding to this site, resulting in downregulation of BAG3 promoter expression. Using BAG3 and T-Ag expression and BAG3 siRNA, it was found that BAG3 and T-Ag had antagonistic effects on the induction of apoptosis, being anti-apoptotic and pro-apoptotic, respectively. The significance of these interactions to the JCV life cycle is discussed.
INTRODUCTION
The human polyomavirus JC (JCV) opportunistically infects the oligodendrocytes and astrocytes of the brain during development of the demyelinating disease progressive multifocal leukoencephalopathy (PML). The pathology of PML is thought to involve the destruction of oligodendrocytes, the myelin-producing cells of the brain, by lytic infection with JCV. In contrast, astrocytes do not undergo lytic infection but rather adopt a ‘bizarre’ morphology, yet are still productively infected as judged by the production of viral capsid protein observed by immunohistochemistry and virions observed by electron microscopy (Del Valle et al., 2008; Mázló et al., 2001). PML occurs primarily in individuals with highly suppressed immune function, especially those with human immunodeficiency virus (HIV)/AIDS (Hou & Major, 2000; Khalili et al., 2006). The JCV genome is a circular, supercoiled DNA, which is small in size (5130 bp) and has two coding regions (early and late) and a non-coding regulatory region (Frisque et al., 1984; Cole, 1996). Infection with JCV is common in childhood after which the virus becomes latent. In circumstances of severe immunosuppression, e.g. HIV/AIDS, JCV can reactivate in the central nervus system (CNS) leading to the destruction of oligodendrocytes, which produce myelin, and to PML (Hou & Major, 2000; Khalili et al., 2006).
We have developed a cell-culture system to study JCV infection using primary human fetal astrocytes, which support viral replication (Radhakrishnan et al., 2003, 2004). During the early phase of infection, JCV T antigen (T-Ag) is expressed and this protein can alter the activities of several cellular transcription factors that control cellular proliferation and apoptosis, including p53 (Krynska et al., 1997), pRb (Krynska et al., 1997) and several others (reviewed by White & Khalili, 2004). Infection of cells by JCV results in a high degree of cellular stress. Thus, in the late stage (14–16 days) of infection, severe cytopathic effect (CPE) is observed, as evidenced by the presence of numerous vacuoles in the cytoplasm, a watery appearance of the cytoplasm, breakdown of the organelles and cell detachment (Frisque & White, 1992). However, we have found that these late-stage-infected cells exhibit only a low level of apoptosis (6 %) as measured by fluorescence-activated cell sorting (FACS) analysis and Western blotting of lamin A/C cleavage (Darbinyan et al., 2007). Similar results were obtained by Seth et al. (2004) who reported that human CNS progenitor-derived astrocyte cell cultures supported progressive JCV infection leading to CPE but not to apoptosis, as measured by capsase-3 labelling or a terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay. To understand this phenomenon better, we have been interested in the changes in cellular pro-apoptotic and anti-apoptotic processes occurring during JCV infection. In addition to the well-known binding of p53 by T-Ag, which was first reported for simian virus 40 (SV40) (Lane & Crawford, 1979; Linzer & Levine, 1979), we recently found that JCV infection induces expression of the anti-apoptotic protein survivin (Piña-Oviedo et al., 2007). We now report that JCV infection modulates the apoptotic regulatory protein BAG3.
BAG3 is a member of the human BAG (Bcl-2-associated athanogene) family of molecular co-chaperone proteins (BAG1–BAG6; Doong et al., 2002; Rosati et al., 2007a). These proteins share a common C-terminal BAG domain, which mediates interaction with the ATPase domain of Hsc70/Hsp70 (Brive et al., 2001), and divergent N termini, the specific features of which confer the different properties and functions on each member (Kabbage & Dickman, 2008). The BAG family is evolutionarily ancient, being found in animals, yeasts and plants (Kabbage & Dickman, 2008). The first BAG family member (BAG1) was cloned based on its binding to the anti-apoptotic Bcl-2 protein (Takayama et al., 1995). Likewise, BAG3 (also known as Bis and CAIR-1) was cloned by its ability to bind to Bcl-2 (Lee et al., 1999).
BAG proteins participate in a wide variety of cellular processes including cell survival, cellular stress response, proliferation, migration and apoptosis (Doong et al., 2002; Kabbage & Dickman, 2008; Rosati et al., 2007a). By interacting with the ATPase domain of Hsc70/Hsp70, BAG proteins modulate cellular responses under physiological and stress conditions. Some BAG proteins, including BAG3, have been demonstrated to inhibit Hsc70/Hsp70 chaperone activity in vitro (Takayama et al., 1999). BAG3 also contains a PXXP proline-rich motif, also found in BAG6, which mediates the interaction of BAG3 with the SH3 domain of PLC-γ (Doong et al., 2000). BAG3 is unique among the BAG proteins in possessing a WW protein–protein interaction domain, the function of which remains unknown.
BAG3 is implicated in the pathogenesis of neoplasia via its ability to regulate stress-induced apoptosis in a pro-survival fashion. This regulation occurs at a number of levels including cytochrome c release, apoptosome assembly and others (Bonelli et al., 2004; Rosati et al., 2007a). In addition, BAG3 binds PLC-γ (Doong et al., 2000) and also binds and synergizes with Bcl-2 in preventing cell death (Lee et al., 1999). Other cellular signalling molecules that have been reported to be regulated by BAG3 include Raf-1, CDK-4 and EGFR (Doong et al., 2003) and also focal adhesion kinase (Kassis et al., 2006). Notably, BAG3 expression can be induced by stress-inducing agents such as high temperatures and heavy metals (Franceschelli et al., 2008; Pagliuca et al., 2003) and by HIV-1 infection (Rosati et al., 2007b). In neuroblastoma cells, FGF-2 treatment activates BAG3 expression and this is mediated by two Egr-1 sites in the BAG3 promoter (Gentilella et al., 2008). We recently reported that downregulation of BAG3 protein using an RNA interference approach sensitized primary microglial cells to caspase-3 activation following HIV-1 infection, suggesting a role for BAG3 in the balance of cell death versus survival during HIV-1 infection (Rosati et al., 2009).
We now report that BAG3 expression is downregulated upon JCV infection through the action of JCV T-Ag on the expression of the BAG3 promoter.
METHODS
JCV infection of astrocytes.
Primary cultures of human fetal astrocytes were prepared and infected with the chimeric Mad-1/SVEΔ virus as described previously (Radhakrishnan et al., 2003, 2004) at an m.o.i. of 1. Cells were freshly prepared and purified for each experiment. Cultures at 5, 9 and 15 days after infection were analysed as representing early, middle and late stages of infection, respectively, together with uninfected control cultures at the same time points.
Cell culture, transfection and plasmids.
U-87 MG human glioblastoma cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10 % fetal bovine serum and transfected using a calcium phosphate precipitation method (Graham & van der Eb, 1973), except for the chromatin immunoprecipitation (ChIP) experiments where FuGENE 6 (Roche) was used according to the manufacturer's instructions. Luciferase reporter plasmids containing the BAG3 promoter (full-length and some of the deletion mutants) have been described previously (Gentilella et al., 2008). For the –85 to +306 BAG3 promoter deletion mutants, a luciferase reporter plasmid was constructed by PCR amplification of a KpnI–HindIII fragment and ligation with KpnI/HindIII-digested pGL3 Luciferase (Promega). The plasmid pcDNA3/zeo/JCVT expressing JCV T-Ag under the control of the human cytomegalovirus (CMV) promoter has been described previously (Lassak et al., 2002). Expression plasmids for AP2α and AP2γ were a kind gift from Dr Ronald J. Weigel, University of Iowa, USA (McPherson & Weigel, 1999). A specific small interfering RNA (siRNA) targeting BAG3 mRNA (5′-AAGGUUCAGACCAUCUUGGAA-3′) and a non-specific siRNA (5′-CAGUCGCGUUUGCGACUGG-3′) were purchased from Dharmacon. The BAG3 siRNA was selected for high specificity and lack of off-target effect at the concentration used (Gentilella et al., 2008). For transient transfection assays, U-87 MG cells plated in six-well plates were transfected with reporter construct alone (0.5 μg) or with expression plasmids (0.5 μg). Importantly, the total amount of DNA in all transfections was normalized with pCMV vector DNA to keep the amount of DNA constant. Cells were harvested after 48 h in reporter lysis buffer (Promega) and luciferase activity was determined with 5–10 μg protein using a dual-luciferase assay kit (Promega) as described previously (Amini et al., 2005). The pRLTK plasmid was the internal control for transfection efficiency.
Immunocytochemistry of JCV-infected cells.
Primary human fetal astrocytes were prepared and seeded on polylysine-coated glass chamber slides and left uninfected or infected with JCV (Mad-1/SVEΔ). At 5 and 15 days post-infection (p.i.), cells were fixed with 4 % paraformaldehyde and analysed by immunocytochemistry as described previously (Darbinyan et al., 2007; Radhakrishnan et al., 2003).
Preparation of protein extracts and immunoblot analysis.
For preparation of whole-cell extracts, transfected or treated cells were washed with cold PBS and solubilized in lysis buffer [50 mM Tris/HCl (pH 7.4), 150 mM NaCl, 0.1 % NP-40 and 1 % protease inhibitors cocktail (Sigma)]. Cell debris was removed by centrifugation for 5 min at 4 °C. Fifty micrograms of protein was eluted with Laemmli sample buffer, heated at 95 °C for 10 min and separated by 10 % SDS-PAGE. For Western blot analysis, protein samples or 50 μl conditioned medium were resolved by SDS-PAGE and transferred to nitrocellulose membranes as described previously (White et al., 2008). Proteins were visualized by using an enhanced chemiluminescence detection system, according to the manufacturer's instructions (ECL+; Amersham).
Antibodies.
The antibodies against the following proteins were used: JCV T-Ag (Pab2; clone PAb416, Oncogene Research Products), α-tubulin (T6074; Sigma), Grb2 (610111; BD Biosciences), caspase-3 (Cell Signalling) and AP2α (Santa Cruz Biotechnology). We have previously described a rabbit polyclonal antibody against JCV agnoprotein and VP1 (Del Valle et al., 2002). Rabbit polyclonal antibody (TOS-2) for Western blotting and mouse monoclonal antibody (AC-1) for immunocytochemistry were used for BAG3 (both from Alexis Biochemicals).
Gel shift assay.
Cells were transfected with and without JCV T-Ag expression plasmid for 48 h. Nuclear proteins were extracted and 10 μg was incubated with 50 000 c.p.m. of a 32P-labelled double-stranded oligonucleotide probe as described previously (Darbinian-Sarkissian et al., 2006; Romagnoli et al., 2008). Probes used in the gel shifts corresponded to the BAG3 promoter AP2-binding site (−146 to –125; 5′-CGCGCCCGCCCGCGGCGACTCC-3′) and Ets-binding site (−104 to –79; 5′-TCGGAAGGGGGAGGGGCGGGAGGAGG-3′).
ChIP assay.
ChIP assays were carried out using a ChIP assay kit (Upstate Biotechnology), as described previously (Darbinian-Sarkissian et al., 2006; Gentilella et al., 2008). Briefly, U-87 MG cells were transfected with and without T-Ag expression plasmid for 48 h. Cells were harvested and treated with 1 % formaldehyde to cross-link the chromatin. Cell lysates (from 106 cells) were sonicated on ice to break the chromatin DNA and treated with AP2 antibody (2 μg). PCR was performed using primers complementary to the BAG3 promoter region harbouring both the putative AP2- and Ets-binding sites. The primer sequences were 5′-GACGGCCCCAGTCCAGCTCG-3′ (forward) and 5′-CTGAGTCATCGGCTATAATCG-3′ (reverse), and generated a 204 bp PCR product. DNA samples were analysed by electrophoresis on 1.2 % agarose gels, stained with ethidium bromide and transferred to Hybond-N nylon membranes (Amersham). The filter was pre-hybridized for 1 h at 42 °C with Ultrahyb (Ambion) and probed using 106 c.p.m. ml−1 of a terminally labelled oligonucleotide containing the Egr-1 binding site labelled with T4 polynucleotide kinase (Roche) and [γ-32P]dATP. After hybridization for 20 h, the blot was rinsed twice with 2× SSC/0.1 % SDS at 42 °C and washed twice with 0.2× SSC/0.1 % SDS at 42 °C for 20 min.
Hydrogen peroxide (H2O2) treatment.
U-87 MG cells were transfected with BAG3, T-Ag and/or empty vector. Equal amounts of DNA were used in each transfection. Alternatively, cells were treated with BAG3 siRNA or non-specific siRNA as described above. After 4 days, cells were treated with 0.5 mM H2O2.
Measurement of caspase-3 activity.
Caspase-3 activity was assayed using the substrate DEVD-aminoluciferin from a Caspase-Glo 3/7 assay kit (Promega) as described previously (White et al., 2008), using a Turner Designs Luminometer TD-20/20 (Promega).
Trypan blue exclusion viability assay.
Trypan blue exclusion was performed as described previously (White et al., 2008) to assess cell viability.
FACS analysis.
Cells were harvested by trypsinization followed by the addition of complete medium. Cells were washed with PBS and fixed in ice-cold 70 % ethanol. After incubation for 24 h at −20 °C, cells were washed with PBS containing 1 % BSA, stained with 10 μg propidium iodide ml−1 in PBS containing 250 mg RNase A ml−1 and incubated at 37 °C for 30 min in the dark before analysis by FACS. Flow cytometry was performed with a Becton Dickinson FACScan flow cytometer. Data were analysed using the ModFitLT v2.0 (PMAC) software.
RESULTS
BAG3 protein level is downregulated during JCV infection
Primary human fetal astrocytes were infected with JCV and protein was extracted from these and uninfected controls at 5, 9 and 15 days p.i. Western blotting was performed for BAG3, JCV T-Ag, VP1, agnoprotein and caspase-3, using α-tubulin as a loading control. BAG3 was downregulated at all time points p.i. (Fig. 1a, lanes 1–4). No change in the level of BAG3 was observed in uninfected control cultures collected at the same time points (Fig. 1a, lanes 5–7). The levels of BAG3 during the time course of infection were quantified by densitometry (Fig. 1b). BAG3 was downregulated approximately 2.5-fold at days 5 and 9 p.i. when T-Ag expression was detectable, and downregulation increased to 4-fold at 15 days p.i., when very high levels of T-Ag were observed. A small amount of cleaved caspase-3 was observed at 15 days p.i. consistent with our earlier observation of a low level of apoptosis (6 %) late in infection (Darbinyan et al., 2007). Next, astrocytes were seeded into poly-l-lysine-treated glass chamber slides, infected with JCV or mock-infected, and fixed for immunocytochemistry at 5 and 15 days p.i. Fig. 1(c) shows the results of immunocytochemistry for BAG3, T-Ag and agnoprotein. Robust labelling of BAG3 was seen in the cytoplasm of uninfected cells at both time points. In the JCV-infected cells, expression of BAG3 was downregulated at 5 days p.i. and even more so at 15 days p.i. Most of the cells in the culture were positive for nuclear T-Ag and agnoprotein.
Fig. 1.

Expression of BAG3 during JCV infection. (a) Primary human fetal astrocytes were infected with JCV (Mad-1/SVEΔ) and harvested at the time points indicated (lanes 1–4). Western blot analysis for BAG3 was performed to monitor cellular BAG3 levels. Western blot analyses for T-Ag, VP1 and agnoprotein (Agno) were performed to monitor viral infection, and α-tubulin was used as a loading control. BAG3 levels were also monitored in uninfected cultures at the same time points (lanes 5–7). Protein sizes are indicated in kDa. (b) The levels of BAG3 and α-tubulin in JCV-infected astrocytes were measured by densitometry of the Western blots in (a). After normalization to α-tubulin, the relative intensities of the BAG3 bands are shown at each time point relative to lane 1 (100 %). (c) In another experiment, immunocytochemistry of primary human fetal astrocytes was performed for BAG3 after infection with JCV or mock infection. Immunocytochemistry of T-Ag and agnoprotein was also performed. Magnification, ×400.
Ectopic expression of JCV T-Ag downregulates BAG3 expression
To determine whether the effect of JCV infection on BAG3 protein level was due to T-Ag, U-87 MG cells were transfected with various amounts of plasmid expressing JCV T-Ag and analysed by Western blotting (Fig. 2a). Note that the total amount of DNA used for each transfection was kept constant by the addition of pCMV vector plasmid. Expression of T-Ag and BAG3 was quantified by densitometry (Fig. 2b). BAG3 was downregulated approximately threefold with 2 μg plasmid.
Fig. 2.
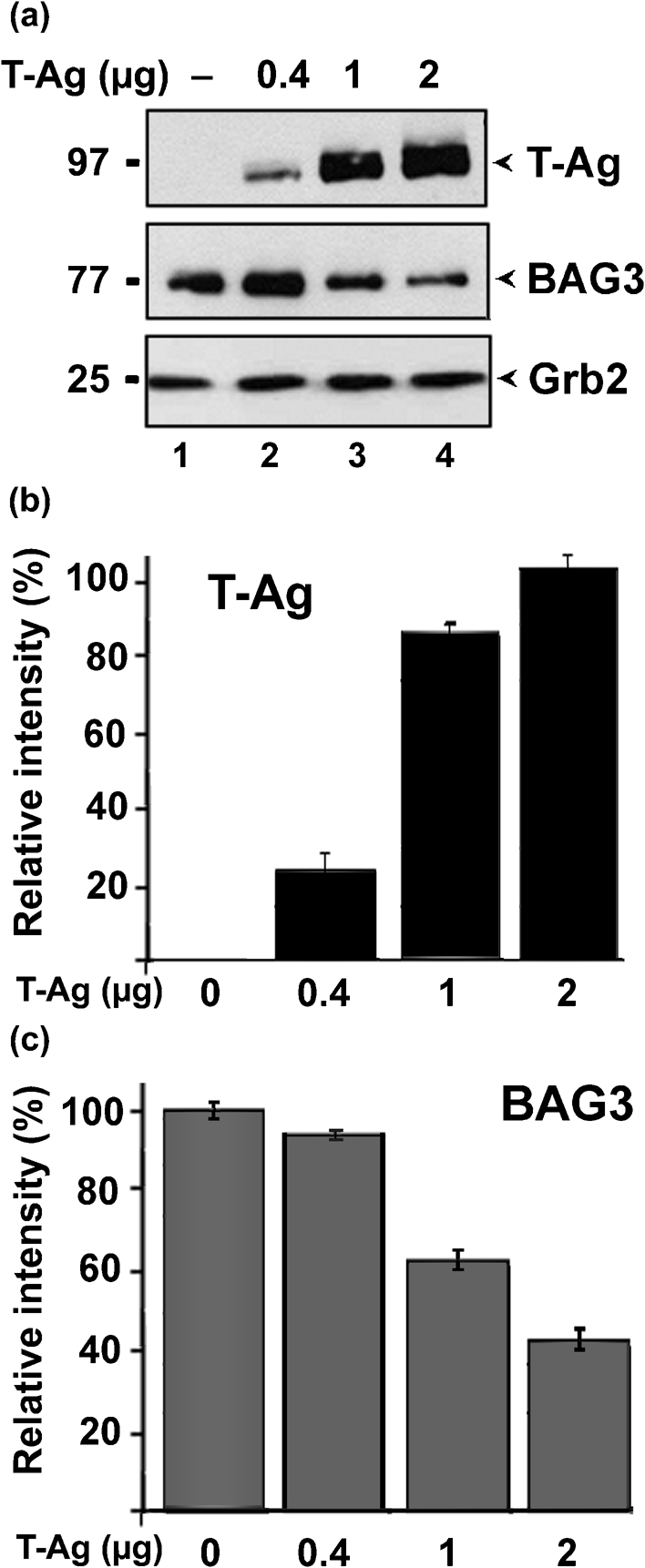
Effect of ectopic JCV T-Ag on BAG3 expression. (a) U-87 MG cells were transfected with various amounts of plasmid expressing JCV T-Ag, as indicated. The amount of DNA used in each transfection was kept constant using pCMV empty plasmid vector DNA. After 48 h, cells were harvested and analysed by Western blot for expression of T-Ag and BAG3. Grb2 served as a loading control. Protein sizes are indicated in kDa. (b, c) The levels of Grb2 and either T-Ag (b) or BAG3 (c) were measured by densitometry of the Western blots in (a). After normalization to Grb2, the relative intensity of the T-Ag or BAG3 bands is shown for each amount relative to the maximal amount detected, i.e. lane 4 or lane 1, respectively, (100 %).
JCV T-Ag downregulates the BAG3 promoter
To determine whether the effect of JCV T-Ag levels occurred at the transcriptional level, U-87 MG cells were transfected with a reporter plasmid containing the BAG3 promoter driving expression of the firefly luciferase reporter gene together with various amounts of plasmid expressing JCV T-Ag (Fig. 3a). Again, the total amount of DNA was constant for each transfection. The activity of the BAG3 promoter was downregulated approximately threefold with 2 μg plasmid. Next, we compared the effect of T-Ag on the activity of the BAG3 promoter with two control plasmids that were either sensitive (JCV late promoter) or insensitive (CMV promoter) to T-Ag regulation. As expected, the activity of the JCV late promoter was upregulated by T-Ag, whilst the CMV promoter was unaffected. Again, the activity of the BAG3 promoter was downregulated by T-Ag (Fig. 3b).
Fig. 3.

Effect of ectopic JCV T-Ag expression on the BAG3 promoter. (a) U-87 MG cells were transfected with a luciferase reporter plasmid containing the full-length BAG3 promoter with or without plasmid expressing JCV T-Ag, as indicated, using pCMV vector plasmid to keep the amount of DNA constant in each transfection. After 48 h, cells were harvested and assayed for luciferase activity. (b) U-87 MG cells were transfected with or without plasmid expressing JCV T-Ag, as indicated, together with reporter plasmids expressing luciferase driven by the full-length BAG3 promoter, the JCV late promoter or the CMV promoter. After 48 h, the cells were harvested and assayed for luciferase activity.
Defining the T-Ag-responsive element of the BAG3 promoter using promoter deletion mutants
A series of deletion mutants was created in the BAG3 promoter. The transcriptional activity of these mutants in driving luciferase expression was examined in U-87 MG cells in the presence and absence of expression plasmid for JCV T-Ag (Fig. 4). Mutants deleted up to nt –205 and –146 had 50 and 57 % relative activity with T-Ag present, respectively, i.e. they were inhibited about twofold by T-Ag. On the other hand, mutants deleted up to nt –85 and –26 had 110 and 114 % relative activity, respectively, i.e. they were largely unaffected by T-Ag. Thus, these mutants defined the region between nt –146 and −85 as being necessary for responsiveness to T-Ag.
Fig. 4.

Deletion analysis of the BAG3 promoter. A series of deletion mutants was created in the human BAG3 promoter as shown. Reporter plasmids containing the full-length and mutant promoters driving the luciferase gene were transfected into U-87 MG cells in the presence or absence of plasmid expressing JCV T-Ag. After 48 h, the cells were harvested and assayed for luciferase activity. For each construct, the percentage activity is given as activity in the presence of T-Ag relative to its absence: 100 × (activity in presence of T-Ag)/(activity in absence of T-Ag). Thus, the lower the percentage, the more sensitive the promoter construct to inhibition by T-Ag.
Implication of the AP2 site in T-Ag regulation of the BAG3 promoter by gel shift and ChIP assays
The T-Ag-sensitive region, defined by the data in Fig. 4, contains predicted binding sites for the transcription factors AP2 and Ets1. Next, we performed gel shift assays using double-stranded oligonucleotides corresponding to these sites with nuclear extracts from cells transfected with and without T-Ag. First, we performed gel shifts with an oligonucleotide (nt −146 to −125) corresponding to the AP2 site. A single distinct band was observed for untransfected cells (Fig. 5a, lane 2) and this band disappeared in the presence of T-Ag (Fig. 5a, lane 3). The band was competed away by the addition of cold AP2 oligonucleotide (Fig. 5a, lanes 4 and 5) but not with non-specific oligonucleotide (Fig. 5a, lanes 6 and 7). The AP2 gel shift was also performed in the presence of antibody specific for AP2α and the band was found to be supershifted (Fig. 5a, lane 8) but not in the presence of normal mouse serum (Fig. 5a, lane 10). These data indicated that JCV T-Ag prevents the binding of AP2 to its site in the BAG3 promoter.
Fig. 5.

Gel shift analysis and ChIP assay of the effect of JCV T-Ag at the AP2 site and Ets site of the BAG3 promoter. (a) A gel shift assay was performed with a probe corresponding to the AP2 site of the BAG3 promoter (nt −146 to −125) using nuclear extracts from U-87 MG cells transfected (+) or not (−) with JCV T-Ag. Unlabelled AP2 competitor DNA (Comp) or a non-specific control DNA (NC) were added as indicated. To some gel shift reactions, α-AP2α antibody or normal mouse serum control (NMS) were added as indicated. The asterisk indicates probe alone without extract. The position of the free probe, AP2–DNA complex and the supershift are shown by a P, arrow and arrowhead, respectively. (b) A gel shift assay was performed with a probe corresponding to the Ets site of the BAG3 promoter (nt −104 to −79) using nuclear extracts from U-87 MG cells transfected (+) or not (−) with JCV T-Ag. Unlabelled Ets competitor DNA (Comp) or a non-specific control DNA (NC) were added as indicated. The asterisk indicates probe alone without extract. The position of free probe and the Ets–DNA complex are indicated by a P and an arrow, respectively. (c) The nuclear extracts used in (a) and (b) were analysed by Western blotting as indicated. (d) A ChIP assay was performed on U-87 MG cells using antibody to AP2α (α-AP2α), Ets (α-Ets) and NMS as indicated.
With regard to the role of Ets, a gel shift assay with an oligonucleotide (nt −104 to −79) corresponding to the Ets site showed a single band that was unaffected by T-Ag expression (Fig. 5b, compare lanes 2 and 3), indicating that Ets is not involved in the effect of T-Ag on the BAG3 promoter. Western blot analysis of the nuclear extracts that were used in these gel shift experiments showed that T-Ag was expressed in the transfected cells and that BAG3 was downregulated as expected (Fig. 5c).
Fig. 5(d) shows a ChIP assay using antibodies to AP2 and Ets. These data confirmed the binding of both transcription factors to the BAG3 promoter. No signal was observed with normal mouse serum (Fig. 5d, lanes 4 and 5). The amount of Ets bound to the BAG3 promoter was unaffected by T-Ag expression (Fig. 5d, compare lanes 8 and 9). In contrast, the amount of AP2 bound to the BAG3 promoter was strongly reduced by T-Ag expression (Fig. 5d, compare lanes 6 and 7). This provided further evidence that T-Ag inhibits the BAG3 promoter through a reduction in AP2 binding to its site in the promoter.
Ectopic expression of AP2 isoforms stimulates expression by the BAG3 promoter and reverses the inhibition caused by JCV T-Ag
U-87 MG cells were transfected with reporter constructs containing the full-length BAG3 promoter (Fig. 6a) or a deletion mutant (nt −26 to +306) promoter (Fig. 6b). Reporter plasmids were transfected alone or with expression plasmids for T-Ag, AP2α and/or AP2γ. The total amount of DNA in each transfection was kept constant. T-Ag inhibited the full-length BAG3 promoter (Fig. 6a, lane 2) whereas both AP2α and AP2γ stimulated transcription (Fig. 6a, lanes 3 and 4, respectively). Both AP2α and AP2γ also reversed T-Ag inhibition of the promoter (Fig. 6a, lanes 5 and 6, respectively). The deletion mutant promoter, which lacked the AP2 site, was unaffected by ectopic expression of T-Ag, AP2α and AP2γ (Fig. 6b). Expression of proteins was verified by Western blotting (Fig. 6c).
Fig. 6.
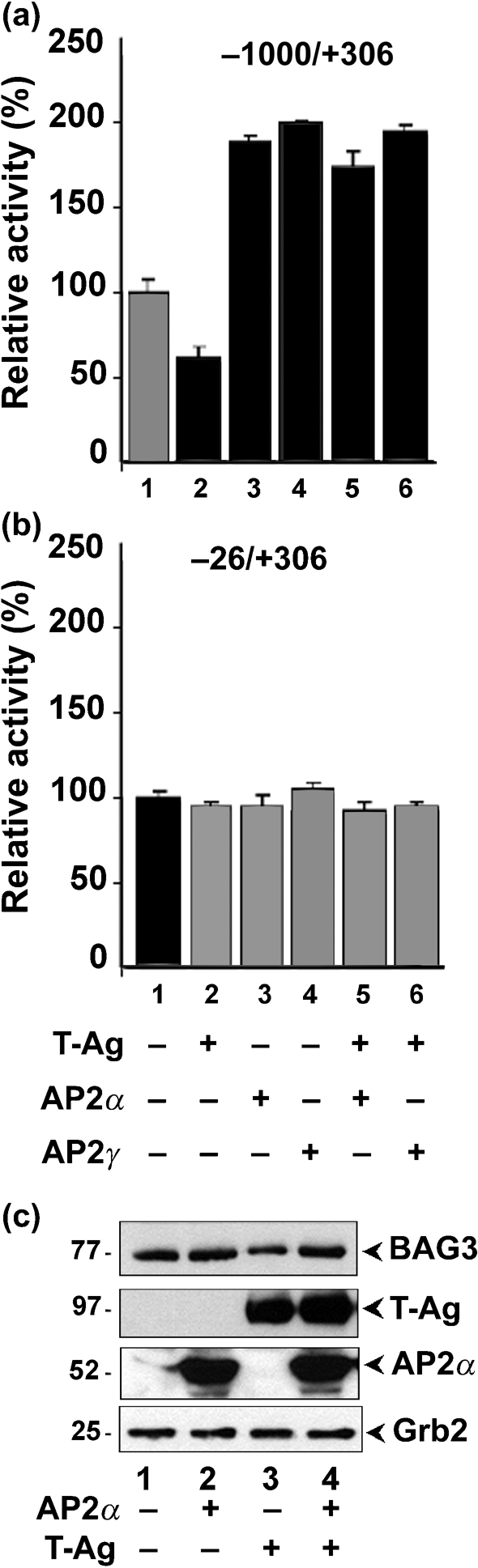
Effects of AP2α, AP2γ and T-Ag on BAG3 promoter activity. (a) U-87 MG cells were transfected with reporter plasmids for the full-length BAG3 promoter (nt −1000 to +306) together with expression plasmids for T-Ag, AP2α and/or AP2γ as indicated. The amount of DNA used in each transfection was constant. (b) U-87 MG cells were transfected with reporter plasmids for deleted BAG3 promoter (nt −26 to +306) together with expression plasmids for T-Ag, AP2α and/or AP2γ as indicated. A constant amount of DNA was used in each transfection. (c) Western blots of the cell extracts. Protein sizes are indicated in kDa.
Role of BAG3 and T-Ag in caspase-3 activation
BAG3 has been shown to participate in a number of cell functions, including a role in the inhibition of apoptosis (Doong et al., 2002; Kabbage & Dickman, 2008; Rosati et al., 2007a). As changes in apoptosis regulation are a common feature of viral infection, we next investigated the regulation of apoptosis using H2O2 as an inducer of apoptosis to unmask a role for BAG3 either by expressing it in the presence of T-Ag (Fig. 7) or by curtailing BAG3 expression by RNA interference (Fig. 8). Caspase-3 is the executioner caspase of apoptosis and thus lies downstream of all apoptotic signalling pathways, i.e. cleavage and activation of caspase-3 is a useful marker for apoptosis. We investigated the effects of T-Ag and BAG3 on the induction of apoptosis by H2O2. U-87 MG cells were transfected with expression plasmids for BAG3 and/or T-Ag (the negative control was transfected with empty vector pCMV alone). After 4 days, cells were treated with or without H2O2 for 2 h. As shown in Fig. 7(a), expression of T-Ag and BAG3 was verified by Western blot with α-tubulin as a loading control (upper three panels). In agreement with Fig. 2, endogenous BAG3 was downregulated by T-Ag expression (Fig. 7a, compare lanes 1 and 2). In contrast, ectopic BAG3 was unaffected by T-Ag expression, as BAG3 was expressed from the CMV promoter, which would not be expected to respond to T-Ag (Fig. 7a, compare lanes 3 and 4).
Fig. 7.

Effects of H2O2, BAG3 expression and/or T-Ag on caspase-3 activation in U-87 MG cells. U-87 MG cells were transfected with plasmid expressing either BAG3 and/or T-Ag from the CMV promoter and/or treated (+) or not (−) with H2O2. Cell extracts were harvested after 4 days. (a) Cell extracts were analysed by Western blotting for BAG3 and T-Ag expression and with antibody to caspase-3 to detect procaspase-3 cleavage/activation. Protein sizes are indicated in kDa. (b) Cells were assayed with a fluorescent capsase-3 substrate assay kit that detects caspase-3/7 activity.
Fig. 8.
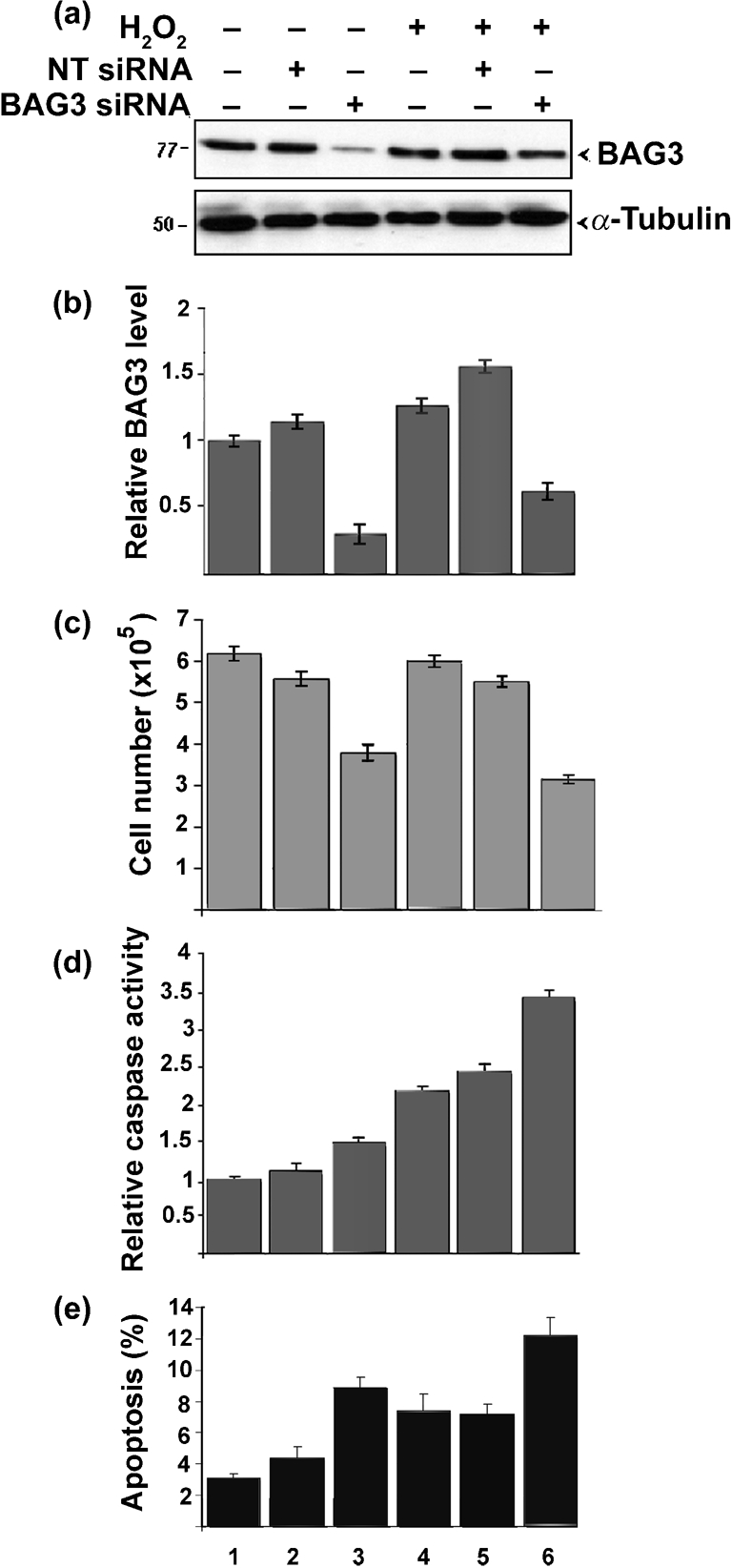
Effects of BAG3 siRNA on cellular responses to H2O2. U-87 MG cells were transfected with BAG3 siRNA or with a non-targeting (NT) siRNA and treated (+) or not (−) with H2O2 as indicated. (a) BAG3 was analysed by Western blotting with α-tubulin serving as a loading control. Protein sizes are indicated in kDa. (b) The level of BAG3 protein normalized to α-tubulin was measured by densitometry. Data were then normalized relative to untreated cells (lane 1), which were taken as 1. (c) Cells were counted with a haemocytometer in the presence of trypan blue and the number of dye-excluding (viable) cells was determined for each condition. (d) Cells were assayed with a fluorescent capsase-3 substrate assay kit that detects caspase-3/7 activity. (e) Cells were labelled with propidium iodide and subject to FACS analysis.
In order to investigate caspase-3 activation, these proteins were also analysed by high-density 12 % PAGE, and caspase-3 cleavage was analysed by Western blotting with α-tubulin as a loading control (Fig. 7a, lower two panels). No caspase-3 was observed in the absence of H2O2 (Fig. 7a, lanes 1–4). In all lanes where H2O2 was added, cleaved active caspase-3 (17/19 kDa) was clearly detectable (Fig. 7a, lanes 5–8), with a particularly strong band for the cells expressing T-Ag (Fig. 7a, lane 7).
The activity of caspase-3 can also be measured using a pro-luminescent substrate specific for caspase-3/7. Similar results were obtained using this method. Activation of caspase-3 was evident in all cultures treated with H2O2 (Fig. 7b, compare lanes 6–10 with lanes 1–5). The strongest activation was seen in H2O2-treated cells expressing T-Ag (Fig. 7b, lane 8, threefold increase). Empty vector had no effect on caspase-3 activation (Fig. 7b, compare lanes 6 and 7), whilst BAG3 cells showed significantly less apoptosis (Fig. 7b, compare lanes 6 and 9). Co-expression of T-Ag reversed the BAG3-mediated reduction in caspase-3 observed in H2O2-treated cells expressing BAG3 (Fig. 7b, compare lanes 9 and 10). The data shown in Fig. 7(b) were obtained using cells that were treated with H2O2 at 4 days post-transfection. The same results were obtained in cultures that were treated at 2, 3, 5 and 6 days post-transfection (data not shown). Thus, in H2O2-treated cells, BAG3 has an anti-apoptotic effect whereas T-Ag sensitizes cells to apoptosis upon stress induction.
Downregulation of BAG3 by RNA interference has a pro-apoptotic effect on H2O2-treated cells
U-87 MG cells were treated with and without siRNA for BAG3 or non-targeting siRNA. Cells were then treated with or without H2O2. As shown in Fig. 8(a, b), the BAG3 protein level in cells treated with BAG3 siRNA was approximately three- to fourfold lower than in untreated cells (Fig. 8a, b, compare lanes 3 and 6 with lane 1), whilst non-targeting siRNA had little effect (Fig. 8a, b, lanes 2 and 5). BAG3 siRNA also reduced cell number (Fig. 8c, lanes 3 and 6). Caspase-3 activity was again induced in H2O2-treated cells (Fig. 8d, lanes 4–6) with the strongest induction in cells that received BAG3 siRNA (Fig. 8d, lane 6). A similar pattern was seen when the extent of apoptosis was measured by FACS analysis (Fig. 8e). Thus, BAG3 siRNA sensitizes H2O2-treated cells to apoptosis.
DISCUSSION
One of the functions of apoptosis is to eliminate cells that have become damaged or infected with viruses. Control of apoptosis is a complex multi-level signalling network containing both pro-apoptotic and anti-apoptotic signal transduction pathways (White & McCubrey, 2001), and viruses have evolved many different strategies to subvert these processes. In the case of JCV, infection leads to rapid and severe damage to the cellular DNA (Darbinyan et al., 2007) and to profound CPE that is observed at later stages of infection. Nevertheless, only a small percentage of cells in the late stage of infection undergo apoptosis, as judged by FACS analysis and the occurrence of lamin A and C cleavage (Darbinyan et al., 2007). Similar results were reported by Seth et al. (2004) using caspase-3 labelling and a TUNEL assay to measure apoptosis. Clearly, there is a conflicting interaction between the virus and the host with respect to the induction of apoptosis. One example of this is the rapid induction of the potent anti-apoptotic protein survivin upon JCV infection, which has been shown to have a role in protecting JCV-infected cells from apoptosis (Piña-Oviedo et al., 2007). Another important regulator of apoptosis is BAG3, which is a member of the BAG family of proteins that participates in a wide variety of cellular processes, including cell survival, the cellular stress response, proliferation and apoptosis (Doong et al., 2002; Kabbage & Dickman, 2008; Rosati et al., 2007a). BAG3 interacts with the ATPase domain of Hsc70/Hsp70, inhibits Hsc70/Hsp70 chaperone activity in vitro (Takayama et al., 1999) and thus modulates the cellular responses to stress conditions. Here, we found that JCV infection resulted in downregulation of the level of BAG3 protein, which was mediated, at least in part, by inhibition of the BAG3 promoter by T-Ag. The involvement of other JCV proteins, such as agnoprotein, is unknown. T-Ag downregulated the BAG3 promoter by inhibiting the binding of the AP2 transcription factor to its site within the BAG3 promoter as judged by gel shift and ChIP assays. Furthermore, the ectopic expression of either AP2α or AP2γ reversed T-Ag-mediated BAG3 promoter inhibition.
This type of mechanism involving inhibition mediated through AP2 is not without precedent. Mitchell et al. (1987) reported that the T-Ag of SV40, which is closely related to JCV, inhibited sequence-specific binding of AP2 to the SV40 and human metallothionein promoters. Furthermore, they reported that SV40 T-Ag inhibited AP2-dependent transcriptional activation of the human metallothionein promoter, which did not involve binding of T-Ag to the human metallothionein promoter DNA but rather resulted from the sequestration of AP2 from its binding site due to protein–protein interaction with SV40 T-Ag (Mitchell et al., 1987). Such a mechanism is also consistent with our data for JCV T-Ag, as shown in Fig. 5, where T-Ag eradicated the binding of AP2 to its cognate DNA site as detected by gel shift and ChIP assays. In another study, the adenovirus E1A protein was shown to repress the type IV collagenase promoter by targeting AP2, and protein–protein interaction of E1A and AP2 was demonstrated by glutathione S-transferase affinity chromatography (Somasundaram et al., 1996).
Interplay between T-Ag and BAG3 in the control of apoptosis was revealed using H2O2 as an apoptotic inducer and caspase-3 cleavage as a marker for apoptosis (Fig. 7). Whilst T-Ag was pro-apoptotic, BAG3 was anti-apoptotic. Furthermore, curtailing BAG3 expression by RNA interference was pro-apoptotic (Fig. 8). It will also be of interest to investigate whether BAG3 can have an effect on JCV transcription, as it has been reported that BAG1 can stimulate JCV early and late transcription through JCV NF-1 sites (Devireddy et al., 2000).
What is the significance of the repression of BAG3 during JCV infection? As described in the Introduction, many other studies have ascribed an anti-apoptotic role to BAG3. This is supported by our data, as described above. Thus, it is possible that downregulation of BAG3 by JCV infection represents a cellular defensive response to viral infection in order to promote apoptosis. A similar phenomenon has been described by Verma et al. (2006), where JCV was found to induce genes involved in the interferon-mediated host defence response. These included signal transducer and activator of transcription-1, interferon stimulating gene-56, myxovirus resistance-1 and 2′-5′-oligoadenylate synthetase (Verma et al., 2006). Thus, JCV infection involves not only gene expression changes that are favourable to the virus, such as survivin induction (Piña-Oviedo et al., 2007), but also changes that reflect a host cellular antiviral response, such as the interferon-mediated host defence response reported by Verma et al. (2006) and downregulation of the anti-apoptotic protein BAG3 reported here. Indeed, such antiviral responses may control JCV replication in immunocompetent hosts and restrain the development of PML.
What is the mechanism of action of BAG3 on the host apoptotic machinery in the context of JCV infection? BAG3 is known to interact via its C-terminal BAG domain with the ATPase domain of Hsp70, and BAG3 inhibits the chaperone activity of Hsp70 in vitro (Takayama et al., 1999). Interestingly, T-Ag also interacts with Hsp70 (Sawai & Butel, 1989) and is involved in a molecular chaperone function via the N-terminal J-domain of T-Ag (DeCaprio, 1999). Indeed, the J-domain of JCV T-Ag can reconstitute the molecular chaperone function of DnaJ in Escherichia coli (Kelley & Georgopoulos, 1997). The molecular details of the chaperone function of T-Ag in human cells and how it may be affected by BAG proteins are unknown. We plan to investigate further the interplay among JCV T-Ag, BAG3 and Hsp70, how this is involved in the viral life cycle of JCV and how this axis may be manipulated therapeutically for treatment of PML.
Acknowledgments
We thank past and present members of the Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. We are grateful to Dr Ronald J. Weigel, University of Iowa, USA, for providing plasmids expressing AP2α and AP2γ. We also wish to thank C. Schriver for editorial assistance. This work was supported by grants awarded by the NIH to M. K. W. and K. K.
References
- Amini, S., Mameli, G., Del Valle, L., Skowronska, A., Reiss, K., Gelman, B. B., White, M. K., Khalili, K. & Sawaya, B. E. (2005). p73 interacts with human immunodeficiency virus type 1 Tat in astrocytic cells and prevents its acetylation on lysine 28. Mol Cell Biol 25, 8126–8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonelli, P., Petrella, A., Rosati, A., Romano, M. F., Lerose, R., Pagliuca, M. G., Amelio, T., Festa, M., Martire, G. & other authors (2004). BAG3 protein regulates stress-induced apoptosis in normal and neoplastic leukocytes. Leukemia 18, 358–360. [DOI] [PubMed] [Google Scholar]
- Brive, L., Takayama, S., Briknarová, K., Homma, S., Ishida, S. K., Reed, J. C. & Ely, K. R. (2001). The carboxyl-terminal lobe of Hsc70 ATPase domain is sufficient for binding to BAG1. Biochem Biophys Res Commun 289, 1099–1105. [DOI] [PubMed] [Google Scholar]
- Cole, C. N. (1996). Polyomavirinae: the viruses and their replication. In Fundamental Virology, 3rd edn, pp. 917–946. Edited by B. N. Fields, D. M. Knipe & P. M. Howley. Philadelphia: Lippincott, Williams & Wilkins.
- Darbinian-Sarkissian, N., Czernik, M., Peruzzi, F., Gordon, J., Rappaport, J., Reiss, K., Khalili, K. & Amini, S. (2006). Dysregulation of NGF-signaling and Egr-1 expression by Tat in neuronal cell culture. J Cell Physiol 208, 506–515. [DOI] [PubMed] [Google Scholar]
- Darbinyan, A., White, M. K., Akan, S., Radhakrishnan, S., Del Valle, L., Amini, S. & Khalili, K. (2007). Alterations of DNA damage repair pathways resulting from JCV infection. Virology 364, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio, J. A. (1999). The role of the J domain of SV40 large T in cellular transformation. Biologicals 27, 23–28. [DOI] [PubMed] [Google Scholar]
- Del Valle, L., Gordon, J., Enam, S., Delbue, S., Croul, S., Abraham, S., Radhakrishnan, S., Assimakopoulou, M., Katsetos, C. D. & Khalili, K. (2002). Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst 94, 267–273. [DOI] [PubMed] [Google Scholar]
- Del Valle, L., White, M. K. & Khalili, K. (2008). Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J Neuropathol Exp Neurol 67, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devireddy, L. R., Kumar, K. U., Pater, M. M. & Pater, A. (2000). BAG-1, a novel Bcl-2-interacting protein, activates expression of human JC virus. J Gen Virol 81, 351–357. [DOI] [PubMed] [Google Scholar]
- Doong, H., Price, J., Kim, Y. S., Gasbarre, C., Probst, J., Liotta, L. A., Blanchette, J., Rizzo, K. & Kohn, E. (2000). CAIR-1/BAG-3 forms an EGF-regulated ternary complex with phospholipase C-γ and Hsp70/Hsc70. Oncogene 19, 4385–4395. [DOI] [PubMed] [Google Scholar]
- Doong, H., Vrailas, A. & Kohn, E. C. (2002). What's in the ‘BAG’? – a functional domain analysis of the BAG-family proteins. Cancer Lett 188, 25–32. [DOI] [PubMed] [Google Scholar]
- Doong, H., Rizzo, K., Fang, S., Kulpa, V., Weissman, A. M. & Kohn, E. C. (2003). CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: accumulation of poly-ubiquitinated Hsp90 client proteins. J Biol Chem 278, 28490–28500. [DOI] [PubMed] [Google Scholar]
- Franceschelli, S., Rosati, A., Lerose, R., De Nicola, S., Turco, M. C. & Pascale, M. (2008). BAG3 gene expression is regulated by heat shock factor 1. J Cell Physiol 215, 575–577. [DOI] [PubMed] [Google Scholar]
- Frisque, R. J. & White, F. A. (1992). The molecular biology of JC virus, causative agent of progressive multifocal leukoencephalopathy. In Molecular Neurovirology, pp. 25–160. Edited by R. P. Roos. Totawa, NJ: Humana Press.
- Frisque, R. J., Bream, G. L. & Cannella, M. T. (1984). Human polyomavirus JC virus genome. J Virol 51, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentilella, A., Passiatore, G., Deshmane, S., Turco, M. C. & Khalili, K. (2008). Activation of BAG3 by Egr-1 in response to FGF-2 in neuroblastoma cells. Oncogene 27, 5011–5018. [DOI] [PubMed] [Google Scholar]
- Graham, F. L. & van der Eb, A. J. (1973). A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Hou, J. & Major, E. O. (2000). Progressive multifocal leukoencephalopathy: JC virus induced demyelination in the immune compromised host. J Neurovirol 6 (Suppl. 2), S98–S100. [PubMed] [Google Scholar]
- Kabbage, M. & Dickman, M. B. (2008). The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci 65, 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis, J. N., Guancial, E. A., Doong, H., Virador, V. & Kohn, E. C. (2006). CAIR-1/BAG-3 modulates cell adhesion and migration by downregulating activity of focal adhesion proteins. Exp Cell Res 312, 2962–2971. [DOI] [PubMed] [Google Scholar]
- Kelley, W. L. & Georgopoulos, C. (1997). The T/t common exon of simian virus 40, JC, and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc Natl Acad Sci U S A 94, 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili, K., Gordon, J. & White, M. K. (2006). The polyomavirus, JCV and its involvement in human disease. Adv Exp Med Biol 577, 274–287. [DOI] [PubMed] [Google Scholar]
- Krynska, B., Gordon, J., Otte, J., Franks, R., Knobler, R., DeLuca, A., Giordano, A. & Khalili, K. (1997). Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem 67, 223–230. [DOI] [PubMed] [Google Scholar]
- Lane, D. P. & Crawford, L. V. (1979). T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- Lassak, A., Del Valle, L., Peruzzi, F., Wang, J. Y., Enam, S., Croul, S., Khalili, K. & Reiss, K. (2002). Insulin receptor substrate 1 translocation to the nucleus by the human JC virus T-antigen. J Biol Chem 277, 17231–17238. [DOI] [PubMed] [Google Scholar]
- Lee, J. H., Takahashi, T., Yasuhara, N., Inazawa, J., Kamada, S. & Tsujimoto, Y. (1999). Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene 18, 6183–6190. [DOI] [PubMed] [Google Scholar]
- Linzer, D. I. & Levine, A. J. (1979). Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52. [DOI] [PubMed] [Google Scholar]
- Mázló, M., Ressetar, H. G. & Stoner, G. L. (2001). The neuropathology and pathogenesis of progressive multifocal leukoencephalopathy. In Human Polyomaviruses: Molecular and Clinical Perspectives, pp. 257–335. Edited by K. Khalili & G. L. Stoner. New York: Wiley.
- McPherson, L. A. & Weigel, R. J. (1999). AP2α and AP2γ: a comparison of binding site specificity and trans-activation of the estrogen receptor promoter and single site promoter constructs. Nucleic Acids Res 27, 4040–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, P. J., Wang, C. & Tjian, R. (1987). Positive and negative regulation of transcription in vitro: enhancer-binding protein AP-2 is inhibited by SV40 T antigen. Cell 50, 847–861. [DOI] [PubMed] [Google Scholar]
- Pagliuca, M. G., Lerose, R., Cigliano, S. & Leone, A. (2003). Regulation by heavy metals and temperature of the human BAG-3 gene, a modulator of Hsp70 activity. FEBS Lett 541, 11–15. [DOI] [PubMed] [Google Scholar]
- Piña-Oviedo, S., Urbanska, K., Radhakrishnan, S., Sweet, T., Reiss, K., Khalili, K. & Del Valle, L. (2007). Effects of JC virus infection on anti-apoptotic protein survivin in progressive multifocal leukoencephalopathy. Am J Pathol 170, 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan, S., Otte, J., Enam, S., Del Valle, L., Khalili, K. & Gordon, J. (2003). JC virus-induced changes in cellular gene expression in primary human astrocytes. J Virol 77, 10638–10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan, S., Gordon, J., Del Valle, L., Cui, J. & Khalili, K. (2004). Intracellular approach for blocking JC virus gene expression by using RNA interference during viral infection. J Virol 78, 7264–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli, L., Sariyer, I. K., Tung, J., Feliciano, M., Sawaya, B. E., Del Valle, L., Ferrante, P., Khalili, K., Safak, M. & White, M. K. (2008). Early growth response-1 protein is induced by JC virus infection and binds and regulates the JC virus promoter. Virology 375, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati, A., Ammirante, M., Gentilella, A., Basile, A., Festa, M., Pascale, M., Marzullo, L., Belisario, M. A., Tosco, A. & other authors (2007a). Apoptosis inhibition in cancer cells: a novel molecular pathway that involves BAG3 protein. Int J Biochem Cell Biol 39, 1337–1342. [DOI] [PubMed] [Google Scholar]
- Rosati, A., Leone, A., Del Valle, L., Amini, S., Khalili, K. & Turco, M. C. (2007b). Evidence for BAG3 modulation of HIV-1 gene transcription. J Cell Physiol 210, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati, A., Khalili, K., Deshmane, S. L., Radhakrishnan, S., Pascale, M., Turco, M. C. & Marzullo, L. (2009). BAG3 protein regulates caspase-3 activation in HIV-1-infected human primary microglial cells. J Cell Physiol 218, 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai, E. T. & Butel, J. S. (1989). Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J Virol 63, 3961–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth, P., Diaz, F., Tao-Cheng, J. H. & Major, E. O. (2004). JC virus induces nonapoptotic cell death of human central nervous system progenitor cell-derived astrocytes. J Virol 78, 4884–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram, K., Jayaraman, G., Williams, T., Moran, E., Frisch, S. & Thimmapaya, B. (1996). Repression of a matrix metalloprotease gene by E1A correlates with its ability to bind to cell type-specific transcription factor AP-2. Proc Natl Acad Sci U S A 93, 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama, S., Sato, T., Krajewski, S., Kochel, K., Irie, S., Millan, J. A. & Reed, J. C. (1995). Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell 80, 279–284. [DOI] [PubMed] [Google Scholar]
- Takayama, S., Xie, Z. & Reed, J. C. (1999). An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem 274, 781–786. [DOI] [PubMed] [Google Scholar]
- Verma, S., Ziegler, K., Ananthula, P., Co, J. K., Frisque, R. J., Yanagihara, R. & Nerurkar, V. R. (2006). JC virus induces altered patterns of cellular gene expression: interferon-inducible genes as major transcriptional targets. Virology 345, 457–467. [DOI] [PubMed] [Google Scholar]
- White, M. K. & Khalili, K. (2004). Polyomaviruses and human cancer: molecular mechanisms underlying patterns of tumorigenesis. Virology 324, 1–16. [DOI] [PubMed] [Google Scholar]
- White, M. K. & McCubrey, J. A. (2001). Suppression of apoptosis: role in cell growth and neoplasia. Leukemia 15, 1011–1021. [DOI] [PubMed] [Google Scholar]
- White, M. K., Amini, S., Khalili, K. & Darbinian, N. (2008). Development of a bidirectional caspase-3 expression system for the induction of apoptosis. Cancer Biol Ther 7, 945–954. [DOI] [PubMed] [Google Scholar]