

Abstract
Development of cancer is a long-term and multistep process which comprises initiation, progression, and promotion stages of carcinogenesis. Conceivably, it can be targeted and interrupted along these different stages. In this context, many naturally occurring dietary compounds from our daily consumption of fruits and vegetables have been shown to possess cancer preventive effects. Phenethyl isothiocyanate (PEITC) and sulforaphane (SFN) are two of the most widely investigated isothiocyanates from the crucifers. Both have been found to be very potent chemopreventive agents in numerous animal carcinogenesis models as well as cell culture models. They exert their chemopreventive effects through regulation of diverse molecular mechanisms. In this review, we will discuss the molecular targets of PEITC and SFN potentially involved in cancer chemoprevention. These include the regulation of drug-metabolizing enzymes phase I cytochrome P450s and phase II metabolizing enzymes. In addition, the signaling pathways including Nrf2–Keap 1, anti-inflammatory NFκB, apoptosis, and cell cycle arrest as well as some receptors will also be discussed. Furthermore, we will also discuss the similarities and their potential differences in the regulation of these molecular targets by PEITC and SFN.
Key words: dietary cancer chemoprevention, NF-kB, Nrf2, phenethyl isothiocyanate, sulforaphane
INTRODUCTION
Carcinogenesis is a long-term process which could take as long as 10–30 years to develop from initiated cells to advanced and metastatic cancer. It is a multistep event comprising three different stages, including initiation, promotion, and progression (1–4). Initiation occurs when cellular insults perturb the normal genetic and or epigenetic events. Promotion follows when cellular signals stimulate the expansion of the initiated cells. Finally, progression ensues when the clonal group of cells continues to expand into malignant tumors (1–4). Increasing evidence from epidemiological and pathological studies suggest that cancer could be prevented or their progression could be stopped (5). In this context, the prevention of cancer by the use of natural or synthetic compounds has been described as chemoprevention (6). Chemopreventive compounds could target the initiation stage of carcinogenesis or by stopping the promotion and progression of cancer or their combinations. Prevention of cancer initiation can be achieved by limiting the exposure of cells to carcinogenic substances via either by inhibiting their activation or by increasing their detoxification and subsequent removal (7). On the other hand, chemopreventive compounds can also suppress progression and promotion of carcinogenesis by interfering with various signaling pathways involving oxidative stress (8), inflammation (9), and cellular proliferation. Finally, these compounds can also induce cell cycle arrest and apoptosis; therefore, they could target all different stages of carcinogenesis, including initiation, promotion, and progression.
Numerous studies have shown that many natural dietary compounds can potently modulate various molecular targets, leading to prevention of cancer initiation, promotion, and progression. In particular, dietary fruits and vegetables have been regarded as rich sources of chemopreventive compounds and are widely investigated due to their low toxicity but significant chemopreventive efficacies (10). Two classes of the most abundant naturally occurring dietary chemopreventive compounds are polyphenolic and the isothiocyanate (ITC)-containing compounds with totally different chemical structures. Polyphenolic compounds are characterized by their phenolic functional groups while the ITC-containing compounds are characterized by sulfur containing N=C=S functional group (Fig. 1). Curcumin, dimethylbenzoylmethane, and green tea polyphenols are some of the commonly studied polyphenolic compounds with chemopreventive effect. Ally isothiocyanate (AITC) from cabbage, mustard, and horseradish; benzyl isothiocyanate (BITC); phenethyl isothiocyanate (PEITC) from watercress and garden cress; and sulforaphane (SFN) from broccoli, cauliflower, brassicas, and kale are commonly used ITC-containing compounds (11) (Table I).
Fig. 1.

Chemical structures of PEITC and SFN. Both PEITC and SFN are isothiocyanates, which are characterized by sulfur containing N=C=S functional group
Table I.
Sources of Isothiocyanates (ITCs) from Dietary Vegetables
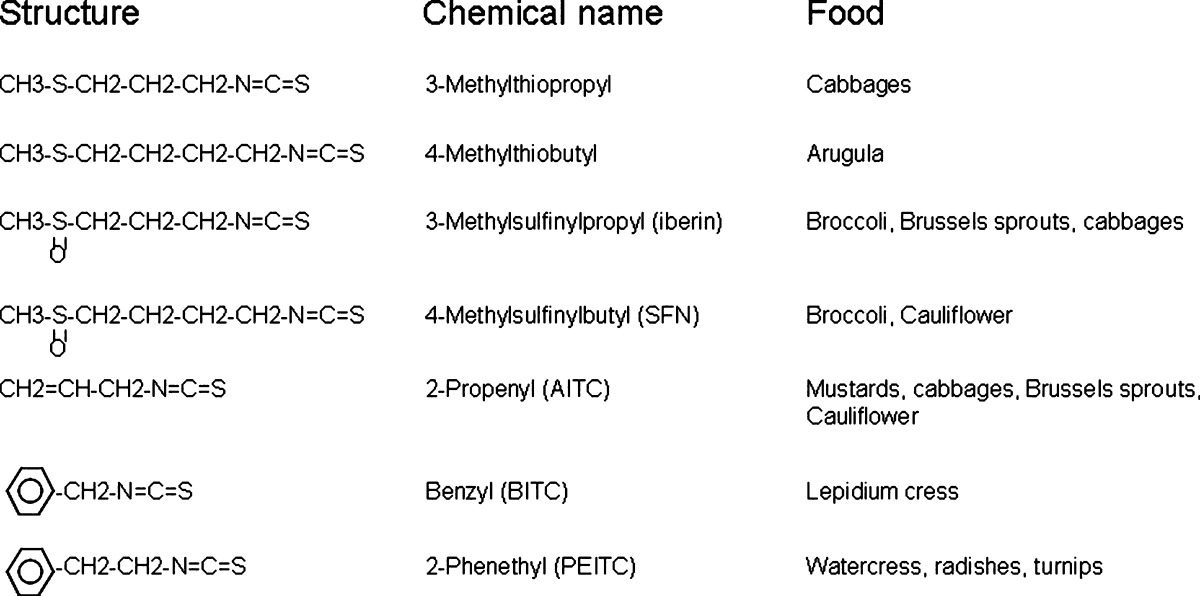
Many of the chemopreventive effects of cruciferous vegetables are attributed to the ITCs rather than their parent moiety, the glucosinolates. The general structures of glucosinolates consist of β-d-thioglucose group, a sulfonated oxime group, and a side chain derived from methionine, phenylalanie, tryptophane, or branch-chained amino acids (12). The glucosinolates are not bioactive and appear to have no chemopreventive effects unless they are converted to ITCs and indole-3 carbinols by hydrolysis catalyzed by myrosinase. Disruption of plant cells during harvesting, processing, or chewing releases myrosinase which comes into contact with glucosinolates and hydrolyzes them to different ITCs (13). It is estimated that in humans, intake of SFN or PEITC can range from 50 to 200 μmol after ingesting 100 g (wet weight) of broccoli and watercress (14,15). In rats, after an oral dose of 50 μmol of SFN, the plasma concentration of SFN can peak at 20 μM at 4 h and decline with a half-life of about 2.2 h. This increase in plasma concentration of SFN is accompanied by an induction of genes that are important in cellular defense mechanisms and cell cycle regulation (16). In mice fed with diets supplemented with 300 (∼5 μmol/day) and 600 ppm (∼10 μmol/day) of SFN for 3 weeks, the steady-state levels of SFN in plasma and intestine reached 124–254 nM and 3–13 nmol/g of tissue (roughly equivalent to about 3–10 μM of total SFN). These concentrations of SFN had been shown to be effective in prevention of adenoma formation in ApcMin± mice (17). On the other hand, plasma concentration of PEITC in rats could reach 9.2 to 42.1 μM after oral dose of 10 and 100 μmol/kg of PEITC (18). These in vivo pharmacokinetic data show that micromolar concentrations of PEITC and SFN are achievable in vivo. In animal and cell culture models, micromole doses or micromolar concentrations of PEITC and SFN have been shown to prevent cancer through different mechanisms both in vivo and in vitro (10, 13). In this review, we will discuss the diverse cellular and molecular targets regulated by PEITC and SFN (Fig. 2) and integrate these targets as potential examples as to how ITCs could prevent carcinogenesis.
Fig. 2.

Potential molecular pathways targeted by PEITC and SFN. PEITC and SFN affect various molecular targets and pathways, including phases I and II DME, NFκB, cell cycle arrest, apoptosis, and receptors
INHIBITION OF PHASE I CYP DMEs
Phase I (Cytochrome P450 (CYP)) drug-metabolizing enzymes (DMEs) are usually involved in oxidation, reduction, or hydrolysis of chemicals including carcinogens. These chemical reactions have been implicated in the bioactivation of carcinogens (conversion of pro-carcinogens to carcinogens) (19). Thus, compounds that could regulate either mRNA transcript levels or activity of CYPs are thought to be important in prevention of chemical-induced carcinogenesis.
The effects of PEITC on the expression and the enzyme activity of CYPs remain elusive and need to be studied further. It has been shown that PEITC induces the expression of several xenobiotic-metabolizing CYPs which could potentially activate carcinogens. In cultured human primary hepatocytes, it was also shown that PEITC dose-dependently upregulated the expression of carcinogen-activating enzymes CYP1A1 and CYP1A2 using quantitative polymerase chain reaction analyses (20). On the other hand, activities of CYPs have been shown to be inhibited by PEITC. Using microsomes from baculovirus-infected insect cells expressing specific human CYP isoforms, it was shown that PEITC competitively inhibited CYP1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4 (21). Activation of CYP has generally been considered as carcinogen activation. The above data indicate that although PEITC induces the CYP transcript levels, it inactivates CYP enzymes as well.
The effect of SFN on the expression and activity of CYPs appears to be more straightforward. SFN dose-dependently inhibited the activities of CYP1A1 and 2B1/2 in rat hepatocytes (22). It has also been shown to be a competitive inhibitor of CYP2E1 in acetone-induced rat liver microsomes (23). In human hepatocytes, SFN decreased CYP3A4 activity by reducing CYP3A4 transcript level (22). Overall, evidence presented so far suggest that SFN can inhibit the activities of several CYPs, thus potentially leading to reduced activation of pro-carcinogens.
INDUCTION OF PHASE II DMEs
The formation of electrophilic reactive metabolites or pro-carcinogens is often catalyzed by phase I DMEs through two-electron oxidation to hydroxylated or epoxidated intermediates (24). These electrophilic metabolites are highly reactive, but cells have protective mechanisms against damages caused by them. One major mechanism is through induction of phase II DMEs. Phase II enzymes are well-known detoxifying enzymes which conjugate endogenous substrates such as glutathione (GSH), glucuronide, and sulfate to the phase I metabolites (25). The transfer of these large polar molecules to phase I metabolites through sulfation, glucuronidation, and conjugation with GSH are known as phase II metabolisms, which would limit further biotransformation of phase I metabolites and result in enhanced elimination and excretion (3). Compounds such as PEITC and SFN which could induce the expression of phase II enzymes are considered to be chemopreventive. Commonly induced phase II enzymes include glutathione S-transferase (GST), NAD(P)H:quinone oxidoreductase (NQO-1), and UDP-glucuronosyltransferase (UGT). GSTs generally catalyze the nucleophilic addition of GSH to electrophilic groups of a broad spectrum of xenobiotics (26) while UGTs catalyze the transfer of glucuronic acid to hydrophobic chemicals, facilitating their detoxification and excretion (27). NQO-1 catalyzes the beneficial two-electron reduction of quinones to hydroquinones, preventing the one-electron reduction of quinones by other quinone reductases that would result in the production of radical species (28).
Numerous studies have shown that PEITC induces phase II enzymes in vitro and in vivo. In an experiment to determine the chemopreventive effect of PEITC against 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP, a heterocyclic amine that is found in cooked meat and may be risk factors for cancer)-induced genotoxicity, it was found that PEITC induced one or more hepatic phase II enzymes, leading to significantly decreased PhIP-DNA adduct levels in all tissues examined (29). Using a microarray approach, several isozymes of GST were shown to be induced by PEITC 12 h after treatment in the mouse liver (30). In agreement with this, a recent study showed that PEITC treatment stimulated hepatic GST activity in rat but not in the lung or kidney (31). Aside from GST, expression of the antioxidant enzyme heme-oxygenase-1 (HO-1) has also been shown to be strongly increased by PEITC treatment in PC-3 cells (32). Importantly, the induction of these phase II/antioxidant enzymes was shown to be NF-E2-related factor-2 (Nrf2)-dependent in these studies. Furthermore, HO-1 and NQO-1 mRNA expression levels in RAW264.7 cells have been found to be induced by PEITC and appear to be associated with anti-inflammatory effects of PEITC (33). Taken these data together, PEITC appears to induce several phase II/antioxidant enzymes that have been shown to have anti-inflammatory and anticancer effects, but the induction of each specific phase II/antioxidant enzyme may be tissue- and cell type-specific.
The same effect of induction of phase II/antioxidant enzymes appears to be commonly observed in various cells and tissues after SFN treatment (34). SFN significantly reduced the level of PhIP-DNA adducts in a dose-dependent manner in hepatocytes possibly due to induction of UGT1A1 and GSTA1 mRNA expression (35). NQO1 and GST activities were increased in forestomach, duodenum, and bladder in rats treated with 40 μmol SFN/Kg/day (36) while a higher dose of 200–1,000 μmol SFN/Kg/day increased NQO1 and GST activities in liver, colon, and pancreas (37). NQO1, total GST, and GST-mu activities in prostate, liver, kidney, and bladder tissues were also increased by SFN (38). The increase in expressions and activities of these phase II enzymes was also observed in vitro. SFN induced GSTA1/2 and GSTP1 mRNA in primary rat hepatocytes, induced GSTA1/2 and GSTM1 mRNA in primary human hepatocytes (22), and increased GST and NQO1 activities in murine HepG1c1c7 cells (39,40). SFN also induced phase II genes in other cell lines (colon and prostate). GSTA1 and UGT1A1 mRNA were shown to be induced in HT-29 and Caco2 cells (41,42) while GSTA1 and NQO-1 mRNA were induced in human prostate cells (43). Interestingly, GSTA1 mRNA has consistently been induced by PEITC in different cell types. The induction of HO-1 by SFN has been elucidated and shown to be Nrf2-dependent in mouse peritoneal macrophages (44). Taken together, it appears that SFN is a very potent inducer of various phase II/antioxidant enzymes, and its induction of GST and NQO1 was consistently found in numerous cell types and tissues.
The induction of phase II enzymes by PEITC and SFN was associated with disruption of Nrf2–Keap1 interactions and increased nuclear translocation of Nrf2. In unstimulated cells, Keap 1 sequesters Nrf2 and bridges it to Cul-3-dependent ubiquitinase for ubiquitination. The ubiquitinated Nrf2 is then targeted for proteosomal degradation. When the cells are stimulated by external insults, the Nrf2–Keap1 complex is disrupted and Nrf2 translocation occurs (45,46). Both PEITC and SFN were shown to disrupt the Nrf2–Keap1 interaction, and the molecular targets modulated will be discussed below (Fig. 3).
Fig. 3.

Molecular targets in Nrf2/phase II enzymes pathway modulated by PEITC and SFN. In unstimulated cells, Keap 1 sequesters Nrf2 and bridges it to Cul-3-dependent ubiquitinase for ubiquitination. The ubiquitinated Nrf2 is then targeted for proteosomal degradation. Both PEITC and SFN can activate Nrf2, although through different mechanisms. PEITC activates Nrf2 through activation of MAPK pathway while SFN modifies thiol group of Keap1 and releases Nrf2 from Keap1 binding
PEITC ACTIVATES NRF2 COUPLED WITH ACTIVATION OF MAPK PATHWAY
Mitogen-activated protein kinase (MAPKs) are characterized as proline-directed serine/threonine kinases which convert extracellular signals into cellular responses through cascades of proteins phosphorylation events. It appears that PEITC activates the MAPK pathway possibly via electrophilic-mediated stress response (47) such as inhibition of phosphatases (48). The activation of MAPK pathway is coupled with the activation of Nrf2 and its dimerization with Mafs. The Nrf2–Mafs complex subsequently binds to the antioxidant/electrophile response element (ARE/EpRE) enhancers that are found in the promoters of many phase II/antioxidant genes. MAPKs, therefore, could serve as cellular stress sensors, bringing homeostatic response and protects against environmental challenges after exposure to xenobioitcs such as PEITC (49).
PEITC induces the expression of ARE-mediated phase II enzymes via the c-Jun N-terminal kinase-1 (JNK1)- and Nrf2-dependent pathways (50). Using HeLa cells that were transiently transfected with different cDNA plasmids, PEITC transcriptionally activated ARE in a dose-dependent manner, and that transient expression of either dominant-negative Nrf2 or dominant-negative JNK1 attenuated PEITC-induced ARE expression. These data indicated that PEITC, at least in HeLa cells, induced ARE activation in part through JNK1 and Nrf2 (50). In addition, when the role of extracellular signal-regulated kinase (ERK) and JNK in the regulation of PEITC-induced and Nrf2-dependent ARE activity in PC-3 cells was studied, PEITC was found to induce ARE activity but the induction was attenuated by inhibition of JNK and ERK signaling, which in part confirmed the importance of JNK signaling in PEITC-induced ARE activity (32). In the same experiment, PEITC was shown to increase the phosphorylation of JNK1/2 and ERK1/2. Using in vitro kinase assays, JNK1 and ERK2 were shown to directly phosphorylate GST-Nrf2 protein (32). The phosphorylated Nrf2 appeared to be released from Keap1 binding and subsequently translocated into the nucleus. These data suggested that the PEITC-induced phosphorylation of JNK and ERK could be important in regulating phosphorylation of Nrf2 and inducing ARE transcriptional activity (32). Interestingly, a recent report indicated a different interpretation of the roles of MAPK on Nrf2. Using mass spectrometry; five serine/threonine residues (S215, S408, S558, T559, and S577) were shown to be major targets of MAPK-mediated phosphorylation. Subsequent abolishment of Nrf2 phosphorylation by mutation of the five identified residues to alanine (A) was shown to have limited effect on the transcriptional activity of Nrf2 (51). However, several potential caveats remain unclear. The five phosphorylation sites were identified under basal conditions when the MAPKs would appear to be mostly inactive. These five sites could be possibly phosphorylated by other nonspecific kinases which would be random and may have no specific significance. It would be interesting to find out whether the phosphorylation of each site by each specific MAPK (JNK, ERK, and p38) would be the same, since each MAPK could phosphorylate Nrf2 at specific sites and may have different functional effects on the activation of Nrf2. Certainly, more studies would be needed to dissect the functional significance of phosphorylation on Nrf2’s biological functions.
SFN MODIFIES KEAP1 AND RELEASES NRF2 FROM KEAP1 BINDING
The mechanism of SFN-induced nuclear translocation of Nrf2 appears to be different from PEITC. It was shown that murine Keap1 formed complexes with Neh2 (Nrf2-ECH homology 2) domain of Nrf2, and they were disrupted dose-dependently by SFN (52). Two groups provided evidence that SFN disrupts the Nrf2–Keap1 complex through Keap1 modifications. One provided spectroscopic evidence that SFN reacted with the thiol groups of Keap1. It was suggested that the most reactive residues of Keap1 (Cys257, Cys273, Cys288, and Cys297) are located in the intervening region between BTB and Kelch repeat domains of Keap1 and are probably the direct sensors of SFN (52). Another group developed a liquid-tandem mass spectrometry method to map the labile SFN adduct sites formed on Keap1 in vitro. They provided direct evidence suggesting that SFN could react with thiol groups of Keap1 to form thionoacyl adducts (53). This specific modification of Keap1 released Nrf2 from its sequestration and activated Nrf2. In addition to phase II enzymes induction, it was shown that selenoprotein gastrointestinal glutathione peroxidase (GI-GPx) induction by SFN was Nrf2-dependent and probably through electrophilic thiol modification of Keap1 (54).
PEITC AND SFN INACTIVATES NFκB PATHWAY
Nuclear factor kappa B (NFκB) could be an important molecular target of cancer chemopreventive compounds PEITC and SFN. The activation of NFκB has been linked to inflammation, cancer cell survival, and progression (9). Thus, inactivation of NFκB by PEITC and SFN could be considered as an important chemopreventive mechanism. NFκB is normally sequestrated by IκB in the cytosol which is subjected to phosphorylation by IκK. Phosphorylation of IκB generally leads to the degradation of IκB, with the concomitant release of NFκB and nuclear translocation of NFκB (Fig. 4). NFκB binds to the promoter of many pro-inflammatory genes, most notably inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and tumor necrosis factor (TNF) among others. These molecules have been shown to be involved in inflammation and cancer development following chronic inflammation (9).
Fig. 4.

Molecular targets in NFκB pathway modulated by PEITC and SFN. Inactivation of NFκB by PEITC and SFN is considered as one important chemopreventive mechanism. NFκB is normally sequestrated by IκB which is subjected to phosphorylation by IκK. Phosphorylation of IκB by IκK increases degradation of IκB and, therefore, increases nuclear translocation of NFκB. Both PEITC and SFN have been shown to inhibit activation of NFκB through inhibiting IκK. In addition, SFN have been shown to decrease DNA binding ability of NFκB
PEITC decreased iNOS and COX-2 protein expression levels leading to reduced secretion of both pro-inflammatory mediators (55). Interestingly, the reduction in both iNOS and COX-2 expression was associated with the inactivation of NFκB resulting from stabilization of IκBα by PEITC. IKK and IκB have been shown to be the molecular targets of PEITC. Several laboratories have shown that SFN share similar mechanisms with PEITC targeting the NFκB pathway. Both PEITC and SFN strongly inhibited LPS-induced NFκB luciferase activity and IκBα phosphorylation in HT-29 cells (56). SFN (20 and 30 μM) and PEITC (5 and 7.5 μM) significantly inhibited NFκB transcriptional activity, nuclear translocation of p65 (a subunit of NFκB), and NFκB-regulated vascular endothelial growth factor, cylcin D1, and Bcl-XL expression in PC-3 C4 cells, which was mediated through potent inhibition of IκKα and IκKβ phosphorylation (57). Again, it appears that SFN and PEITC converge in the inhibition of IκBα phosphorylation and degradation, therefore, leading to a decrease of nuclear translocation of p65 and activation of NFκB (57).
On the other hand, there are reports that SFN did not interfere with LPS-induced degradation of IκB nor NFκB nuclear translocation in RAW 264.7 cells, but it reduced DNA binding ability of NFκB directly. Two mechanisms have been proposed. One mechanism suggests that SFN interacts with the thiol groups by dithiocarbamate formation and directly binds to essential Cys residues of NFκB subunits, thereby decreasing their DNA binding abilities (58). Another mechanism suggests SFN interacts with glutathione and other redox regulators like thioredoxin or Ref-1 which in turn indirectly interferes with NFκB-DNA binding ability. Thioredoxin/thioredoxin reductase (TrxR) system was a key redox mechanism regulating NFκB DNA binding and that SFN was identified as a novel inhibitor of TrxR enzymatic activity in vitro (59). These findings further emphasize the importance of redox modulation and thiol reactivity in the regulation of NFκB-dependent transcription by SFN. The importance of redox modulation and thiol reactivity was further supported by another model: RANKL-induced osteoclastogenesis which is mediated through NFκB. It was shown that SFN selectively inhibited NFκB activation induced by RANKL through interaction with thiol groups of NFκB. However, the NFκB inhibition was blocked by the use of reducing agents (60), suggesting that the redox modulation could be important in regulating thiol reactivity of NFκB to SFN and, thus, its inactivation. Apart from NFκB, other targets (coactivators of NFκB) could also be potentially affected by SFN. These include CCAAT-enhancer-binding proteins, cAMP response element binding protein, and activator protein-1 (AP-1) which were identified to be inhibited and responsible for SFN-mediated COX-2 downregulation (61).
PEITC AND SFN INDUCE APOPTOSIS
Apoptosis or programmed cell death is important in controlling unlimited cell proliferation (62). There are compelling evidence to support PEITC and SFN as potent inducers of apoptosis both in vitro and in vivo. The different molecular targets or mechanisms in the apoptosis pathway affected by PEITC and SFN, therefore, deserve some discussions (Fig. 5).
Fig. 5.

Molecular targets in apoptosis and cell cycle arrest pathways modulated by PEITC and SFN. Although both PEITC and SFN are potent inducers of apoptosis, the initial signal that triggers apoptosis has been suggested to be different. SFN increases ROS generation and disrupts mitochondrial membrane potential, which leads to cytosolic release of cytochrome c and apoptosis. On the other hand, PEITC binds to tubulin, increases the cleavage of caspases 8 and 9, and triggers apoptosis. Both PEITC and SFN activate Chk2 and inactivate Cdk1, leading to cell cycle arrest. Cyclins A, D, and E are also downregulated by PEITC
SFN induces apoptosis in both HeLa and HepG2 cells as evidenced by the formation of apoptotic bodies and the accumulation of the cells in sub-G1 phase (63). SFN affects classical molecular targets involved in the apoptosis pathways including downregulation of anti-apoptotic Bcl-2 and Bcl-XL, upregulation of pro-apoptotic Bax expression, proteolytic activation of caspase-3, and the degradation/cleavage of poly (ADP-ribose) polymerase (63). In addition, it was shown that Bax activation, downregulation of IAP family proteins (cIAP1, cIAP2, and XIAP), and apoptotic protease activating factor-1 induction are also involved in the regulation of SFN-induced cell death (64). One interesting study using Jurkat T-leukemia cells suggested that cell vulnerability to SFN-mediated apoptosis was dependent on cell-cycle mechanisms. It was shown that these cells were most sensitive to SFN-induced apoptosis in the G1 phase, less sensitive in the G2/M phase, and least sensitive during the S phase (65).
The upstream activities or initial signals activated by SFN are more complicated. Using PC-3 and DU145 human prostate cancer cells as model systems, it was demonstrated that the initial signal for SFN-induced apoptosis is possibly from reactive oxygen species (ROS) (66). SFN resulted in ROS generation and disruption of mitochondrial membrane potential, which leads to cytosolic release of cytochrome c and apoptosis in PC3 cells. Pretreatment with N-acetylcysteine or overexpression of catalase block cytochrome c release and apoptosis, indicating the importance of ROS-mediated cytochrome c release in SFN-induced apoptosis pathway (66). Apart from ROS, MAPK pathway has also been reported to be activated by SFN in PC-3 cells (67). Results indicated that SFN activates AP-1 which requires the activation of the ERK and JNK signaling pathways and is involved in the regulation of cell death elicited by SFN in PC-3 cells (67).
PEITC has been shown to be a potent inducer of apoptosis (68). A 24-h exposure of PC-3 cells to 10 μM PEITC resulted in about 56% and 44% decrease in the levels of antiapoptotic proteins Bcl-2 and Bcl-XL, respectively (69). Treatment of cells with PEITC also resulted in cleavage of procaspase-3, procaspase-9, and procaspase-8. Moreover, the PEITC-induced apoptosis was significantly attenuated in the presence of general caspase inhibitor and specific inhibitors of caspase-8 and caspase-9, suggesting the involvement of both caspase-8- and caspase-9-mediated pathways in apoptosis induction by PEITC (69). The initial signals activated by PEITC were investigated. Recent evidence suggests that apart from ROS, there may be other signals also involved in PEITC’s induction of apoptosis. It was suggested that PEITC is a more potent inducer of apoptosis, but SFN is a more potent activator of ROS and oxidative damages in A549 cells, implicating that ROS and oxidative damages may probably not trigger PEITC-induced apoptosis in these cells (70,71). Instead, binding of PEITC to tubulin could be the initial trigger for PEITC-induced apoptosis. Relative tubulin-binding affinities of ITCs were measured, and PEITC was shown to bind more avidly to tubulin than SFN. This higher tubulin affinity of PEITC appears to correlate with its higher potency of induction of apoptosis (70,71).
On the other hand, translation inhibition has been reported to be another possible mechanism of PEITC-induced apoptosis (72). Using pharmacologically achievable concentrations, PEITC treatment of human colorectal cancer HCT-116 cells and human prostate cancer PC-3 cells caused an increase in eukaryotic translation initiation factor 4E (eIF4E)-binding protein (4E-BP1) expression and inhibition of 4E-BP1 phosphorylation (72). The net effect was a reduction in the availability of eIF4E for translation initiation. Ectopic expression of eIF4E prevented PEITC-induced translation inhibition and apoptosis. This was the first report to show PEITC modulates availability of eIF4E for translation initiation through modification of its binding protein (4E-BP1), thus leading to inhibition of cap-dependent protein translation (72).
PEITC AND SFN INDUCE CELL CYCLE ARREST
The cell cycle arrest effect of PEITC was first investigated in 1993 (73); 10 μM AITC, 2.5 μM BITC, or PEITC caused the accumulation of cells at G2/M phase in HeLa cells with inhibition of cell growth to 41–79% of the control (73). In 2003, it was reported that exposure of cancer cells to either AITC, BITC, or PEITC for only 3 h was long enough for the inhibition of cell growth regardless of the origin of cancer cells and even in drug-resistant cells that overexpressed multidrug resistance-associated protein-1 or P-glycoprotein-1 (74). In contrast, the growth inhibitory effect of SFN on these cells was highly time-dependent (74). Inhibition of cell growth by SFN follows a biphasic pattern in human colon cancer cell lines: transient exposure of human HT-29 colon carcinoma cells to SFN for up to 6 h resulted in reversible G2/M cell cycle arrest and cytostatic growth inhibition, whereas a minimum continuous exposure time of 12 h irreversibly arrested cells in G2/M phase and subsequently induced apoptosis (75). These findings suggest the possibility of more complex regulations of cell cycle arrest induced by SFN than PEITC.
In HT-29 cells, G1 cell cycle arrest was found to be induced by PEITC through the downregulation of cyclin A, D, and E (76). On the other hand, the PEITC-induced G2/M phase cell cycle arrest in PC-3 cells was associated with a >80% reduction in the protein levels of cyclin-dependent kinase 1 (Cdk1), cell division cycle 25C (Cdc25C), and accumulation of Tyr(15) phosphorylated (inactive) Cdk1 (69). The PEITC-induced decrease of Cdk1 and Cdc25C protein levels was probably through increased proteosome-mediated degradation since cell cycle arrest was significantly blocked with pretreatment of PC-3 cells with proteasome inhibitor lactacystin (69). Apart from Cdk and Cdc25C, G2/M DNA damage checkpoint Chk2 could be another molecular target involved in PEITC-induced cell cycle arrest (Fig. 5). In Caco2 cells, BITC and PEITC inhibited DNA synthesis with IC50 of 5.1 and 2.4 μmol/L, respectively (77). Both of these compounds caused an accumulation of cells in the G2/M phase of the cell cycle which was due in part to the phosphorylation and activation of Chk2 and sustained G2/M phase cell cycle arrest that was maintained through upregulation of p21 (77).
Like PEITC, SFN caused an irreversible arrest of G2/M phase of the cell cycle with possibly similar mechanisms (Fig. 5). The cell cycle arrest was associated with a significant decrease in protein levels of cyclin B1, cell division cycle Cdc25B, Cdc25C, and an accumulation of Tyr-15-phosphorylated (inactive) Cdk1 (78). In addition, SFN treatment also resulted in a rapid and sustained phosphorylation of Cdc25C at Ser-216 which was a result of Chk2 activation, leading to translocation of Cdc25C from the nucleus to the cytoplasm and increased binding to 14-3-3beta. Transient knockout of Chk2 in PC-3 cells with Chk2-specific small interfering RNA duplexes significantly attenuated SFN-induced G2/M arrest. Furthermore, Chk2(−/−) HCT116 human colon cancer cells were significantly more resistant to G2/M arrest induced by SFN as compared with the wild type HCT116 cells, indicating that Chk2-mediated phosphorylation of Cdc25C could be the major regulator in irreversible G2/M arrest by SFN (78). Thus, studies have consistently showed the link of Chk2 activation to cell cycle arrest induced by PEITC and SFN.
Epigenetic regulation of cell cycle arrest genes by PEITC and SFN has also been described recently (79). Histone deacetylases (HDACs) inhibition has been shown as one possible mechanism of PEITC-mediated growth attenuation in human prostate cancer cells (79). PEITC was found to be an inhibitor of HDACs which was associated with an increase in p21 expression in prostate cancer cells (79). PEITC treatment resulted in cell cycle arrest and a p53-independent upregulation of the inhibitors of Cdks, including p21 and p27 in LNCap cells. Chromatin immunoprecipitation revealed that the induction of p21 gene was associated with PEITC-induced hyperacetylated histones which allowed modification of histone methylation for chromatin unfolding, consequently permitting the transcription activation of the p21 gene (79). Similarly, SFN and its metabolites appear to inhibit HDAC activity and increase acetylated histones bound to the p21 promoter (80). Collectively, these findings suggest that inhibition of HDAC could be a common mechanism of PEITC and SFN.
RECEPTORS AS MOLECULAR TARGETS FOR PEITC AND SFN
PEITC and SFN have been shown to regulate endogenous receptors in some cells, leading to their potential chemopreventive effect. In the model of testosterone (hormone)-induced prostatic cell growth, PEITC negated the growth via downregulation of androgen receptor (AR) (81). This repression of AR in prostate cancer cells by PEITC is significant in cancer chemoprevention since abnormalities of the AR, such as overexpression, have been postulated to be related to the hormone-independent growth of prostate cancer (81). The question as to how PEITC regulates AR was subsequently investigated. It was found that PEITC mediates dual effects on AR, both at the transcriptional and the post-translational level (81). At the transcriptional level, the AR transcript level was reduced via inhibition of the transcription factor Sp1 (which transactivates AR promoter), and at the post-translational level, AR protein degradation was accelerated (81). Correspondingly, there is a report suggesting that SFN regulates the expression of estrogen receptor alpha (ERα) in human breast cancer cells (82). The inhibition of ERα expression in MCF-7 was due in part to inhibition of ERα mRNA transcription as well as increased-proteosome-mediated degradation (82). These data suggest that SFN could potentially inhibit cancer cell proliferation caused by aberrant hormone ER receptor expression in MCF-7 cells.
SFN could sensitize cells to apoptosis through upregulation of death receptors (83, 84). It was found that SFN enhanced TRAIL-induced apoptosis in human osteosarcoma cells (Saos2 and MG63) and hepatoma cells. The sensitization of SFN was shown to be mediated through upregulation of death receptor 5 (DR5) mRNA and protein expressions. Thus, SFN could sensitize cancer cells to apoptosis through DR5 upregulation which presents another potential mechanism of chemoprevention (83,84). Using proteomics approach, recently some novel targets of SFN were identified and characterized (85). Human colon adenocarcinoma cell (Caco-2) was treated with 5 μM SFN for 48 h and serotonin receptor 5-HT was downregulated to undetectable levels as compared to the controls (85). It was suggested that the activation of neurotransmitter receptors followed by initiation of cyclic AMP signaling might be crucial events in colon carcinoma progression. Thus, the effect of SFN on serotonin receptor 5-HT could be yet another potential chemopreventive target that could be utilized.
CONCLUSION
PEITC and SFN are multitargeted chemopreventive agents which exert their effects through activation or inhibition of various cellular signaling pathways. In this review, we have discussed the mechanisms of action of PEITC and SFN on some well-established molecular targets such as phase I/II DMEs, Nrf2/Keap1, IκB/NFκB, Chk2, caspases, and receptors. We also discuss the similarities and differences in some of the mechanism of actions between PEITC and SFN. Some of the recent findings regarding the triggering of cell cycle arrest and apoptosis by PEITC and SFN are also discussed. The increased understanding of the in vitro and in vivo cancer chemopreventive activities of these dietary cancer chemopreventive compounds and their molecular targets would allow us to better predict their efficacies in different types of human cancers by single and/or combination strategies.
Acknowledgement
We thank all the members in Dr. Tony Kong’s lab for their help in the discussion and preparation of this manuscript. We also thank Drs. Siwang Yu (Beijing University, People Republic of China) and Young-Sam Keum (Hormel Institute, Austin, MN) for their helpful discussions. This study was supported in part by Institutional Funds and by R01-CA073674, R01-CA094828 and R01-CA118947 to Dr Ah-Ng Tony Kong from the National Institutes of Health (NIH).
Abbreviations/acronyms
- ARE/EpRE
Antioxidant/electrophile response element
- COX-2
Cyclooxygenase-2
- CYP
Cytochrome P450
- DME
Drug-metabolizing enzyme
- ERK
Extracellular signal-regulated kinase
- GST
Glutathione-S-transferase
- HO-1
Heme-oxygenase 1
- IAP
Inhibitor of apoptosis
- iNOS
Inducible nitric oxide synthase
- ITCs
Isothiocyanates
- IκB
Inhibitor of kappa B
- IκK
IκB kinase
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen-activated protein kinase
- NFκB
Nuclear factor kappa B
- NQO
NAD(P)H:quinone oxidoreductase
- Nrf2
NF-E2-related factor-2
- PEITC
Penethyl isothiocyanate
- RANKL
Receptor activator of NFκB ligand
- ROS
Reactive oxygen species
- RT-PCR
Reverse-transcription polymerase chain reaction
- SFN
Sulforaphane
- TNF
Tumor necrosis factor
- UGT
UDP-glucuronosyltransferase
References
- 1.Wattenberg LW. Inhibition of chemical carcinogen-induced pulmonary neoplasia by butylated hydroxyanisole. J Natl Cancer Inst. 1973;50:1541–1544. doi: 10.1093/jnci/50.6.1541. [DOI] [PubMed] [Google Scholar]
- 2.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. doi: 10.1016/S0065-230X(08)60265-1. [DOI] [PubMed] [Google Scholar]
- 3.Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol Sci. 2005;26(6):318–326. doi: 10.1016/j.tips.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Smart RC. Chemical carcinogenesis. In: Hodgson E, editor. A textbook of modern toxicology. 3. Hoboken: Wiley; 2004. pp. 240–242. [Google Scholar]
- 5.Weinstein IB. Cancer prevention: recent progress and future opportunities. Cancer Res. 1991;51:5080–5085. [PubMed] [Google Scholar]
- 6.Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids) Fed Proc. 1976;35:1332–1338. [PubMed] [Google Scholar]
- 7.Hanausek M, Walaszek Z, Slaga TJ. Detoxifying cancer causing agents to prevent cancer. Integr Cancer Ther. 2003;2:139. doi: 10.1177/1534735403002002005. [DOI] [PubMed] [Google Scholar]
- 8.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 9.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 10.Jeong WS, Kong AN. Chemopreventive functions of isothiocyanates. Drug News Perspect. 2005;18:445–451. doi: 10.1358/dnp.2005.18.7.939350. [DOI] [PubMed] [Google Scholar]
- 11.IARC . Cruciferous vegetables, isothiocyanates and indoles. Lyon: IARC; 2004. [Google Scholar]
- 12.Kliebenstein DJ, Kroymann J, Mitchell-Olds T. The glucosinolate–myrosinase system in an ecological and evolutionary context. Curr Opin Plant Biol. 2005;8:264–271. doi: 10.1016/j.pbi.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Hayes JD, Kelleher MO, Eggleston IM. The cancer chemopreventive actions of phytochemicals derived from glucosinolates. Eur J Nutr. 2008;47(Suppl 2):73–88. doi: 10.1007/s00394-008-2009-8. [DOI] [PubMed] [Google Scholar]
- 14.Chung FL, Morse MA, Eklind KI, Lewis J. Quantitation of human uptake of the anticarcinogen phenethyl isothiocyanate after a watercress meal. Cancer Epidemiol Biomarkers Prev. 1992;1:383–388. [PubMed] [Google Scholar]
- 15.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer Epidemiol Biomarkers Prev. 2001;10:501–508. [PubMed] [Google Scholar]
- 16.Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. 2004;310:263–271. doi: 10.1124/jpet.103.064261. [DOI] [PubMed] [Google Scholar]
- 17.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27:2038–2046. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- 18.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res. 2005;22:1658–1666. doi: 10.1007/s11095-005-7097-z. [DOI] [PubMed] [Google Scholar]
- 19.Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14:473–486. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Gross-Steinmeyer K, Stapleton PL, Liu F, Tracy JH, Bammler TK, Quigley SD, et al. Phytochemical-induced changes in gene expression of carcinogen-metabolizing enzymes in cultured human primary hepatocytes. Xenobiotica. 2004;34:619–632. doi: 10.1080/00498250412331285481. [DOI] [PubMed] [Google Scholar]
- 21.Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos. 2001;29:1110–1113. [PubMed] [Google Scholar]
- 22.Mahéo K, Morel F, Langouët S, Kramer H, Le Ferrec E, Ketterer B, et al. Inhibition of cytochromes P-450 and induction of glutathione S-transferases by sulforaphane in primary human and rat hepatocytes. Cancer Res. 1997;57:3649–3652. [PubMed] [Google Scholar]
- 23.Barcelo S, Gardiner JM, Gescher A, Chipman JK. CYP2E1-mediated mechanism of anti-genotoxicity of the broccoli constituent sulforaphane. Carcinogenesis. 1996;17:277–282. doi: 10.1093/carcin/17.2.277. [DOI] [PubMed] [Google Scholar]
- 24.Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 25.Kensler TW. Chemoprevention by inducers of carcinogen detoxication enzymes. Environ Health Perspect. 1997;105(Suppl 4):965–970. doi: 10.2307/3433311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pool-Zobel B, Veeriah S, Böhmer FD. Modulation of xenobiotic metabolising enzymes by anticarcinogens – focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesis. Mutat Res. 2005;591:74–92. doi: 10.1016/j.mrfmmm.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 27.Saracino MR, Lampe JW. Phytochemical regulation of UDP-glucuronosyltransferases: implications for cancer prevention. Nutr Cancer. 2007;59:121–141. doi: 10.1080/01635580701458178. [DOI] [PubMed] [Google Scholar]
- 28.Vasiliou V, Ross D, Nebert DW. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum Genomics. 2006;2(5):329–335. doi: 10.1186/1479-7364-2-5-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dingley KH, Ubick EA, Chiarappa-Zucca ML, Nowell S, Abel S, Ebeler SE, et al. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutr Cancer. 2003;46:212–221. doi: 10.1207/S15327914NC4602_15. [DOI] [PubMed] [Google Scholar]
- 30.Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci. 2006;79:1944–1955. doi: 10.1016/j.lfs.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 31.Konsue N, Ioannides C. Tissue differences in the modulation of rat cytochromes P450 and phase II conjugation systems by dietary doses of phenethyl isothiocyanate. Food Chem Toxicol. 2008;46:3677–3683. doi: 10.1016/j.fct.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 32.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, et al. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther. 2006;5:1918–1926. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- 33.Cheung KL, Khor TO, Kong AN. Synergistic effect of combination of phenethyl isothiocyanate and sulforaphane or curcumin and sulforaphane in the inhibition of inflammation. Pharm Res. 2009;26:224–231. doi: 10.1007/s11095-008-9734-9. [DOI] [PubMed] [Google Scholar]
- 34.Dinkova-Kostova AT, Fahey JW, Wade KL, Jenkins SN, Shapiro TA, Fuchs EJ, et al. Induction of the phase 2 response in mouse and human skin by sulforaphane-containing broccoli sprout extracts. Cancer Epidemiol Biomarkers Prev. 2007;16:847–851. doi: 10.1158/1055-9965.EPI-06-0934. [DOI] [PubMed] [Google Scholar]
- 35.Bacon JR, Williamson G, Garner RC, Lappin G, Langouët S, Bao Y. Sulforaphane and quercetin modulate PhIP-DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis. 2003;24:1903–1911. doi: 10.1093/carcin/bgg157. [DOI] [PubMed] [Google Scholar]
- 36.Munday R, Munday CM. Induction of phase II detoxification enzymes in rats by plant-derived isothiocyanates: comparison of allyl isothiocyanate with sulforaphane and related compounds. J Agric Food Chem. 2004;52:1867–1871. doi: 10.1021/jf030549s. [DOI] [PubMed] [Google Scholar]
- 37.Matusheski NV, Jeffery EH. Comparison of the bioactivity of two glucoraphanin hydrolysis products found in broccoli, sulforaphane and sulforaphane nitrile. J Agric Food Chem. 2001;49:5743–5749. doi: 10.1021/jf010809a. [DOI] [PubMed] [Google Scholar]
- 38.Jones SB, Brooks JD. Modest induction of phase 2 enzyme activity in the F-344 rat prostate. BMC Cancer. 2006;6:62. doi: 10.1186/1471-2407-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prestera T, Talalay P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1995;92:8965–8969. doi: 10.1073/pnas.92.19.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang ZQ, Chen C, Yang B, Hebbar V, Kong AN. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003;72:2243–2253. doi: 10.1016/S0024-3205(03)00101-2. [DOI] [PubMed] [Google Scholar]
- 41.Basten GP, Bao Y, Williamson G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis. 2002;23:1399–1404. doi: 10.1093/carcin/23.8.1399. [DOI] [PubMed] [Google Scholar]
- 42.Svehlíková V, Wang S, Jakubíková J, Williamson G, Mithen R, Bao Y. Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells. Carcinogenesis. 2004;25:1629–1637. doi: 10.1093/carcin/bgh169. [DOI] [PubMed] [Google Scholar]
- 43.Brooks JD, Paton VG, Vidanes G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol Biomarkers Prev. 2001;10:949–954. [PubMed] [Google Scholar]
- 44.Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem Pharmacol. 2008;76:967–973. doi: 10.1016/j.bcp.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 47.Kong AN, Owuor E, Yu R, Hebbar V, Chen C, Hu R, et al. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metab Rev. 2001;33:255–271. doi: 10.1081/DMR-120000652. [DOI] [PubMed] [Google Scholar]
- 48.Chen YR, Han J, Kori R, Kong AN, Tan TH. Phenylethyl isothiocyanate induces apoptotic signaling via suppressing phosphatase activity against c-Jun N-terminal kinase. J Biol Chem. 2002;277:39334–39342. doi: 10.1074/jbc.M202070200. [DOI] [PubMed] [Google Scholar]
- 49.Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- 50.Keum YS, Owuor ED, Kim BR, Hu R, Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC) Pharm Res. 2003;20:1351–1356. doi: 10.1023/A:1025737622815. [DOI] [PubMed] [Google Scholar]
- 51.Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4:e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol. 2005;18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 54.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohé R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose P, Won YK, Ong CN, Whiteman M. Beta-phenylethyl and 8-methylsulphinyloctyl isothiocyanates, constituents of watercress, suppress LPS induced production of nitric oxide and prostaglandin E2 in RAW 264.7 macrophages. Nitric Oxide. 2005;12:237–243. doi: 10.1016/j.niox.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res. 2004;21:661–670. doi: 10.1023/B:PHAM.0000022413.43212.cf. [DOI] [PubMed] [Google Scholar]
- 57.Xu C, Shen G, Chen C, Gélinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 58.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhäuser C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 59.Heiss E, Gerhäuser C. Time-dependent modulation of thioredoxin reductase activity might contribute to sulforaphane-mediated inhibition of NF-kappaB binding to DNA. Antioxid Redox Signal. 2005;7:1601–1611. doi: 10.1089/ars.2005.7.1601. [DOI] [PubMed] [Google Scholar]
- 60.Kim SJ, Kang SY, Shin HH, Choi HS. Sulforaphane inhibits osteoclastogenesis by inhibiting nuclear factor-kappaB. Mol Cells. 2005;20:364–370. [PubMed] [Google Scholar]
- 61.Woo KJ, Kwon TK. Sulforaphane suppresses lipopolysaccharide-induced cyclooxygenase-2 (COX-2) expression through the modulation of multiple targets in COX-2 gene promoter. Int Immunopharmacol. 2007;7:1776–1783. doi: 10.1016/j.intimp.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 62.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 63.Park SY, Kim GY, Bae SJ, Yoo YH, Choi YH. Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol Rep. 2007;18:181–187. [PubMed] [Google Scholar]
- 64.Choi S, Lew KL, Xiao H, Herman-Antosiewicz A, Xiao D, Brown CK, et al. D, L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis. 2007;28:151–162. doi: 10.1093/carcin/bgl144. [DOI] [PubMed] [Google Scholar]
- 65.Fimognari C, Lenzi M, Sciuscio D, Cantelli-Forti G, Hrelia P. Cell-cycle specificity of sulforaphane-mediated apoptosis in Jurkat T-leukemia cells. In Vivo. 2007;21:377–380. [PubMed] [Google Scholar]
- 66.Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, Lee YJ, Xiao H, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280:19911–19924. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- 67.Xu C, Shen G, Yuan X, Kim JH, Gopalkrishnan A, Keum YS, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–445. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 68.Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–1367. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 69.Xiao D, Johnson CS, Trump DL, Singh SV. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol Cancer Ther. 2004;3:567–575. [PubMed] [Google Scholar]
- 70.Mi L, Chung FL. Binding to protein by isothiocyanates: a potential mechanism for apoptosis induction in human nonsmall lung cancer cells. Nutr Cancer. 2008;60(Suppl 1):12–20. doi: 10.1080/01635580802381287. [DOI] [PubMed] [Google Scholar]
- 71.Mi L, Xiao Z, Hood BL, Dakshanamurthy S, Wang X, Govind S, et al. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem. 2008;283:22136–22146. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu J, Straub J, Xiao D, Singh SV, Yang HS, Sonenberg N, et al. Phenethyl isothiocyanate, a cancer chemopreventive constituent of cruciferous vegetables, inhibits cap-dependent translation by regulating the level and phosphorylation of 4E-BP1. Cancer Res. 2007;67:3569–3573. doi: 10.1158/0008-5472.CAN-07-0392. [DOI] [PubMed] [Google Scholar]
- 73.Hasegawa T, Nishino H, Iwashima A. Isothiocyanates inhibit cell cycle progression of HeLa cells at G2/M phase. Anticancer Drugs. 1993;4:273–279. doi: 10.1097/00001813-199304000-00021. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther. 2003;2:1045–1052. [PubMed] [Google Scholar]
- 75.Pappa G, Bartsch H, Gerhäuser C. Biphasic modulation of cell proliferation by sulforaphane at physiologically relevant exposure times in a human colon cancer cell line. Mol Nutr Food Res. 2007;51:977–984. doi: 10.1002/mnfr.200700115. [DOI] [PubMed] [Google Scholar]
- 76.Cheung KL, Khor TO, Yu S, Kong AN. PEITC induces G1 cell cycle arrest on HT-29 cells through the activation of p38 MAPK signaling pathway. AAPS J. 2008;10:277–281. doi: 10.1208/s12248-008-9032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Visanji JM, Duthie SJ, Pirie L, Thompson DG, Padfield PJ. Dietary isothiocyanates inhibit Caco-2 cell proliferation and induce G2/M phase cell cycle arrest, DNA damage, and G2/M checkpoint activation. J Nutr. 2004;134:3121–3126. doi: 10.1093/jn/134.11.3121. [DOI] [PubMed] [Google Scholar]
- 78.Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, et al. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004;279:25813–25822. doi: 10.1074/jbc.M313538200. [DOI] [PubMed] [Google Scholar]
- 79.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33:375–380. [PubMed] [Google Scholar]
- 80.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 81.Beklemisheva AA, Feng J, Yeh YA, Wang LG, Chiao JW. Modulating testosterone stimulated prostate growth by phenethyl isothiocyanate via Sp1 and androgen receptor down-regulation. Prostate. 2007;67:863–870. doi: 10.1002/pros.20472. [DOI] [PubMed] [Google Scholar]
- 82.Ramirez MC, Singletary K. Regulation of estrogen receptor alpha expression in human breast cancer cells by sulforaphane. J Nutr Biochem. 2009;20:195–201. doi: 10.1016/j.jnutbio.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 83.Matsui TA, Sowa Y, Yoshida T, Murata H, Horinaka M, Wakada M, et al. Sulforaphane enhances TRAIL-induced apoptosis through the induction of DR5 expression in human osteosarcoma cells. Carcinogenesis. 2006;27:1768–1777. doi: 10.1093/carcin/bgl015. [DOI] [PubMed] [Google Scholar]
- 84.Kim H, Kim EH, Eom YW, Kim WH, Kwon TK, Lee SJ, et al. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of DR5. Cancer Res. 2006;66:1740–1750. doi: 10.1158/0008-5472.CAN-05-1568. [DOI] [PubMed] [Google Scholar]
- 85.Mastrangelo L, Cassidy A, Mulholland F, Wang W, Bao Y. Serotonin receptors, novel targets of sulforaphane identified by proteomic analysis in Caco-2 cells. Cancer Res. 2008;68:5487–5491. doi: 10.1158/0008-5472.CAN-07-6171. [DOI] [PubMed] [Google Scholar]