

Abstract
The monoclonal antibody market continues to witness an impressive rate of growth and has become the leading source of expansion in the biologic segment within the pharmaceutical industry. Currently marketed monoclonal antibodies target a diverse array of antigens. These antigens are distributed in a variety of tissues such as tumors, lungs, synovial fluid, psoriatic plaques, and lymph nodes. As the concentration of drug at the proximity of the biological receptor determines the magnitude of the observed pharmacological responses, a significant consideration in effective therapeutic application of monoclonal antibodies is a thorough understanding of the processes that regulate antibody biodistribution. Monoclonal antibody distribution is affected by factors such as molecular weight, blood flow, tissue and tumor heterogeneity, structure and porosity, target antigen density, turnover rate, and the target antigen expression profile.
Key words: biodistribution, monoclonal antibodies, pharmacokinetics and pharmacodynamics, tumor penetration
INTRODUCTION
Similar to their small molecule counterparts, biodistribution of antibody-based therapeutics is a key consideration that can be modulated to impact the ensuing in vivo pharmacological effect(s). Understanding factors associated with antibody biodistribution allow for the intelligent design of therapeutic candidates that colocalize within the relevant effect compartment. Manipulation of candidate biodistribution is particularly useful in the development of next-generation antibody-based therapies. Given increasing competition among validated therapeutic targets such as tumor necrosis factor (TNF), epidermal growth factor receptor, vascular endothelial growth factor, and CD20, novel attributes may be engineered for the design of the next-generation candidates to permit commercial differentiation against the marketed predecessors. Differences in biodistribution between antibody-based therapeutics to the same target can result in a competitive advantage for one product versus another, and therefore, next-generation leads often are centered on leveraging such improvements to gain market penetration. For example, the current cadre of anti-TNF agents illustrates various biodistribution-related properties that may differentiate the next-generation antibody-based therapeutics. While full-length immunoglobulin G (IgG) anti-TNF agents are primarily distributed within the blood stream (30–80 mL/kg) (1–3), smaller IgG-derived competitors, namely etanercept, a dimeric fusion protein consisting of the extracellular ligand-binding protein of p75 TNF receptor linked to human IgG1 Fc, appear to distribute within tissues to a greater extent (0.1 to 0.2 L/kg) (4,5). Since tissue distribution of an anti-TNF agent may lead to greater efficacy in certain autoimmune/inflammatory indications, some next-generation anti-TNF agents have utilized lower molecular weight compounds with greater biodistribution properties to compete against the marketed predecessors (i.e., anti-TNF domain antibody with an apparent volume of distribution of 0.3 to 0.5 L/kg (6)).
Additionally, a diverse set of processes regulates biodistribution of antibodies into tumors as the anatomical and physiological properties of solid tumors are highly different from those observed with normal tissues (7–9). Heterogeneous and leaky tumor vasculature, rapid tumor growth, elevated interstitial fluid pressure (IFP), tumor necrosis, and tissue porosity can introduce challenges for antibody biodistribution into tumors that may be irrelevant to other tissues. Therefore, a thorough understanding of the processes that regulate antibody biodistribution in health and disease will be necessary for the effective application of antibody therapeutics. A unique characteristic of antibody function is the exquisite specificity for the interaction with targeted antigens (soluble or cell-associated); hence, antigen expression and antigen density can impact antibody pharmacokinetics (PK), pharmacodynamics (PD), and biodistribution (10–16). All currently approved intact antibodies are of the IgG class of either IgG1, IgG2, IgG4, or murine IgG2a isotypes. Thus, this article will focus on evaluating factors that regulate IgG antibody biodistribution in normal and tumor tissues.
OVERVIEW OF FACTORS IMPACTING ANTIBODY BIODISTRIBUTION
Antibody Structure
Each IgG molecule is a large glycoprotein with molecular mass of ~150 kDa containing two identical heavy chains (50 kDa each) and two identical light chains (25 kDa each) linked together by interchain disulfide bonds. Antibody structure has evolved to accommodate the diverse antigen-binding specificities through diversity in the variable region sequence. The antigen-binding site is formed by the intertwining of the light VL and heavy VH variable domains. Each V domain contains three short stretches of peptide known as the complementarity determining regions (CDRs); the CDRs are the major determinants of antigen-binding affinity and specificity. The light chain contains one constant domain: CL. The heavy chain contains three constant domains: CH1, CH2, and CH3. The CH2 and CH3 domains allow interactions of the IgG molecule with various components of the immune system by either binding C1q or Fcγ receptors on the immune effector cells. These same variable and constant domains of the molecule can also regulate IgG distribution, elimination, and activity (17–19).
General Biodistribution Mechanisms
Distribution defines the reversible transfer of molecules from one location to another within the body. In general, active and/or passive transport processes govern the movement of molecules across tissue membranes. Active transport requires an energy source, such as a biological carrier, to facilitate the transport of the drug. This carrier-mediated transport can proceed from regions of low concentrations to high concentrations via the action of a receptor or a carrier. In contrast, passive transfer involves simple diffusion of drug and is impacted by factors such as tissue permeability, surface area, concentration, and pressure gradients across the membrane.
Movement of molecules from blood to body tissues depends on various factors as related to (a) drug size, polarity, lipophilicity, and charge, (b) membrane porosity and structure, (c) blood flow characteristics, as well as (d) concentration and pressure gradients. Passive diffusion across membranes depends on factors such as tissue permeability, membrane surface area, and concentration and pressure gradients of drug across the membrane. In general, the rate of distribution may be limited by blood flow (perfusion rate limited) or the permeability of the membrane (permeability rate limited). In the first case, the cell membrane presents no barrier to drug distribution, as it is observed with highly lipophilic drugs, and the rate of transfer is dependent on drug molecules’ transit time through the tissue as well as the tissue flow rate characteristics (Fig. 1 a). In contrast, as it is expected with highly polar and charged molecules diffusing across tightly knitted membranes, distribution would be dependent on membrane thickness and physiological structure (Fig. 1 b). Additionally, the process of convection may facilitate transvascular transport of macromolecules. Contrary to diffusion that is proportional to the concentration gradient (Fig. 1 d), convection will depend on the pressure gradient (hydrostatic pressure − osmotic pressure) across vascular and interstitial compartments (Fig. 1 c). The proportionality constant that relates vascular leakage to pressure gradient is referred to as the hydraulic conductivity (20).
Fig. 1.

Movement of molecules from blood to body tissues. In general, the rate of distribution may be limited by blood flow (perfusion-rate limited, a) or the membrane permeability (permeability-rate limited, b). Mechanisms of transvascular transport: convection, due to the pressure gradient (c), and diffusion, primarily due to the concentration gradient (d)
For IgG antibodies with similar physical and structural properties (charge, polarity), the transcapillary transport across the blood capillary beds occurs mainly via diffusion and/or convection and, hence, will mainly depend on the capillary endothelium and the underlying basement membrane structure. In general, there are four types of blood capillaries that could facilitate the vascular transport of macromolecules such as IgG monoclonal antibodies (21). First, the continuous capillaries are distributed in structures such as connective tissue, skin, muscle, and a variety of other tissues throughout the body. The continuous capillaries have fully formed membranes, and the endothelial cells form an almost uninterrupted lining rich in tight junctions. The second type, the fenestrated capillaries, is found in the gastrointestinal tract, various glands, and renal glomeruli. The membranes of the fenestrated capillaries contain clefts of about 30 to 80 nm in diameter. Third, the sinusoids have clefts of about 100 nm and are found in organs such as liver, spleen, and tissues like bone marrow. Monoclonal antibodies can freely travel through the sinusoidal clefts. Diffusion of large molecular weight substances such as dextran and IgG (both 150 kDa) in normal tissues was reported using an in vivo rabbit model (20,22). Finally, in brain, the capillary endothelium and the underlying basement membrane structure are composed of tight-junction capillaries, and their intracellular junctions are closed by a belt of tight junctions (21,23).
Pharmacokinetic Concepts
The process of distribution can be characterized by a pharmacokinetic parameter known as the volume of distribution (V). As an independent physiological parameter, distribution volume can be influenced by physiological variables such as binding in blood/plasma and tissue, partition into fat, body composition, and body size (24). Volume of distribution of an antibody relates to volume of plasma or blood (Vp), the volume of tissue (VT), and the tissue-to-plasma partitioning (kP). Under linear conditions, IgG antibodies are primarily distributed into the plasma compartment and the extravascular fluid following intravascular administration in animals or humans (10,13,14). However, as binding to plasma or tissue targets can influence antibody distribution, it is not surprising that density and expression of the target antigen or, alternatively, active transport processes such as uptake by neonatal Fc receptor (FcRn) may also impact antibody biodistribution.
Role of FcRn in Antibody Pharmacokinetics and Distribution
FcRn functions both to protect IgG from elimination and influence IgG tissue distribution (for a review, see (13)). FcRn is expressed ubiquitously throughout the body in a wide variety of adult tissues. The protective role of FcRn, a major histocompatibility complex class I-related receptor, in regulation of IgG homeostasis was postulated by Brambell et al. (25,26). Earlier investigations demonstrated that the clearance rate of IgG was greatly dependent on its serum concentration, whereas the concentration impact on clearance did not apply to other immunoglobulin subclasses such as immunoglobulin M and immunoglobulin A (13,27). Longer IgG half-lives were reported at lower IgG concentrations consistent with Brambell’s hypothesis for the protective role of FcRn in IgG homeostasis. The impact of serum IgG concentrations on IgG clearance was also demonstrated in experimental animal models (28,29). Administration of a large dose of purified human IgG (2 g/kg) in mice resulted in a rapid decrease (>60%) in baseline IgG1 and IgG3 mouse serum concentrations (29). However, normal variation in endogenous IgG levels will not impact the elimination rate or distribution of therapeutic antibodies; likewise, the usual therapeutic doses of monoclonal antibodies are not expected to increase total IgG levels to the point that IgG biodistribution and elimination is affected (13). Additionally, the role of FcRn in biodistribution of IgG antibodies was examined recently (30). Lower tissue exposures were observed in FcRn knockout (KO) mice when compared to wild-type animals. However, in the majority of the tissues examined, the ratio of tissue-to-plasma exposure between wild-type and KO animals was not significantly different, suggesting that tissue distribution was primarily due to transvascular transport processes such as convection and/or diffusion (Fig. 1) (30). In some tissues such as skin and muscle, a more prominent role for FcRn in antibody distribution was observed, and in these tissues, exposure profiles did not appear to decline, in parallel with the plasma exposure. This observation is consistent with the structural property of these tissues comprising mainly of a continuous capillary structure with an uninterrupted lining rich in tight junctions—a microenvironment where processes such as convection or diffusion may not be effective due to the larger molecular size of IgG antibodies.
Impact of Affinity on Antibody Elimination and Distribution
Soluble Antigens
In addition to FcRn, three classes of Fc receptors (FcγRs) for IgG interactions have been identified in humans; these receptors are expressed by various phagocytic cells, such as monocytes, macrophages, neutrophils and eosinophils, and other cells of the immune system, including B and T cells, as well as platelets. Human FcγRs bind IgGs with varying degrees of affinity, ranging from low (>10−7 M) for FcγRII (CD32), medium (= 10−7 M) for FcγRIII (CD16), to high (10−8 to 10−9 M) for FcγRI (CD64) and can regulate IgG activity and elimination (31–33). Recent findings highlight that Fcγ receptors expressed on macrophages or dendritic cells can trigger the internalization of captured immune complexes, which leads to degradation and the clearance of the antigen–antibody complexes (Fig. 2a). In the simplest case, the in vivo steady-state concentrations of endogenous proteins are regulated by their rates of production and elimination. Changes in either antigen production or degradation rates could directly impact the in vivo steady-state concentrations of the antigen. Following administration of therapeutic doses of monoclonal antibodies in vivo, steady-state concentrations are generally achieved following administration of >4 to 5 doses when dosing frequency is less than or equal to the in vivo antibody elimination half-life. Although free concentrations of antigen are suppressed following administration of the antibody doses, a simultaneous increase in the antibody–antigen complex is observed (34,35). The magnitude of the in vivo increases in antibody–antigen complex concentrations will be dependent on (a) the turnover rate of the antigen (i.e., antigen synthesis and clearance rates) relative to that observed for the antibody and (b) the elimination rate of the antibody–antigen complex due to interaction with Fcγ receptors (Fig. 2). For example, omalizumab is a humanized IgG1 antibody that inhibits binding of immunoglobulin E (IgE) to its high affinity receptor, FcεRI (36,37). Omalizumab was shown to lower free IgE levels in a dose- and baseline IgE-dependent manner along with simultaneous increases in the antibody-bound complexes (38,39). In vitro studies indicated that omalizumab formed a variety of complexes at various antibody/IgE molar ratios (40). Indeed, due to target-mediated disposition of omalizumab, as related to formation of large complexes with IgE, nonlinear pharmacokinetics was reported for the free antibody at doses below 0.5 mg/kg (41,42). As formation of these complexes in vivo could be impacted by the antibody dose and affinity (more stable complexes can form with higher affinity antibodies), it is possible that increases in antibody–antigen binding in circulation may translate into lower serum and tissue exposure when a more rapid clearance of the complex is anticipated. This hypothesis is supported by previous experimental data demonstrating formation of large immune complexes in mice following simultaneous administration of a therapeutic antibody with its anti-idiotypic counterpart (43). As suggested by theoretical simulations shown in Fig. 2b, when the clearance of the complex is increased by 10-fold relative to the clearance of the free antibody, a proportional decrease in the plasma exposure is predicted. The predicted decrease in serum exposure is more pronounced when antibody affinity is higher for the target antigen (Fig. 2b relative to c). These theoretical simulations project the potential impact of binding affinity on antibody exposure and distribution.
Fig. 2.

Fcγ receptor-mediated clearance of antibody–antigen (i.e., omalizumab–IgE) complexes (a). Theoretical impact of antibody affinity and antibody–antigen clearance (CLComplex) relative to free antibody clearance (solid line CLComplex=CLAntibody; dashed line CLComplex=x2CLAntibody; dotted line CLComplex=x10CLAntibody) on the free antibody serum concentration–time profiles at a simulated affinity of 200 pM (b) and 1,000 pM (c). Theoretical impact of changes in affinity on antibody exposure was evaluated using a bimolecular interaction PK–PD model. The model accounted for free antibody PK (CL and volume of distribution), bimolecular interactions between antigen and antibody, and the elimination of free antigen. The impact of changes in antibody affinity and clearance of antibody–antigen complex on antibody exposure was evaluated
Cell-Associated Antigens
In addition to tumor properties (outlined in next section), penetration of antibodies into tumors can be influenced by factors such as antibody affinity, antigen internalization, and antibody metabolism by the tumor. A number of comprehensive reviews have recently addressed this topic in detail (44–47). Under nonsteady-state conditions, an inverse relationship between antibody affinity and tumor penetration has been predicted (48). This inverse relationship is termed the “binding-site barrier” hypothesis and can be offset by factors such as dose and antibody elimination half-life. For antibody fragments with rapid clearance rates (t1/2 ≈ minutes for single-chain variable fragment; hours for fragments with antigen binding) and under nonsteady-state conditions, tumor penetration is predicted to be highly influenced by antibody affinity (45–47). However, full-length antibodies generally have a long elimination half-life and are administered frequently, i.e., weekly, biweekly, or monthly, where steady-state serum concentrations are achieved. Under steady-state conditions, which is a condition generally met in clinical practice, binding equilibrium between antibody and antigen is achieved rapidly, and the movement of antibody through the tumor can be governed by antigen turnover rate, antigen density, as well as the antibody concentration gradient (45–48). In general, cell-associated antigens undergo internalization at constitutive rates, ranging from minutes to days. After binding to cell-associated antigens, antibodies are internalized at a similar rate as the antigen. The internalized complex then is transferred to the lysosome where it undergoes degradation. The newly synthesized antigen following resurfacing can then interact with the unbound antibody. Again under nonsteady-state conditions, it is predicted that tumor exposure can be reduced by the rate of antigen recycling (45–48).
Factors Impacting Tumor Distribution
One of the major challenges in cancer therapy is the hampered ability of large molecules to penetrate tumors efficiently. This limitation is thought to be the result of the architecture and physiology of solid tumors. The poor uptake of antibodies by tumors is the result of slow diffusion rates and the long distances required for diffusional transport in poorly vascularized tumors. This compromised uptake is further exacerbated by virtue of the fact that tumors often lack functional lymphatics (49–52), which can lead to increased levels of IFP (50,53,54). An increase in IFP is likely to reduce convection and thereby inhibit the uptake of antibodies (7,49). Small tumors (or micrometastases), on the other hand, tend to exhibit a more uniform circulatory system and lower interstitial pressure; as a result, antibody drug delivery is anticipated to be more efficient under these conditions.
Additionally, most tumor blood vessels have an irregular diameter and an abnormal branching pattern. For example, Hori and colleagues have shown that terminal arterioles in rat hepatoma or lung carcinomas approximated 50 branches per 0.1 mm2, as compared to 14 per 0.1 mm2 in normal tissues (55). Interestingly, a similar phenomenon occurs in leukemia: irregular and complex branching of microvessels has been observed in leukemic bone marrow, compared with the single, straight microvessels without branching in normal bone marrow (56).
Tumor endothelial cells, although present on most if not all tumor vessels, are actively dividing, unlike the endothelium within normal tissues (57). Tumor blood vessels often do not contain smooth muscle cells, in contrast to normal endothelium, and are characterized by either a discontinuous or absent basement membrane (58–60). Furthermore, tumor endothelial cells also do not form a normal monolayer and therefore do not exhibit normal barrier function. The cells are disorganized and irregularly shaped, and the tumor vessel wall may be lined with only cancer cells or a “mosaic” layer of cancer and endothelial cells (61). These defects render tumor blood vessels leaky (62–64). In fact, studies indicate that tumor vessels are an order of magnitude more leaky than normal vessels. The pore cutoff size (indicating the largest particles that can cross vessels walls) is measured in hundreds of nanometers in tumors, as compared to tens of nanometers for normal endothelium (58,61,65). Also, blood vessels near the center of the tumor are compressed due to the dysregulated growth of tumor cells in a confined space, ultimately impeding blood flow. Pericytes, contractile cells that stabilize vessel walls and participate in the regulation of blood flow, have been reported to exhibit an abnormal association with tumor endothelium and display altered expression of marker proteins (60). While pericytes are present on most tumor vessels, studies indicate that these cells exhibit a loose association with endothelial cells and have cytoplasmic processes that extend deep into the tumor tissue (60). The composition and structure of the extracellular matrix is another contributing factor that can retard the movement of molecules within the tumor (66–68).
As a consequence of the poorly organized vasculature in solid tumors, blood flow is sluggish with unstable rheology. Hence, delivery of oxygen and nutrients to cells that are distant from functional blood vessels is limited, resulting in significant hypoxia and accumulation of products of metabolism, including lactic and carbonic acid, which decrease the pH in this microenvironment (69,70). These oxygen- and nutrient-deprived regions are often necrotic and localized to the central regions of the tumor; as a result of the limited and abnormal blood flow rates in necrotic and seminecrotic regions, efficient drug delivery is severely hindered. For example, the laboratory of Rakesh Jain determined in rodent models that a continuously supplied monoclonal antibody could take several months to reach a uniform concentration in a tumor that measured 1 cm in radius and lacked a blood supply in its center (49,50).
It is also postulated that the aforementioned barriers may hinder the migration of immune effector cells to the tumor (71). In the context of antibody therapy, targeting of malignant cells within a poorly vascularized tumor requires infiltration of immune effector cells and tumor cell killing via Fc receptor-mediated mechanisms such as antibody-dependent cellular cytotoxicity (ADCC). Thus, in addition to poor antibody penetration in this microenvironment, the reduced migration of immune effector cells could adversely impact antibody efficacy.
BIODISTRIBUTION OF MARKETED ANTIBODIES
Antibody Distribution in Lung
Respiratory syncytial virus (RSV) is the main viral respiratory pathogen causing hospitalization in infants and young children worldwide (72). Anti-RSV antibodies have proven highly effective in the prevention of respiratory tract infection and reduction of infant hospitalization caused by this pathogen (72). Palivizumab is the first humanized monoclonal antibody currently marketed in the USA effective in prevention of RSV infection when administered systemically (73). As RSV infection directly impacts the respiratory tract in infants, biodistribution of anti-RSV antibodies to lungs is a critical requirement for pulmonary viral load neutralization following systemic administration (Fig. 3a). Under steady-state serum concentrations, linear distribution of IgG antibodies into lung was reported previously (74). In cynomolgus monkeys, dose-dependent increases in concentrations of mepolizumab, an IgG1 antibody, in bronchoalveolar lavage fluid (BALF) were observed and reported to be approximately between 500- and 1,000-fold less than the steady-state plasma concentrations of the antibody (74). Similar findings have been reported for anti-RSV antibodies (75,76). As improvements in antibody affinity and PK half-life can directly impact viral neutralization as shown in animal models of RSV infection, more potent second-generation anti-RSV antibodies such as motavizumab (MEDI-524) are currently in late-stage clinical development (76,77). Motavizumab is an IgG1 anti-RSV antibody that offers approximately a 20-fold higher in vitro potency for viral neutralization as compared to palivizumab (77). Additionally, prophylaxis studies in cotton rats revealed an approximately 5-fold reduction in required serum concentrations (~8 versus 40 μg/mL) as compared to palivizumab for >2log10 decrease in lung viral load (Fig. 3b), the desired clinical endpoint in RSV immune prophylaxis in infants. Following single dose administration of MEDI-524 in monkeys, the partition coefficient for antibody distribution into BALF 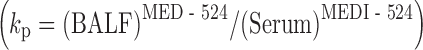 on days 4 and 24 postantibody administration were reported to be between 0.001 and 0.002, reflecting approximately >1,000-fold lower effective concentrations (>8 ng/mL) of MEDI-524 in the effect compartment relative to that observed in serum (Fig. 3b). Additionally, improvement in PK half-life via enhancement of FcRn binding for MEDI-524-YTE resulted in further increases in BALF antibody concentrations (75). A 10-fold improvement in FcRn binding resulted in approximately a 4-fold increase in MEDI-524-YTE circulating half-life relative to that observed for MED-524 in monkeys (Fig. 3c). Consistent with the increase in serum exposure, proportional increases in the FcRn-engineered antibody concentrations in BALF were also observed (75). However, when the BALF concentrations were normalized to serum antibody concentrations, no significant difference in antibody concentrations was observed relative to MEDI-524 (75). These results are consistent with previous data reported in FcRn KO mice highlighting the involvement of transvascular transport processes such as convection and/or diffusion in MEDI-524 distribution into the lung compartment (30).
on days 4 and 24 postantibody administration were reported to be between 0.001 and 0.002, reflecting approximately >1,000-fold lower effective concentrations (>8 ng/mL) of MEDI-524 in the effect compartment relative to that observed in serum (Fig. 3b). Additionally, improvement in PK half-life via enhancement of FcRn binding for MEDI-524-YTE resulted in further increases in BALF antibody concentrations (75). A 10-fold improvement in FcRn binding resulted in approximately a 4-fold increase in MEDI-524-YTE circulating half-life relative to that observed for MED-524 in monkeys (Fig. 3c). Consistent with the increase in serum exposure, proportional increases in the FcRn-engineered antibody concentrations in BALF were also observed (75). However, when the BALF concentrations were normalized to serum antibody concentrations, no significant difference in antibody concentrations was observed relative to MEDI-524 (75). These results are consistent with previous data reported in FcRn KO mice highlighting the involvement of transvascular transport processes such as convection and/or diffusion in MEDI-524 distribution into the lung compartment (30).
Fig. 3.

Biodistribution of anti-RSV antibodies to the lung compartment is a critical requirement for pulmonary viral load neutralization following systemic administration (a). Relationships between viral neutralization and MEDI-524 in serum (adapted from (75)) and estimated BALF concentrations assuming a k P of 0.001 (b). Serum concentration–time profiles for MEDI-524 and MEDI-524-YTE following a single-dose administration in monkeys generated from pharmacokinetic parameters estimate reported previously (75) (c)
Antibody Distribution in the Arthritic Joint
Antibodies have been utilized successfully in the management of various inflammatory diseases such as RA, psoriasis, and Crohn’s disease. As inhibition of the target antigen in synovium or psoriatic skin is necessary for induction of the pharmacological effect, antibody penetration into these compartments is a critical requirement for therapeutic efficacy. The importance of suppression of cytokines such as TNF, interleukin (IL)-17, and IL-12/23 in synovium during treatment of RA has recently been demonstrated, and the role of the synovial membrane expression of these cytokines as predictive of joint damage and disease progression is now well established (78–82). In line with these observations, effective inhibition of cytokines such as IL-17 and TNF allows rapid control of the inflammatory manifestations of RA and retards cartilage and bone destruction. Previous studies reported approximately >5-fold lower concentrations of IgG antibodies in synovial fluid in human RA patients (83). Due to lower synovial concentrations relative to serum following systemic administration of antibodies, both affinity and pharmacokinetic half-life are among the critical factors that could impact the clinical dose, dosing frequency, and the extent and duration of synovial antigen suppression. This conclusion is supported by a review of recently marketed anti-TNF antibodies (Table I). Currently, a number of intact IgG antibodies have been successfully used in the management of RA, psoriasis, or Crohn’s disease by targeting TNF. Although these antibodies bind to and neutralize TNF, they share few similarities with respect to their affinity, pharmacokinetics, recommended dose, and dosing frequency (Table I) (1,84–87). It is evident that improvements in antibody affinity and pharmacokinetic half-life directly influence the performance of these molecules in the clinic; the most recent anti-TNF antibody marketed in the USA, golimumab, has the highest potency and the longest PK half-life relative to other anti-TNF antibodies with the lowest required effective clinical dose and dosing frequency(Table I) (87).
Table I.
Intact Anti-TNF IgG Antibodies Currently Marketed in the USA for Treatment of Immune-Mediated Inflammatory Disorders
The theoretical impact of improvements in antibody half-life and affinity on the suppression profile of a circulating antigen can be evaluated using a bimolecular interaction PK–PD model (10–14). The model accounts for antibody PK, bimolecular interactions between antigen and antibody, and the elimination of free antigen. As observed with anti-TNF antibodies, computer projections predict that improvements in antibody PK and affinity favorably impact the antigen suppression profiles in serum and synovium (Fig. 4). As shown, improvements in both antibody half-life and affinity results in more pronounced (40% to 70%) and prolonged antigen suppression following a monthly dosing regimen, i.e., once monthly dosing of a 50- to 60-mg dose versus a 40-mg dose administered twice monthly (Fig. 4a). In line with the lower required dose and less frequent dosing frequency observed with golimumab, the profiles describing the theoretical impact of improvements in antibody affinity on antigen suppression in synovial fluid are also shown in Fig. 4b.
Fig. 4.

a Theoretical improvements in area under the antigen suppression curves (Y axis) following changes in both antibody half-life (from 12 to 18 days) and affinity (from 10 to 2 pM). These improvements resulted in a more pronounced (40% to 70%) and prolonged antigen suppression following a monthly administration of 40- to 70-mg doses relative to administration of a 40-mg dose twice monthly with antibody affinity of 10 pM and half-life (t 1/2) of 12 days (not shown). Theoretical impact of improvements in antibody affinity on antigen suppression time profiles in synovial fluid is shown in b. Simulations were generated using a bimolecular interaction PK–PD model as described for Fig. 2
Antibody Distribution in Brain
The capillary endothelium and the underlying basement membrane structure in brain are composed of tight-junction capillaries, and their intracellular junctions are closed by a belt of tight junctions (21,23). Under the conditions where the integrity of blood–brain barrier (BBB) is intact, the presence of tight junctions prevents the transcellular route for diffusion of antibodies across the capillary. In general, antibody penetration into brain (cerebrospinal fluid (CSF)/serum partitioning) has been reported to be >0.1% in animal models and in human patients (88,89). These data are in line with the distribution of endogenous IgG subclasses in normal volunteers ranging from 800- to 1,000-fold less in CSF relative to that reported in serum (Fig. 5) (90). Similarly, these data were reproduced following administration of weekly doses of rituximab in patients with central nervous system (CNS) lymphoma (the antibody in CSF was reproducibly detected at concentrations that were 1,000-fold less than that observed in serum). Therefore, alternative routes of administration, i.e., intrathecal route, were proposed to overcome the poor penetration of rituximab into the brain for treatment of CNS lymphoma (89).
Fig. 5.

Concentrations of endogenous IgG subclass in the CSF and sera of normal subjects (n = 7). The line in each box represents the median and the box shows the range. Data adapted from (90)
FcRn is shown to be expressed in the CNS endothelium and choroid plexus (91). It is hypothesized that FcRn may play a key role in limiting CNS inflammation in pathological situations such as bacterial infection where IgG can move into the CNS down its sharp concentration gradient (23,92). Under these conditions, FcRn may mediate reverse transcytosis of IgG from the CNS back into circulation. Evidence in support of this hypothesis has been established following direct intracerebral injection of IgG antibodies where IgG molecules rapidly transported back into the circulation via an FcRn-mediated process (93). Indeed, this mechanism has been exploited to reduce amyloid plaque burden in the experimental model of Alzheimer’s in rodents (94,95). Recently, the design of a trifunctional fusion construct allowed the application of a new antibody-based therapeutic engineered to cross the BBB in both directions for experimental treatment of Alzheimer’s disease (95). The reverse transcytosis of IgG by FcRn may further provide a potential mechanism for therapeutic efficacy of IgG antibodies in the treatment of Alzheimer’s disease (see below).
In conditions where the integrity of the BBB is compromised, antibodies have been utilized for treatment of CNS diseases via the systemic route. Recently, bevacizumab in combination with standard chemotherapy was approved for treatment of patients with recurrent glioblastoma (96–98). The exact underlying mechanism(s) for the encouraging responses observed in this patient population is not well understood (97,99). Glioblastoma is a highly vascularized tumor, and anti-angiogenic therapy has proven effective in reduction of vasogenic edema observed in these patients. In the highly vascularized glioblastoma, a dramatic increase in the permeability of BBB blood vessels was observed, and BBB breakdown was reported by the changes of the molecular compositions of both the endothelial tight junctions and the basal lamina structure (100). Similarly, BBB impairment has been identified as a stable characteristic in patients with mild to moderate Alzheimer’s disease. For example, the CSF/plasma IgG ratio was shown to be correlated with CSF-albumin index, a marker for evaluation of BBB integrity, in a manner indicating that peripheral IgG had greater access to the CNS in patients with an impaired BBB (101). In line with these data, bapineuzumab, a humanized monoclonal antibody that acts on the nervous system with potential therapeutic value for the treatment of Alzheimer's disease, is currently in late-stage clinical development (102).
Antibody Distribution in Lymph Nodes, Spleen, and Bone Marrow
Monoclonal antibodies can freely travel through the sinusoidal clefts found in organs such as liver, spleen, and bone marrow. The sinusoids have clefts of approximately 100 nm in diameter allowing antibody movement and biodistribution. For example, rituximab is a chimeric antibody that contains the human IgG1 Fc kappa constant region and murine variable regions reactive with the membrane-associated human CD20 antigen. Rituximab binds to the CD20 antigen found on the surface of normal and malignant B lymphocytes and is currently approved for treatment of relapsed or refractory low-grade or follicular B cell non-Hodgkin’s lymphoma (103–105). CD20 is a 35–37-kDa nonglycosylated tetra-spanning membrane protein expressed exclusively by B lymphocytes (104). CD20 is expressed at the late pre-B cell stage and is upregulated and expressed at high levels on most normal and malignant B lineage cells before being downregulated in terminally differentiated plasma cells. Effective distribution and hence depletion of B cells in organs such as lymph nodes, bone marrow, and spleen is a critical requirement for successful clinical application of B cell depleting agents such as anti-CD20 antibodies. In vivo binding of rituximab to CD20 positive B cells results in depletion of B cells. Rituximab induces B cell death primarily through complement-dependent lysis and antibody-dependent cellular toxicity effector mechanisms and to a lesser degree via cellular apoptosis (106). Administration of rituximab results in rapid and sustained depletion of circulating and tissue B cells in humans and monkeys (106–109). The strong evidence for biodistribution of rituximab in critical organs such as lymph nodes and bone marrow was recently determined via evaluation of tissue B cell depletion under steady-state antibody serum concentrations (110). Following administration of three weekly doses of rituximab to monkeys where steady-state serum concentrations were achieved, dose-dependent depletion of CD20 positive B cells in spleen, bone marrow, and lymph nodes was observed, underscoring antibody penetration/distribution into these critical organs (Fig. 6). Interestingly, when a second-generation anti-CD20 antibody with enhanced pro-apoptotic activity was administered in monkeys, a more pronounced B cell depletion profile in the same tissues was observed (110). The more efficient B cell depletion observed with the second-generation anti-CD20 antibody was attributed to improved ADCC and apoptosis induction triggered by this antibody and not due to differences in tissue distribution or immunogenicity (110).
Fig. 6.

Dose-dependent depletion of CD20+ B lymphocytes in cynomolgus monkey tissues following administration of three weekly doses of rituximab (tissues were collected and analyzed 3 days after administration of the third dose on day 15). The method is described in (110). Ax. LN. axillary lymph nodes, Mes. LN. mesenteric lymph nodes, Ing. LN. inguinal lymph nodes
CONCLUSIONS
As the concentration of drug at the proximity of the biological receptor determines the magnitude of the observed pharmacological responses, understanding factors associated with antibody biodistribution allows for the intelligent design of therapeutic candidates that colocalize within the intended effect compartment. Improvements in pharmacokinetic half-life via engineering of antibody affinity for FcRn can directly influence the dosing frequency when linear distribution to the effect compartment is anticipated. Additionally, under the conditions where binding is impacted by affinity (i.e., where antigen concentrations are less than or close to KD), improvements in affinity can greatly impact dose and dosing frequency. As manipulation of antibody biodistribution can particularly be useful in the development of next-generation antibody-based therapies, novel strategies such as multifunctional antibody constructs or antibody fragments and domains should evolve to allow for the smart design of future antibody-based therapeutics.
Acknowledgments
The material presented in this article was partly used as teaching aid during the Fourth and Fifth Annual Protein Engineering Summit, PEGS (2008, and 2009), and the Molecular Medicine TriMolecular Conference, MMTC, (2009).
References
- 1.Humira. Prescribing Information: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3930B1_02_B-Abbott-Humira%20Prescribing%20Info.pdf. 2009.
- 2.Mascelli MA, Zhou H, Sweet R, Getsy J, Davis HM, Graham M, et al. Molecular, biologic, and pharmacokinetic properties of monoclonal antibodies: impact of these parameters on early clinical development. J Clin Pharmacol. 2007;47(5):553–565. doi: 10.1177/0091270006298360. [DOI] [PubMed] [Google Scholar]
- 3.Xu Z, Vu T, Lee H, Hu C, Ling J, Yan H, et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-{alpha} human monoclonal antibody, in patients with psoriatic arthritis. J Clin Pharmacol. 2009;49(9):1056–1070. doi: 10.1177/0091270009339192. [DOI] [PubMed] [Google Scholar]
- 4.Nestorov I, Zitnik R, DeVries T, Nakanishi AM, Wang A, Banfield C. Pharmacokinetics of subcutaneously administered etanercept in subjects with psoriasis. Br J Clin Pharmacol. 2006;62(4):435–445. doi: 10.1111/j.1365-2125.2006.02581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou H. Clinical pharmacokinetics of etanercept: a fully humanized soluble recombinant tumor necrosis factor receptor fusion protein. J Clin Pharmacol. 2005;45(5):490–497. doi: 10.1177/0091270004273321. [DOI] [PubMed] [Google Scholar]
- 6.Tomlinson IM. Next-generation protein drugs. Nat Biotechnol. 2004;22(5):521–522. doi: 10.1038/nbt0504-521. [DOI] [PubMed] [Google Scholar]
- 7.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47(12):3039–3051. [PubMed] [Google Scholar]
- 8.Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6(4):559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- 9.Jain RK, Gerlowski LE. Extravascular transport in normal and tumor tissues. Crit Rev Oncol Hematol. 1986;5(2):115–170. doi: 10.1016/s1040-8428(86)80023-3. [DOI] [PubMed] [Google Scholar]
- 10.Tabrizi M, Roskos LK. Exposure–response relationships for therapeutic biologic products. In: Meibohem B, editor. Pharmacokinetics and pharmacodynamics of biotech drugs. Wiley: New York; 2006. pp. 295–327. [Google Scholar]
- 11.Tabrizi M, Suria H. Application of translational biomarkers in development of antibody-based therapeutics. Drug Discov. 2009;5(1): 2–6.
- 12.Tabrizi MA, Bornstein GG, Klakamp SL, Drake A, Knight R, Roskos L. Translational strategies for development of monoclonal antibodies from discovery to the clinic. Drug Discov Today. 2009;14(5–6):298–305. doi: 10.1016/j.drudis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–88. doi: 10.1016/S1359-6446(05)03638-X. [DOI] [PubMed] [Google Scholar]
- 14.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 15.Levy G. Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248–252. doi: 10.1038/clpt.1994.134. [DOI] [PubMed] [Google Scholar]
- 16.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–532. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 17.Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer's perspective. Drug Discov Today. 2007;12(21–22):898–910. doi: 10.1016/j.drudis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24(1):19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 20.Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50(3 Suppl):814s–819s. [PubMed] [Google Scholar]
- 21.Weinstein JN, van Osdol W. The macroscopic and microscopic pharmacology of monoclonal antibodies. Int J Immunopharmacol. 1992;14(3):457–463. doi: 10.1016/0192-0561(92)90176-l. [DOI] [PubMed] [Google Scholar]
- 22.Clauss MA, Jain RK. Interstitial transport of rabbit and sheep antibodies in normal and neoplastic tissues. Cancer Res. 1990;50(12):3487–3492. [PubMed] [Google Scholar]
- 23.Hawkins BT, Davis TP. The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 24.Gibaldi M, Koup JR. Pharmacokinetic concepts—drug binding, apparent volume of distribution and clearance. Eur J Clin Pharmacol. 1981;20(4):299–305. doi: 10.1007/BF00618781. [DOI] [PubMed] [Google Scholar]
- 25.Brambell FW. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet. 1966;2(7473):1087–1093. doi: 10.1016/s0140-6736(66)92190-8. [DOI] [PubMed] [Google Scholar]
- 26.Ghetie V, Ward ES. FcRn: the MHC class I-related receptor that is more than an IgG transporter. Immunol Today. 1997;18(12):592–598. doi: 10.1016/s0167-5699(97)01172-9. [DOI] [PubMed] [Google Scholar]
- 27.Waldmann TA. Variations in the metabolism of immunoglobulins measured by turnover rates. In: Merler E, editor. Immunoglobulins: biological aspects and clinical uses. Washington, DC: National Academy of Sciences; 1970. pp. 33–51. [Google Scholar]
- 28.Bleeker WK, Teeling JL, Hack CE. Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood. 2001;98(10):3136–3142. doi: 10.1182/blood.v98.10.3136. [DOI] [PubMed] [Google Scholar]
- 29.Hansen RJ, Balthasar JP. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood. 2002;100(6):2087–2093. [PubMed] [Google Scholar]
- 30.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 31.de Haas M. IgG-Fc receptors and the clinical relevance of their polymorphisms. Wien Klin Wochenschr. 2001;113(20–21):825–831. [PubMed] [Google Scholar]
- 32.Rascu A, Repp R, Westerdaal NA, Kalden JR, van de Winkel JG. Clinical relevance of Fc gamma receptor polymorphisms. Ann N Y Acad Sci. 1997;815:282–295. doi: 10.1111/j.1749-6632.1997.tb52070.x. [DOI] [PubMed] [Google Scholar]
- 33.Woof JM, Burton DR. Human antibody–Fc receptor interactions illuminated by crystal structures. Nat Rev. 2004;4(2):89–99. doi: 10.1038/nri1266. [DOI] [PubMed] [Google Scholar]
- 34.Margolin K, Gordon MS, Holmgren E, Gaudreault J, Novotny W, Fyfe G, et al. Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol. 2001;19(3):851–856. doi: 10.1200/JCO.2001.19.3.851. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol. 2007;63(5):548–561. doi: 10.1111/j.1365-2125.2006.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang TW. The pharmacological basis of anti-IgE therapy. Nat Biotechnol. 2000;18(2):157–162. doi: 10.1038/72601. [DOI] [PubMed] [Google Scholar]
- 37.Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354(25):2689–2695. doi: 10.1056/NEJMct055184. [DOI] [PubMed] [Google Scholar]
- 38.Busse WW. Anti-immunoglobulin E (omalizumab) therapy in allergic asthma. Am J Respir Crit Care Med. 2001;164(8 Pt 2):S12–S17. doi: 10.1164/ajrccm.164.supplement_1.2103026. [DOI] [PubMed] [Google Scholar]
- 39.Milgrom H, Fick RB, Jr, Su JQ, Reimann JD, Bush RK, Watrous ML, et al. Treatment of allergic asthma with monoclonal anti-IgE antibody. rhuMAb-E25 Study Group. N Engl J Med. 1999;341(26):1966–1973. doi: 10.1056/NEJM199912233412603. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Lester P, Builder S, Shire SJ. Characterization of complex formation by humanized anti-IgE monoclonal antibody and monoclonal human IgE. Biochemistry. 1995;34(33):10474–10482. doi: 10.1021/bi00033a020. [DOI] [PubMed] [Google Scholar]
- 41.Putnam WS, Li J, Haggstrom J, Ng C, Kadkhodayan-Fischer S, Cheu M, et al. Use of quantitative pharmacology in the development of HAE1, a high-affinity anti-IgE monoclonal antibody. AAPS J. 2008;10(2):425–430. doi: 10.1208/s12248-008-9045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johansson SG, Haahtela T, O'Byrne PM. Omalizumab and the immune system: an overview of preclinical and clinical data. Ann Allergy Asthma Immunol. 2002;89(2):132–138. doi: 10.1016/S1081-1206(10)61928-X. [DOI] [PubMed] [Google Scholar]
- 43.Johansson A, Erlandsson A, Eriksson D, Ullen A, Holm P, Sundstrom BE, et al. Idiotypic–anti-idiotypic complexes and their in vivo metabolism. Cancer. 2002;94(4 Suppl):1306–1313. doi: 10.1002/cncr.10301. [DOI] [PubMed] [Google Scholar]
- 44.Rudnick SI, Adams GP. Affinity and avidity in antibody-based tumor targeting. Cancer Biother Radiopharm. 2009;24(2):155–161. doi: 10.1089/cbr.2009.0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60(12):1421–1434. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29(2):57–61. doi: 10.1016/j.tips.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thurber GM, Zajic SC, Wittrup KD. Theoretic criteria for antibody penetration into solid tumors and micrometastases. J Nucl Med. 2007;48(6):995–999. doi: 10.2967/jnumed.106.037069. [DOI] [PubMed] [Google Scholar]
- 48.Graff CP, Wittrup KD. Theoretical analysis of antibody targeting of tumor spheroids: importance of dosage for penetration, and affinity for retention. Cancer Res. 2003;63(6):1288–1296. [PubMed] [Google Scholar]
- 49.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 50.Jain RK. The Eugene M, Landis Award Lecture 1996. Delivery of molecular and cellular medicine to solid tumors. Microcirculation. 1997;4(1):1–23. doi: 10.3109/10739689709148314. [DOI] [PubMed] [Google Scholar]
- 51.Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2(4):266–276. doi: 10.1038/nrc778. [DOI] [PubMed] [Google Scholar]
- 52.Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324–4327. [PubMed] [Google Scholar]
- 53.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 54.Milosevic MF, Fyles AW, Wong R, Pintilie M, Kavanagh MC, Levin W, et al. Interstitial fluid pressure in cervical carcinoma: within tumor heterogeneity, and relation to oxygen tension. Cancer. 1998;82(12):2418–2426. doi: 10.1002/(sici)1097-0142(19980615)82:12<2418::aid-cncr16>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 55.Hori K, Suzuki M, Tanda S, Saito S. In vivo analysis of tumor vascularization in the rat. Jpn J Cancer Res. 1990;81(3):279–288. doi: 10.1111/j.1349-7006.1990.tb02562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, Folkman J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150(3):815–821. [PMC free article] [PubMed] [Google Scholar]
- 57.Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. BioEssays. 1991;13(1):31–36. doi: 10.1002/bies.950130106. [DOI] [PubMed] [Google Scholar]
- 58.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62(18):5381–5385. [PubMed] [Google Scholar]
- 59.McDonald DM, Foss AJ. Endothelial cells of tumor vessels: abnormal but not absent. Cancer Metastasis Rev. 2000;19(1–2):109–120. doi: 10.1023/a:1026529222845. [DOI] [PubMed] [Google Scholar]
- 60.Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160(3):985–1000. doi: 10.1016/S0002-9440(10)64920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 62.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 63.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95(8):4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 66.Brown E, McKee T, diTomaso E, Pluen A, Seed B, Boucher Y, et al. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med. 2003;9(6):796–800. doi: 10.1038/nm879. [DOI] [PubMed] [Google Scholar]
- 67.Davies C de L, Berk DA, Pluen A, Jain RK. Comparison of IgG diffusion and extracellular matrix composition in rhabdomyosarcomas grown in mice versus in vitro as spheroids reveals the role of host stromal cells. Br J Cancer. 2002;86(10):1639–1644. doi: 10.1038/sj.bjc.6600270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–2503. [PubMed] [Google Scholar]
- 69.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 70.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49(16):4373–4384. [PubMed] [Google Scholar]
- 71.Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9(3):253–266. doi: 10.1007/BF00046364. [DOI] [PubMed] [Google Scholar]
- 72.Malley R, DeVincenzo J, Ramilo O, Dennehy PH, Meissner HC, Gruber WC, et al. Reduction of respiratory syncytial virus (RSV) in tracheal aspirates in intubated infants by use of humanized monoclonal antibody to RSV F protein. J Infect Dis. 1998;178(6):1555–1561. doi: 10.1086/314523. [DOI] [PubMed] [Google Scholar]
- 73.Wu H, Pfarr DS, Losonsky GA, Kiener PA. Immunoprophylaxis of RSV infection: advancing from RSV-IGIV to palivizumab and motavizumab. Curr Top Microbiol Immunol. 2008;317:103–123. doi: 10.1007/978-3-540-72146-8_4. [DOI] [PubMed] [Google Scholar]
- 74.Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, Maleeff BE, et al. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy Clin Immunol. 2001;108(2):250–257. doi: 10.1067/mai.2001.116576. [DOI] [PubMed] [Google Scholar]
- 75.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281(33):23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 76.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368(3):652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 77.Wu H, Pfarr DS, Tang Y, An LL, Patel NK, Watkins JD, et al. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J Mol Biol. 2005;350(1):126–144. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 78.Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, et al. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort) Arthritis Rheum. 2006;54(4):1122–1131. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 79.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103(9):1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double- blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371(9625):1675–1684. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 81.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Westacott CI, Barakat AF, Wood L, Perry MJ, Neison P, Bisbinas I, et al. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr Cartil. 2000;8(3):213–221. doi: 10.1053/joca.1999.0292. [DOI] [PubMed] [Google Scholar]
- 83.Choy EH, Connolly DJ, Rapson N, Jeal S, Brown JC, Kingsley GH, et al. Pharmacokinetic, pharmacodynamic and clinical effects of a humanized IgG1 anti-CD4 monoclonal antibody in the peripheral blood and synovial fluid of rheumatoid arthritis patients. Rheumatology (Oxford, England) 2000;39(10):1139–1146. doi: 10.1093/rheumatology/39.10.1139. [DOI] [PubMed] [Google Scholar]
- 84.Kievit W, Fransen J, Oerlemans AJ, Kuper HH, van der Laar MA, de Rooij DJ, et al. The efficacy of anti-TNF in rheumatoid arthritis, a comparison between randomised controlled trials and clinical practice. Ann Rheum Dis. 2007;66(11):1473–1478. doi: 10.1136/ard.2007.072447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Enbrel. Prescribing Information: http://www.enbrel.com/pdf/enbrel_pi.pdf. 2009.
- 86.Remicade. Prescribing Information: http://www.medversation.com/medversation/assets/PI_MedGuides_External/REMICADE_PI.pdf. 2009.
- 87.SIMPONI. Prescribing Information: http://www.centocoraccessone.com/centocoraccessone/assets/simponi/SIMPONI.pdf. 2009.
- 88.Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24(9):1733–1744. doi: 10.1007/s11095-007-9324-2. [DOI] [PubMed] [Google Scholar]
- 89.Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood. 2003;101(2):466–468. doi: 10.1182/blood-2002-06-1636. [DOI] [PubMed] [Google Scholar]
- 90.Kaschka WP, Theilkaes L, Eickhoff K, Skvaril F. Disproportionate elevation of the immunoglobulin G1 concentration in cerebrospinal fluids of patients with multiple sclerosis. Infect Immun. 1979;26(3):933–941. doi: 10.1128/iai.26.3.933-941.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood–brain barrier. J Neurochem. 2002;81(1):203–206. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 92.Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol dis. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood–brain barrier. J Neuroimmunol. 2001;114(1–2):168–172. doi: 10.1016/s0165-5728(01)00242-9. [DOI] [PubMed] [Google Scholar]
- 94.Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, et al. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood–brain barrier neonatal Fc receptor. J Neurosci. 2005;25(50):11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boado RJ, Zhang Y, Zhang Y, Xia CF, Pardridge WM. Fusion antibody for Alzheimer's disease with bidirectional transport across the blood–brain barrier and Abeta fibril disaggregation. Bioconjug Chem. 2007;18(2):447–455. doi: 10.1021/bc060349x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Avastin. Prescribing Information: http://www.gene.com/gene/products/information/pdf/avastin-prescribing.pdf. 2009.
- 97.Chamberlain MC. Bevacizumab plus irinotecan in recurrent glioblastoma. J Clin Oncol. 2008;26(6):1012–1013. doi: 10.1200/JCO.2007.15.1605. [DOI] [PubMed] [Google Scholar]
- 98.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Marcello J, Reardon DA, Quinn JA, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25(30):4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 99.Tate MC, Aghi MK. Biology of angiogenesis and invasion in glioma. Neurotherapeutics. 2009;6(3):447–457. doi: 10.1016/j.nurt.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rascher G, Fischmann A, Kroger S, Duffner F, Grote EH, Wolburg H. Extracellular matrix and the blood–brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin. Acta Neuropathol. 2002;104(1):85–91. doi: 10.1007/s00401-002-0524-x. [DOI] [PubMed] [Google Scholar]
- 101.Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood–brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology. 2007;68(21):1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bapineuzumab. http://www.wyeth.nl/Portals/0/downloads/pers/persberichten/Bapi%20P2%20FINAL%20 -- %2028July08.pdf. 2008.
- 103.Nadler LM, Korsmeyer SJ, Anderson KC, Boyd AW, Slaughenhoupt B, Park E, et al. B cell origin of non-T cell acute lymphoblastic leukemia a model for discrete stages of neoplastic and normal pre-B cell differentiation. J Clin Invest. 1984;74(2):332–340. doi: 10.1172/JCI111428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rosenthal P, Rimm IJ, Umiel T, Griffin JD, Osathanondh R, Schlossman SF, et al. Ontogeny of human hematopoietic cells: analysis utilizing monoclonal antibodies. J Immunol. 1983;131(1):232–237. [PubMed] [Google Scholar]
- 105.Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol. 1980;125(4):1678–1685. [PubMed] [Google Scholar]
- 106.McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 107.Vugmeyster Y, Howell K. Rituximab-mediated depletion of cynomolgus monkey B cells in vitro in different matrices: possible inhibitory effect of IgG. Int Immunopharmacol. 2004;4(8):1117–1124. doi: 10.1016/j.intimp.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 108.Vugmeyster Y, Howell K, Bakshl A, Flores C, Canova-Davis E. Effect of anti-CD20 monoclonal antibody, Rituxan, on cynomolgus monkey and human B cells in a whole blood matrix. Cytometry A. 2003;52(2):101–109. doi: 10.1002/cyto.a.10030. [DOI] [PubMed] [Google Scholar]
- 109.Vugmeyster Y, Howell K, McKeever K, Combs D, Canova-Davis E. Differential in vivo effects of rituximab on two B-cell subsets in cynomolgus monkeys. Int Immunopharmacol. 2003;3(10–11):1477–1481. doi: 10.1016/S1567-5769(03)00147-4. [DOI] [PubMed] [Google Scholar]
- 110.Bornstein GG, Queva C, Tabrizi M, van Abbema A, Chavez C, Wang P, et al. Development of a new fully human anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. Invest New Drugs. 2009. doi:10.1007/s10637-009-9291-z. [DOI] [PubMed]