

Abstract
MicroRNAs (miRNAs) regulate the expression of multiple proteins in a dose dependent manner. We hypothesized that increased expression of miRNAs encoded on chromosome 21 (chr 21) contribute to the leukemogenic role of trisomy 21. The levels of chr 21 miRNAs were quantified by qRT-PCR in four types of childhood ALL characterized by either numerical (trisomy or tetrasomy) or structural abnormalities of chr 21. Suprisingly high expression of the hsa-mir-125b-2 cluster, consisting of three miRNAs, was identified in leukemias with the structural ETV6/RUNX1 abnormality and not in ALLs with trisomy 21. Manipulation of ETV6/RUNX1 expression and chromatin immunoprecipitation studies demonstrated that the high expression of the miRNA cluster is an event independent of the ETV6/RUNX1 fusion protein. Overexpression of hsa-mir-125b-2 conferred a survival advantage to Ba/F3 cells following IL-3 withdrawal or a broad spectrum of apoptotic stimuli through inhibition of caspase 3 activation. Conversely, knockdown of the endogenous miR-125b in the ETV6/RUNX1 leukemia cell line REH increased apoptosis after Doxorubicin and Staurosporine treatments. P53 protein levels were not altered by miR-125b. Together these results suggest that the expression of hsa-mir-125b-2 in ETV6/RUNX1 ALL provides survival advantage to growth inhibitory signals in a p53 independent manner.
Keywords: Acute lymphoblastic leukemia (ALL), ETV6/RUNX1, TEL/AML1, microRNA
Introduction
Aberrations in chromosome 21 are commonly found in childhood ALL. The high-hyperdiploid ALL (HHD ALL) 1, 2, the most common type of childhood ALL with numerical chromosomal aberrations, is characterized by 3–4 copies of chr 21 and a variable presence of other specific chromosomal trisomies. The markedly increased incidence of ALL in children with Down Syndrome (DS) 3 strongly suggests that trisomy 21 is leukemogenic, but the chr 21 genes involved in these leukemias are presently unknown. A newly discovered rare subtype of childhood ALL is characterized by an intrachromosomal amplification of a small region within the long arm of chr 21 (iAMP21) around the RUNX1 locus 4. In contrast to ETV6/RUNX1 and HHD ALL, it is associated with poor prognosis. The most common structural chromosomal aberration in childhood ALL occurring in approximately 25% of pediatric B-cell precursor ALL fuses the RUNX1 (AML1) gene on chr 21 with the ETV6 (TEL) gene on chr 12 5, 6. Although ETV6/RUNX1 causes a prenatal preleukemic clonal expansion, additional genetic events are required for evolution of leukemia 7, 8.
MicroRNAs (miRNAs) are 20–25 nucleotide long non-coding RNAs that play a vital role in the regulation of protein expression 9. Currently there are five validated mature miRNAs encoded on human chr 21 (http://microrna.sanger.ac.uk Figure 1A). Hsa-mir-99a, hsa-let-7c and hsa-mir-125b-2 are clustered together in the same intron of the C21ORF34 gene on 21q.21.1 (NC_000021.7) (hsa-mir-125b-2 cluster). Hsa-mir-125b-1, the homologue of C. elegans lin-4, has been shown to be expressed in solid tumors and associated with enhanced cell proliferation and survival 10–12. An insertion of hsa-mir-125b-1 into the IGH locus was described in a patient with B cell precursor ALL suggesting its involvement in the leukemic process 13. It has been shown to be expressed in myeloid malignancies and to block myeloid differentiation 14. Recently it was also reported to protect from apoptosis through negative regulation of p53 in zebrafish, human neuroblastoma cells and lung fibroblasts but not in mouse cells15. Hsa-mir-155 has been linked to B cell development. We and others have shown its increased expression in lymphomas16–19. Strikingly, transgenic expression of miR-155 induced pre-B cell leukemias and lymphomas in mice20, thus miR-155 seems to be a potent oncogene.
Figure 1. Expression of miRNAs encoded on Chromosome 21 in different leukemia samples.
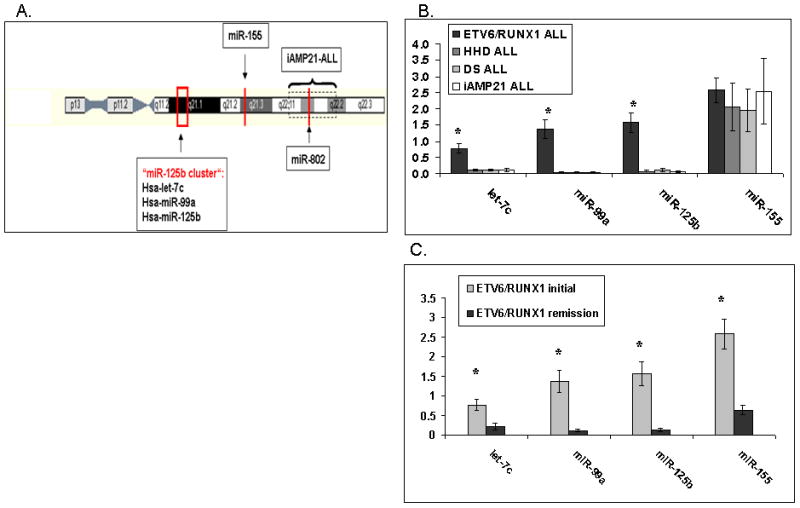
(A) Schematic representation of the location of miRNAs encoded on chr 21. The level of the mature hsa-miR-802 could not be measured because of the lack of an appropriate qRT-PCR based assay. (B–C) MiRNA expression was determined by qRT-PCR using the TaqMan® MicroRNA Assays from Applied Biosystems. The numbers on the Y axis represent relative expression levels of mature miRNA normalized to an internal control (RNU43). Standard errors are indicated. Asterisk marks significance (T-TEST). (B) Expression of the chr 21 miRNAs in diagnostic bone marrow samples of different ALL subtypes: ETV6/RUNX1 (24 patients), Down Syndrome (DS) (10 patients), High Hyperdiploid (10 patients), iAMP21 (7 patients) P<0.05 (ANOVA). (C) Comparison of miRNA expression between diagnosis and remission samples of ETV6/RUNX1 leukemias P<0.005 (T-TEST).
The mechanisms of miRNA function suggest that their activity should be correlated with their dosage and that each miRNA may regulate multiple targets21. Thus we hypothesized that miRNAs contribute to the oncogenic effects of chromosomal trisomies, a situation in which a small change (×1.5) in genomic dosage results in profound effects. This hypothesis is strengthened by our recent observations demonstrating a general increase in expression of multiple genes from the trisomic chromosomes22. Thus, we expected that miRNAs from chr 21 would be over-expressed in HHD and DS ALL (3–4 copies of chr 21) relative to ETV6/RUNX1 and iAMP21 leukemias.
Here we report the surprising observations that unlike most of the genes encoded on chr 21, the expression of the hsa-mir-125b-2 cluster does not correlate with gene copy number but rather is highly expressed in ETV6/RUNX1 ALL. Additional studies suggest that the expression of the hsa-miR-125b-2 cluster is an independent event in ETV6/RUNX1 ALL, conferring survival advantage under growth inhibitory and apoptotic conditions in a p53 independent manner.
Materials and Methods
Patients
RNA was derived from diagnostic or remission bone marrow samples of childhood ALL patients obtained with informed consent. Diagnostic bone marrow samples contained at least 80% lymphoblasts. The samples were anonymized before shipping except for the information on the genetic subgroup. The study was approved by the IRB of the Israeli Health Ministry and Sheba Medical Center.
The rest of the material and methods are detailed in a supplementary file
Results
Hsa-mir-125b-2 cluster is highly expressed in ETV6/RUNX1 leukemias
We measured the expression of the mature forms of four miRNAs encoded on chr 21 (hsa-mir-99a; hsa-let-7c; hsa-mir-125b-2 and hsa-mir-155) by qRT-PCR in RNA derived from diagnostic bone marrow samples of four diverse subtypes of ALL with structural or numerical aberrations of chr 21: ETV6/RUNX1 ALL (n=24), HHD ALL (n=10), DS ALL (n=10) and iAMP21 ALL (n=7). Surprisingly, extra copies of chr 21 did not contribute to the expression level of these miRNAs (Figure 1B). While hsa-mir-155 is similarly expressed between the different ALLs, the other miRNAs hsa-mir-99a, hsa-let-7c, and hsa-mir-125b-2 are up-regulated in the ETV6/RUNX1 ALL, the subtype of leukemia with a structural aberration of chr 21. The expression was derived from the leukemic blasts as it was not observed in remission samples (Figure 1C). To ensure that the observed results were not due to the normalization to a single internal control (RNU 43), we have extended the miRNA profiling using TaqMan® Low Density miRNA arrays (TLDA, ABI) normalizing to two additional internal controls (RNU6 and RNU48). As can be seen in supplementary Figure 1, the TLDA results are similar to the results obtained with the singleplex miRNA expression profiling.
Hsa-mir-125b-2 cluster is not a target of the ETV6/RUNX1 fusion protein
To test if mir-125b-2 is a target of ETV6/RUNX1 we performed overexpression experiments in two mouse hematopoietic progenitor cell systems: Ba/F3 cells and primary mouse embryonic fetal liver progenitors It has been previously shown that ectopic expression of ETV6/RUNX1 in these progenitors induces proliferation of B cell progenitors, similarly to the preleukemia observed in humans 23 (Supplementary Figure 3). In neither of these cells the expression level of the mmu-mir-125b-2 cluster was affected by the fusion protein (Supplementary Figure 4). Since overexpression in mouse cells may not represent the relevant model, we have knocked down ETV6/RUNX1 in the human REH ALL cell line with siRNA oligonucleotides (oligos) directed against the fusion part of this translocation (Supplementary Figure 5). Two third silencing efficiency of ETV6/RUNX1 but not RUNX1 was observed by qRT-PCR and by western blots 5 days after the first round of transfection (Supplementary Figure 6 and Figure 2A and 2B respectively). Figure 2C demonstrates that the expression of the hsa-mir-125b-2 cluster was not influenced by the knockdown of the ETV6/RUNX1 fusion protein. To further examine if ETV6/RUNX1 protein binds to RUNX1 sites on the vicinity of the hsa-mir-125b-2 cluster we performed Chromatin Immunoprecipitation (ChIP) analysis. There are three potential RUNX1 binding sites in the hsa-mir-125b-2 cluster, one upstream of each miRNA (supplementary Figure 7). As a positive control we examined the promoter of granzyme B (GZMB), a RUNX1 regulated protein24. Figure 2D clearly shows that while the RUNX1 binding site upstream granzyme B (GZMB) is occupied by the ETV6/RUNX1 and RUNX1 proteins, the putative RUNX1 binding sites of the hsa-mir-125b-2 cluster are not bound by the fusion protein.
Figure 2. The hsa-mir-125b-2 cluster is not a target of the ETV6/RUNX1 fusion protein shown by knockdown of ETV6/RUNX1 in REH cells.

REH cells were transfected with siRNA directed against the fusion part of the ETV6 and RUNX1 genes (see methods). (A) Western blot analysis of ETV6/RUNX1 knockdown in REH cells: Scr- scramble siRNA, 15- mixture of siRNAs 1 and 5. Two independent experiments are shown (B) A histogram showing quantification of the average ETV6/RUNX1 protein level as detected by two independent western blots, determined by the ImageJ software (C) No significant change in expression levels of the hsa-mir-125b-2 cluster could be observed in REH cells after knockdown of ETV6/RUNX1 as determined by qRT-PCR. An average of three independent experiments is shown. Expression levels are normalized to an internal control (RNU43). (D) QRT-PCR of ChIP analysis with anti-ETV6, anti-RUNX1, anti-IgG (served as negative control) or anti-TBP (served as positive control – see M&M). Granzyme B (GZMB) was used as positive control for the binding of RUNX1. The Y-axis indicates the enrichment normalized to GAPDH from sample and input. Three independent experiments were performed. Standard errors are indicated.
Taken together these experiments suggest that the hsa-mir-125b-2 cluster is not a direct target of ETV6/RUNX1, but rather an independent event occurring during leukemogenesis.
Hsa-miR-125b-2 has a pro-survival effect under growth inhibitory conditions
To test the hypothesis that the expression of the hsa-mir-125b-2 cluster contributes to survival and growth of lymphoid progenitors we performed forced expression experiments in IL-3 dependent Ba/F3 cells. Transformation of these pro-B cells is commonly used to identify activating mutations of kinases25. We decided to focus on hsa-mir-125b-2 because of previous studies implicating a role of this miRNA in human cancers10, 14, 26, 27. In addition the endogenous mmu-miR-99a and let-7c are expressed in Ba/F3 cells whereas miR-125b is not expressed. Furthermore, the sequence of the mature miR-125b is identical in human and mouse. We transduced Ba/F3 cells with an empty retroviral vector, a vector expressing hsa-mir-125b-2 or a construct encoding hsa-mir-125b-2 mutated in the seed region (see methods, supplementary Figure 8) and confirmed the expression of the mature miRNAs by northern blot analysis (Figure 3A). No differences in growth were observed under steady state conditions (not shown). However, Ba/F3 cells expressing hsa-mir-125b-2 were highly resistant to a transient removal of IL-3. Upon reintroduction of IL-3 to the growth medium only hsa-mir-125b2 transduced Ba/F3 cells, but not cells transduced with an empty vector or a mutated hsa-mir-125b-2, resumed their normal growth (Figure 3B). To examine the mechanism of resistance to IL-3 withdrawal we performed a cell cycle analysis of Ba/F3 cells transduced with the different constructs before and after IL-3 withdrawal. 24 hours after IL-3 deprivation most of the hsa-mir –125b-2 transfected cells were arrested in G1 (71.7%) while most of the control Ba/F3 cells were apoptotic (73% in subG1 phase, Figure 4A). This relative protection from apoptosis conferred by hsa-mir-125b-2 was evident at different time points during the first 24 hours of IL-3 deprivation (Figure 4B). The anti-apoptotic activity of hsa-mir-125b-2 was associated with a marked inhibition of caspase 3 activation and the cleavage of its substrate PARP (Figure 4C).
Figure 3. Hsa-mir-125b-2 confers growth advantage in Ba/F3 cells after transient IL-3 deprivation.

Ba/F3 cells were transduced with an empty vector, a vector expressing mir-125b-2 or mutated mir-125b-2. (A) Northern blot analysis showing representative clones over-expressing hsa-mir-125b-2 or mutated hsa-mir-125b-2. (B) Ba/F3 cells transduced with either empty vector, a vector expressing hsa-mir-125b-2 or mutated hsa-mir-125b-2 were cultured in the absence of IL-3 for 24 hours. After 24 hours IL-3 was added to the medium and cell proliferation was measured by MTT every 24 hours. The graph shows an average of at least three different clones and three experiments. Standard errors are indicated. Experiments were repeated 5 times.
Figure 4. Hsa-mir-125b-2 has an anti-apoptotic effect in Ba/F3 cells.

(A and B) Quantification of apoptosis by measuring the subG1 fraction of Ba/F3 cells stained with Propidum Iodide, comparing wildtype cells, cells over-expressing hsa-mir-125b-2 and cells over-expressing mutated mir-125b-2. (A) A representative example before (time 0) and 24 hours after IL-3 withdrawal. (B) Kinetics of apoptosis with time after IL-3 withdrawal. Asterisks marks significance of p<0.005 as calculated by ANOVA. Standard errors are indicated. (C) Western blot analysis showing lack of activation of caspase 3 and cleavage of PARP following removal of IL-3 in Ba/F3 cells over-expressing hsa-mir-125b-2 in comparison to wildtype Ba/F3 and those over-expressing mutated hsa-mir-125b-2. Experiments were repeated 5 times.
To examine how broad the anti-apoptotic phenotype observed in Ba/F3 cells over-expressing miR-125b is beyond the growth factor weaning, we exposed the cells to different apoptotic stimuli. Supplementary Figure 9 summarizes the percentage of living cells as been quantified by FACS analysis of Annexin negative and 7AAD negative staining of four different treatments. Since cytokine survival pathways involve activation of multiple kinases, we examined the effect of miR 125b on apoptosis induced by three kinase inhibitors with decreasing specificity: JAK Inhibitor I, AG490 and Staurosporine. Ba/F3 transduced with hsa-mir-125b-2 were markedly resistant to each of these inhibitors in comparison with Ba/F3 cells transduced with mutated miR or with empty vector. Mild, but statistically significant resistance was also observed after treatment with Doxorubicin. Thus miR125b provides survival advantage in response to multiple pro-apoptotic stimuli.
To test if the survival advantage observed in Ba/F3 cells over-expressing miR-125b is relevant to ETV6/RUNX1 leukemia, we used REH cells, a cell line derived from a patient with ETV6/RUNX1 leukemia that highly expresses miR-125b. We knocked down the endogenous miR-125b by using LNA oligos labeled with FAM (Figure 5A) and measured the level of living cells of the FAM positive population (transfected with the LNA oligo) after treatment with either Doxorubicin or Staurosporine (see M&M). Figure 5B and C show that upon knocking down miR-125b, the REH cells become more sensitive to Staurosporine and Doxorubicin treatments respectively. These results indicate that the endogenous mir-125b-2 provides partial protection from apoptosis induced by those agents similarly to our observation in Ba/F3 cells.
Figure 5. The endogenous hsa-miR-125b protects ETV6/RUNX1 human leukemia cells from apoptosis.

(A) QRT-PCR analysis of the knockdown efficiency of hsa-miR-125b in REH cells using a specific miR-125b antisense LNA oligo or a control oligo. Y axis represents relative expression to the internal control RNU43. Eight independent experiments are shown. (B and C) Knockdown of miR-125b in REH cells increases the sensitivity to Staurosporine or Doxorubicin treatments respectively as indicated by decreased percentage of living cells (Annexin/7AAD negative). Data are means ± SD of four independent experiments for each treatment. *P<0.05 as calculated by T-TEST.
A recent publication by Le et al.15 suggested that miR-125b is a negative regulator of zebrafish and human (but not mouse) p53. We therefore asked if the effects of miR-125b on apoptosis of Ba/F3 and REH cells could be explained by its regulation of p53 protein levels. Thus, we checked the levels of p53 and its main cellular protein target, p21, both under steady state conditions and during apoptotic stress in both human and mouse models. The binding site of miR-125b on the 3′UTR of p53 is different between mouse and human particularly in the “seed” area, an area considered to be the most significant for the recognition of miRNAs and their targets (Figure 6A). Therefore, it was not surprising that in the mouse Ba/F3 model we have not observed any changes in the level of p53 neither under steady state conditions nor under apoptotic conditions of IL-3 withdrawal (data not shown) or Doxorubicin treatment (Figure 6B and C). Surprisingly, knocking down miR-125b in the human REH cells did not alter p53 protein levels or p21 levels. Furthermore, treating the REH cells with Doxorubicin resulted in similar elevation of p53 levels in the cells transfected with a control LNA oligo or a miR-125b antisense LNA oligo, suggesting that p53 is not a target of miR-125b in these cells. This is further substantiated in the lack of alteration in the levels of p21 (Figure 6D and F). Similarly there was no effect of miR-125b knockdown on p53 or p21 levels upon treatment with Staurosporine (Figure 6E and F). Thus p53 levels are not regulated by miR-125b in REH cells.
Figure 6. P53 is not a target of miR-125b in Ba/F3 and REH cells.
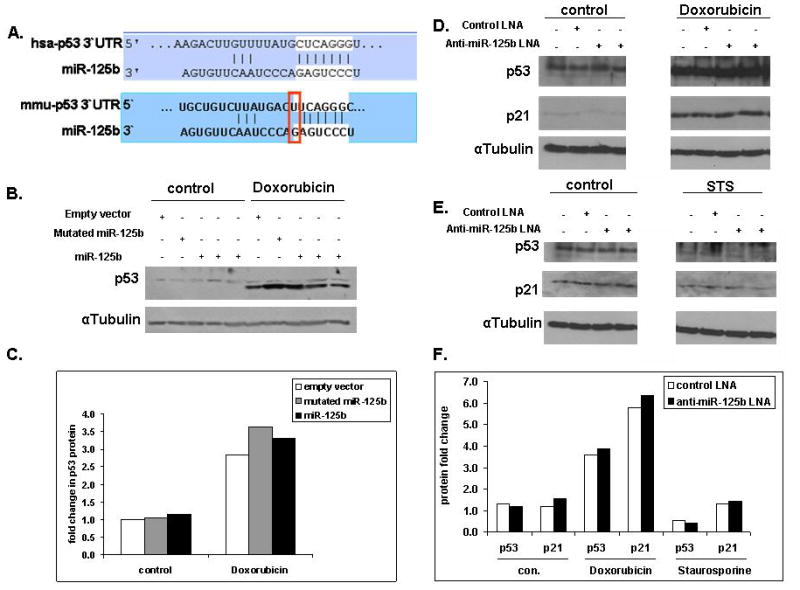
(A) MiR-125b binding site in the 3′UTR of the human and mouse p53 as predicted by TargetScan. Marked in white is the “seed” area of miR-125b, also marked is the single different nucleotide in the potential miR-125b binding site in the 3′UTR of the human and mouse p53. (B) Overexpression of miR-125b in mouse Ba/F3 cells does not down-regulate endogenous p53 levels. A representative western blot of endogenous p53 in Ba/F3 cells transduced with either the empty vector or the vector expressing mutated miR-125b or miR-125b. The cells were treated for 4 hours with 0.2 μg/ml Doxorubicin. (C) Quantification of p53 protein level determined by western blot using the ImageJ software, normalized to αTubulin and presented as fold change relative to the level of p53 in Ba/F3 cells transduced with an empty vector. (D and E) Endogenous human p53 and p21 level following endogenous miR-125b knockdown in the human REH cells. Following the knockdown the cells were treated with either 0.05 μg/ml Doxorubicin (D) or Staurosporine (E). (F) Quantification of p53 and p21 protein levels determined by western blot using the ImageJ software, normalized to αTubulin level and presented as fold change relative to p53 or p21 levels respectively in REH cells transfected with a control LNA oligo.
Discussion
Here, we describe that expression of the hsa-mir-125b-2 cluster residing on chr 21 characterizes ETV6/RUNX1 leukemias compared to other “chromosome 21” leukemias. We demonstrate that this miRNA cluster is not regulated by the ETV6/RUNX1 fusion protein and therefore hypothesize that its expression is an independent event occurring during the evolution of these leukemias. We further demonstrate by overexpression and knockdown studies that mir-125b-2 provides a survival advantage by suppressing apoptosis and caspase 3 activation in response to growth inhibitory conditions.
This study was prompted by the hypothesis, that increased expression of chr 21 miRNAs may explain the leukemogenic role of trisomy 21 in ALL of Down Syndrome (DS) and in sporadic HHD leukemias (which uniformly contain either 3 or 4 copies of chromosome 21). This hypothesis was based on our previous observations that trisomies are usually associated with increased expression of multiple genes from the trisomic chromosomes22 and by increased expression in DS AML of the miRNAs belonging to the hsa-mir-125b-2 cluster 28. Furthermore, increased expression of all chr 21 miRNAs has been recently reported in fetal heart and fetal hippocampus of DS patiens29. Our findings that the expression of those miRNAs in B cell precursor ALLs was not correlated to gene dosage but rather to the leukemia subtype were therefore unexpected. Increased expression levels of the hsa-mir-125b-2 cluster were observed in ETV6/RUNX1 ALL, while hsa-miR-155 was similarly expressed in all subtypes. Furthermore, the expression of the hsa-mir-125b-2 cluster was specific to the leukemic cells, as it was not observed in remission samples.
ETV6/RUNX1 is the most common translocation in childhood ALL. Similarly to HHD ALL, these are B cell precursor leukemias with excellent prognosis on contemporary treatment protocols30. The translocation is necessary for the initiation of a preleukemic clone but insufficient for the evolution of leukemia7, 8, 31. Little is known about the acquired somatic oncogenic events that promote the progression of an ETV6/RUNX1 preleukemic clone into a frank leukemia. The observation that the hsa-mir-125b-2 cluster is not regulated by the ETV6/RUNX1 fusion protein, suggests that its overexpression might be such an independent progression event.
MiRNAs have been shown to be dysregulated in cancer in tissue and cancer type-specific patterns. While one miRNA acts as an oncogene in one type of cancer, the same miRNA serves as a tumor suppressor in another. In breast cancer miR-125b has been found to suppress ERBB2 (HER2) and ERBB3 (HER3) expression26 and thus functions as tumor suppressor. Conversely, a recent study suggested that miR-125b enhances growth and survival of prostate cancer and glioma cells12, 27. To test the hypothesis that the increased expression of mir-125b-2 in ETV6/RUNX1 leukemia has a similar pro-survival role in hematopoietic cells we utilized the IL-3 dependent pro-B Ba/F3 cells. These cells are considered as a standard screening tool to identify leukemia kinase oncogenes25, 32–34. Functional analysis of hsa-mir-125b-2 in Ba/F3 cells demonstrated that this miRNA conferred growth factor independence by blocking apoptosis induced by IL3 withdrawal through delayed activation of caspase 3.
Cytokines such as IL3 promote cell survival through activation of JAK/STAT and related signal transduction pathways35–38. The role of mir125b in protecting from death caused by silencing of pro-survival kinase regulated pathways is demonstrated by the relative resistance it endowed Ba/F3 cells to three kinase inhibitors in decreasing specificity from JAK inhibitor 1 to Staurosporine. However the anti-apoptotic effect of miR-125b is not limited to kinases as modest but consistent and statistically significant resistance to apoptosis was also conferred to cells treated by the DNA damaging drug Doxorubicin. The anti-apoptotic phenotype was not an artifact of miRNA overexpression in Ba/F3 cells. Its relevance to ETV6/RUNX1 leukemias is suggested by the sensitization to apoptosis induced by Staurosporine and Doxorubicin after knockdown of the endogenous miR-125b in REH cells.
Like most of the miRNAs, the precise targets of miR-125b are unknown. MiRNAs are believed to regulate many targets and currently bioinformatic target prediction algorithms are rather limited 39. A recent study suggested that miR-125b reduces the level of the pro-apoptotic protein BAK1 27 thus blocking apoptosis. Though, we have not seen any reduction in BAK1 in either Ba/F3 or HEK293T cells over-expressing hsa-mir-125b-2 (data not shown). We also have not detected any observable changes in several related pro-apoptotic proteins including BIM, BID, BAX and MCL-1 (data not shown). The recent report demonstrating that p53 is negatively regulated by miR-125b15 is intriguing since p53 is a major regulator of apoptosis in response to a variety of stresses including Doxorubicin. However our experiments do not support that report. Similarly to the observations by Le et al, and consistent with the lack of conservation of the miR-125b seed region in the 3′UTR region of mouse p53, we have not seen any alteration of the mouse p53 protein levels in Ba/F3 cells over-expressing miR-125b. Moreover the very efficient knockdown conferred by specific LNA oligos in REH cells did not lead to any alterations in the human p53 levels or its target p21 before or after treatment with Doxorubicin. Thus, at least in these hematopoietic cells p53 is not regulated by miR-25b.
Two provoking recent proteomic studies demonstrate that single miRNAs induce very small alterations in a large number of proteins, and affect functional pathways by acting as sensitive “Rheostats”40, 41. While such small alterations putatively induced by miR-125b in proteins that regulate cell survival may not be significant under steady state conditions, they could provide growth advantage during periods of growth factor deprivation.
The pathogenesis of ETV6/RUNX1 leukemias has been a mystery. In vivo modeling of ETV6/RUNX1 leukemias by many laboratories has been proven experimentally challenging and resulting at most in a preleukemic condition31. A recent analysis of a “knock-in” mouse model in which ETV6/RUNX1 was expressed from the ETV6 promoter has demonstrated an increase in hematopoietic stem cells (HSC). Intriguingly the HSC expressing the fusion protein were markedly more sensitive to apoptosis induced by cytokine withdrawal42. We propose that hsa-mir-125b-2 and possibly the two other miRNAs in this cluster may collaborate with ETV6/RUNX1 in the leukemogenic process by providing survival advantage under growth inhibitory conditions. It would be interesting to test this hypothesis in that mouse model.
Supplementary Material
Acknowledgments
We appreciate the funding by the German Israeli Foundation (to SI and AB), National Institute of Health RO110282006 (SI), Mr. Curtis Katz (SI), JNF UK (SI), Claudine Galli Foundation (NG), Converging Technologies program (LH), Elterninitiative Kinderkrebsklinik e.V. Duesseldorf (AB) and Leukemia Research Fund, UK (OW and MM). We thank J. Crispino, G. Lavi and Y. Sidi for providing reagents; M. Oren for fruitful discussions and provision of reagents; the Department of Pediatric Hematology and Oncology, Dr. von Haunersches Kinderspital, Munich, Germany, for providing clinical samples; members of SI and AB laboratories for helpful discussions and, in particular, P. Landgraf for his excellent criticism and advice.
Financial support: National Institute of Health R01 CA120772-01A2 (SI), German Israeli Foundation (SI & AB), The Wolfson Foundation (SI, NS), Leukemia Research Fund UK (OW and MM), Curtis Katz (SI), MSM0021620813 and MZO00064203 (JT and MZ), Jewish National Fund, UK (SI), Claudine Galli Foundation (NG), Converging Technologies program (LH), Elterninitiative Kinderkrebsklinik Duesseldorf (AB)
Footnotes
Authors’ contributions: SI, NG, VB and AB designed experiments. NG, VB, IL. MM, OW, MZ, JT, LE, NS, AN designed and performed experiments. LH analyzed data. NG, VB, AB, SI wrote the paper.
“Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)”
References
- 1.Heerema NA, Sather HN, Sensel MG, Zhang T, Hutchinson RJ, Nachman JB, et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (> 50 chromosomes) J Clin Oncol. 2000 May;18(9):1876–87. doi: 10.1200/JCO.2000.18.9.1876. [DOI] [PubMed] [Google Scholar]
- 2.Moorman AV, Richards SM, Martineau M, Cheung KL, Robinson HM, Jalali GR, et al. Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood. 2003 Oct 15;102(8):2756–62. doi: 10.1182/blood-2003-04-1128. [DOI] [PubMed] [Google Scholar]
- 3.Hasle H. Pattern of malignant disorders in individuals with Down’s syndrome. Lancet Oncol. 2001 Jul;2(7):429–36. doi: 10.1016/S1470-2045(00)00435-6. [DOI] [PubMed] [Google Scholar]
- 4.Strefford JC, van Delft FW, Robinson HM, Worley H, Yiannikouris O, Selzer R, et al. Complex genomic alterations and gene expression in acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Proc Natl Acad Sci U S A. 2006 May 23;103(21):8167–72. doi: 10.1073/pnas.0602360103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004 Apr 8;350(15):1535–48. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 6.Romana SP, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, et al. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995;85(12):3662–70. [PubMed] [Google Scholar]
- 7.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003 Sep;3(9):639–49. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 8.Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008 Jan 18;319(5861):336–9. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- 9.Ambros V. The functions of animal microRNAs. Nature. 2004 Sep 16;431(7006):350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 10.Lee YS, Kim HK, Chung S, Kim KS, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005 Apr 29;280(17):16635–41. doi: 10.1074/jbc.M412247200. [DOI] [PubMed] [Google Scholar]
- 11.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005 Aug 15;65(16):7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 12.Xia HF, He TZ, Liu CM, Cui Y, Song PP, Jin XH, et al. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell Physiol Biochem. 2009;23(4–6):347–58. doi: 10.1159/000218181. [DOI] [PubMed] [Google Scholar]
- 13.Sonoki T, Iwanaga E, Mitsuya H, Asou N. Insertion of microRNA-125b-1, a human homologue of lin-4, into a rearranged immunoglobulin heavy chain gene locus in a patient with precursor B-cell acute lymphoblastic leukemia. Leukemia. 2005 Nov;19(11):2009–10. doi: 10.1038/sj.leu.2403938. [DOI] [PubMed] [Google Scholar]
- 14.Bousquet M, Quelen C, Rosati R, Mansat-De Mas V, La Starza R, Bastard C, et al. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J Exp Med. 2008 Oct 27;205(11):2499–506. doi: 10.1084/jem.20080285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009 Apr 1;23(7):862–76. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004 Feb;39(2):167–9. doi: 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 17.van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer. 2003 May;37(1):20–8. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- 18.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005 Mar 8;102(10):3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005 Oct;207(2):243–9. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 20.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006 May 2;103(18):7024–9. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends in neurosciences. 2009 Apr;32(4):199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Hertzberg L, Betts DR, Raimondi SC, Schafer BW, Notterman DA, Domany E, et al. Prediction of chromosomal aneuploidy from gene expression data. Genes Chromosomes Cancer. 2007 Jan;46(1):75–86. doi: 10.1002/gcc.20391. [DOI] [PubMed] [Google Scholar]
- 23.Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with pre-leukemic activity. Blood. 2004 May 15;103(10):3890–6. doi: 10.1182/blood-2003-10-3695. [DOI] [PubMed] [Google Scholar]
- 24.Starkova J, Madzo J, Cario G, Kalina T, Ford A, Zaliova M, et al. The identification of (ETV6)/RUNX1-regulated genes in lymphopoiesis using histone deacetylase inhibitors in ETV6/RUNX1-positive lymphoid leukemic cells. Clin Cancer Res. 2007 Mar 15;13(6):1726–35. doi: 10.1158/1078-0432.CCR-06-2569. [DOI] [PubMed] [Google Scholar]
- 25.Frohling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007 Dec;12(6):501–13. doi: 10.1016/j.ccr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007 Jan 12;282(2):1479–86. doi: 10.1074/jbc.M609383200. [DOI] [PubMed] [Google Scholar]
- 27.Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu M, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A. 2007 Dec 11;104(50):19983–8. doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klusmann J-H, Janikova K, Li Z, Orkin SH, Reinhardt D. Chromosome 21-Encoded miR-125b and Its Role in the Development of Myeloid Leukemia in Children with Down’s Syndrome. ASH Annual Meeting Abstracts; 2007 November 16; 2007. p. 716. [Google Scholar]
- 29.Kuhn DE, Nuovo GJ, Martin MM, Malana GE, Pleister AP, Jiang J, et al. Human chromosome 21-derived miRNAs are overexpressed in down syndrome brains and hearts. Biochem Biophys Res Commun. 2008 Jun 6;370(3):473–7. doi: 10.1016/j.bbrc.2008.03.120. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006 Jan 12;354(2):166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 31.Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest. 2009 Apr;119(4):826–36. doi: 10.1172/JCI36428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato T, Toki T, Kanezaki R, Xu G, Terui K, Kanegane H, et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. Br J Haematol. 2008 May;141(5):681–8. doi: 10.1111/j.1365-2141.2008.07081.x. [DOI] [PubMed] [Google Scholar]
- 33.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004 Dec;6(6):587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 34.Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffe M, Mayeux P, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000 Mar 15;95(6):2076–83. [PubMed] [Google Scholar]
- 35.McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Basecke J, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008 Apr;22(4):708–22. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- 36.Ihle JN, Gilliland DG. Jak2: normal function and role in hematopoietic disorders. Curr Opin Genet Dev. 2007 Feb;17(1):8–14. doi: 10.1016/j.gde.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Liu CB, Itoh T, Arai K, Watanabe S. Constitutive activation of JAK2 confers murine interleukin-3-independent survival and proliferation of BA/F3 cells. J Biol Chem. 1999 Mar 5;274(10):6342–9. doi: 10.1074/jbc.274.10.6342. [DOI] [PubMed] [Google Scholar]
- 38.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008 Oct 25;372(9648):1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 39.Hon LS, Zhang Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol. 2007;8(8):R166. doi: 10.1186/gb-2007-8-8-r166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008 Sep 4;455(7209):64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008 Sep 4;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 42.Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, et al. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell stem cell. 2009 Jul 2;5(1):43–53. doi: 10.1016/j.stem.2009.04.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.