

Summary
Murine models of β-thalassaemia have been used to test therapeutic globin gene vectors. However, the level of γ-globin expression necessary to achieve full phenotypic correction in these models is unclear. In order to address this issue, we carried out breeding and transplantation studies in murine models of β-thalassaemia intermedia (Hbbth–3/+) and severe β-thalassaemia major (Hbbth–3/Hbbth–3) using transgenic lines expressing various levels of human γ-globin. Expression of γ-globin RNA at a modest 7–14% of total α-globin RNA resulted in the selective survival of HbF(+) erythrocytes, a fivefold increase in total HbF, and a phenotypic improvement in the β-thalassaemia intermedia model. Full normalisation of erythrocyte indices in this model required γ-globin RNA expression at 27% of α-globin, resulting in an average 40% (6.8 g/dl) HbF. Studies using the homozygous Hbbth–3 model of lethal β-thalassaemia major demonstrated that even this high level of γ-globin expression, for reasons related to the function of the hybrid globin tetramers, could only prolong, but not fully support, survival. Taken together, these results indicate that only the heterozygous Hbbth–3 model of β-thalassaemia intermedia can be reliably used for the pre-clinical assessment of γ-globin gene therapy vectors, as well as other means of γ-globin gene induction.
Keywords: haemoglobin, thalassaemia, molecular, animal model
Murine models of β-thalassaemia and sickle cell disease developed over the past decade have been used extensively to study the patho-physiology of, and potential therapies for, these diseases (Sadelain et al, 2004). In particular, such models have been used to assess the functional properties of recombinant virus vectors for variants of human β-globin and γ-globin designed to treat the β-chain haemoglobinopathies. Most of these studies have focused on one of three murine models of relatively mild β-thalassaemia intermedia or sickle cell disease (May et al, 2000, 2002; Pawliuk et al, 2001; Imren et al, 2002; Person et al, 2003; Hanawa et al, 2004), while only one study has focused on a murine model of severe β-thalassaemia major (Rivella et al, 2003). Likewise, most of these studies have also focused on the use of recombinant virus vectors for wild type (WT) or variant forms of human β-globin (May et al, 2000, 2002; Pawliuk et al, 2001; Imren et al, 2002), while relatively few have focused on the use of vectors for human γ-globin (Person et al, 2003; Hanawa et al, 2004). These and other studies (Person et al, 2001) have provided some information correlating the level of β-globin or γ-globin gene expression with the level of phenotypic improvement in these models. However, several critical questions remain regarding the level of human globin gene expression, and especially γ-globin gene expression that is necessary to improve or normalise the phenotypic sequela in these thalassaemia models. For example, it is unclear whether expression of γ-globin confers the same degree of selective RBC survival in murine models of β-thalassaemia as that observed in patients with equivalent disease. The amount of γ-globin expression that is necessary to fully normalise the wide spectrum of erythrocytic indices in murine models of β-thalassaemia has also remained unresolved. Finally, no studies have been reported to date on the ability of human γ-globin to rescue the murine model of lethal β-thalassaemia major characterised by a complete absence of β-chains.
In the studies described here, we sought to address these questions in order to better characterise and justify the use of these models as a means of validating candidate gene transfer vectors for human γ-globin prior to their use in clinical trials. For these studies, we focused on the th-3 murine model of β-thalassaemia (Yang et al, 1995). In this model, both the Hbb-b1 and Hbb-b2 adult β-globin genes are deleted in cis. Homozygous Hbbth–3/Hbbth–3 mice die perinatally, while heterozygous Hbbth–3/+ mice backcrossed to C57BL/6J exhibit a phenotype of β-thalassaemia intermedia with decreased haematocrit and haemoglobin (Hb) content, increased reticulocytes, and abnormal red blood cell (RBC) morphology.
We carried out a series of studies crossing Hbbth–3/+ mice to transgenic lines expressing various levels of human γ-globin. This included lines expressing a wider range of γ-globin and a more comprehensive analysis of RBC indices than previously reported (Person et al, 2001). We also included a comparison with non-thalassaemic controls in order to accurately assess the degree of selective RBC survival/expansion afforded by γ-globin expression in this thalassaemic environment. In a separate series of studies, we carried out breeding and fetal liver cell (FLC) transplantation studies so as to assess the ability of the human γ-globin transgenes to rescue the lethal phenotype in the Hbbth–3/Hbbth–3 model of β-thalassaemia major. The results of theses studies are compared to similar studies in murine models of β-thalassaemia with human β-globin and γ-globin, as well as to clinical observations of patients with varying degrees of β-thalassaemia and elevated levels of HbF.
Materials and methods
Mouse strains
Wild-type C57BL/6J mice and thalassaemic Hbbth–3/+ mice (Yang et al, 1995) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Thalassaemic Hbbth–3/+ mice are heterozygous for a deletion encompassing both the Hbb-b1 and Hbb-b2 adult β-globin genes in cis, and thus fail to express all β-like globins from this allele. The transgenic lines 1279 and 135/4-2 have been previously described (Enver et al, 1989; Constantoulakis et al, 1991; Stamatoyannopoulos et al, 1993). As shown in the top panel of Fig 1, line 1279 (herein labelled γlo) contains three copies in cis of a transgene consisting of a 2.5-kb composite of the human β-globin locus control region (termed a μLCR) linked to a coding cassette for human Aγ-globin extending −1348 bp 5′ from the transcriptional start site. This includes a putative stage-specific silencer that results in minimal transgene expression in adults. The bottom panel of Fig 1 shows that line 135/4-2 (herein labelled γhi) contains nine copies in cis of a transgene similar to that in line 1279, except that the Aγ-globin gene promoter is truncated to −382, effectively deleting the stage-specific silencer. The Hbbth–3/+ line and the two transgenic lines were backcrossed to C57BL/6J for more than 10 generations. Combinations of β-thalassaemia and γ-globin transgenic lines were then generated by appropriate crosses. Offspring were typed for the Hbbth–3 allele by polymerase chain reaction (PCR) analysis for the residual hprt gene from the knock-out cassette using the following primers: 5′-GGCAAAGGATGTGATACGTGGAAG-3′ and 5′-CCAGTTTCACTAATGACACAAACATG-3′. These results were confirmed by quantitative real-time PCR analysis for murine β-globin using the following primers: β-globin, 5′-GTCATCACCGAAGCCTGATT-3′ and 5′-TGTCTGTTTCTGGGGTTGTG-3′; Aire (internal control), 5′-CACACAGGCAATTTGTCCAC-3′ and 5′-AGAGGAAGCGGGAAAGTTGT-3′. This latter approach was also used to confirm the genotype of homozygous Hbbth–3/Hbbth–3 fetuses. Offspring were typed for the γ-globin transgenes by immunofluorescent analysis (described below) to identify transgene-positive individuals, and real-time PCR to differentiate between heterozygotes and homozygotes using the following primers: γ-globin, 5′-TGGCTAAA CTCCACCCATGGGTTG-3′ and 5′-CCAGAAGCGAGTGTGTGGAACTGCT-3′; Aire (internal control) described above. All animal studies were carried out in accordance with accepted standards and were approved by the University of Washington Institutional Animal Care and Use Committee.
Fig 1.

γ-Globin transgenic lines. The γ-globin expression cassettes in the transgenic mouse lines 1279 (Enver et al, 1989; Constantoulakis et al, 1991), and 135/4-2 (Stamatoyannopoulos et al, 1993), include a 2.5 kb locus control region (μLCR) containing DNase hypersensitive sites 1–4 from the human β-globin locus linked to a coding cassette for human Aγ-globin that includes either a −1348 bp 5′-promoter region (line 1279) or a −382 bp 5′-promoter region (line 135/4-2). The line 1279 allele (denoted γlo) contains three transgene copies in cis, while the line 135/4-2 allele (denoted γhi) contains nine copies in cis. Grey boxes, μLCR; filled and open boxes, Aγ-globin exons and introns respectively; heavy line: 5′- and 3′-regions flanking the Aγ-globin coding region.
Transgene expression analysis
Total RNA prepared from peripheral blood was analysed by RNase protection as previously described (Stamatoyannopoulos et al, 1993), using the following probes: pT7Mα (128) linearised with HindIII to give a 128 bp protected fragment within exon 1 of the mouse α-globin gene; and pT7Aγ (170) linearised with BstEII to give a 170 bp protected fragment within exon 2 of the human Aγ-globin gene. A total of 50 ng RNA was hybridised overnight at 48°C with 106 cpm of each radiolabelled probe. A pilot experiment confirmed that the probe was in excess under these conditions. After digestion with RNase A and T1, the protected fragments were separated on 6% polyacrylamide 8 mol/l urea gels, and autoradiography was performed without intensifying screens. Signal intensities were quantified by Phosphorimager, and expression levels of the human Aγ-globin transgenes were calculated as a percentage of total mouse α-globin. For immunofluorescent analysis, 3 μl of peripheral blood was fixed in 4% formaldehyde for 30 min at room temperature, permeabilised by sequential washes in cold acetone, stained with a phycoerythrin-conjugated monoclonal antibody for HbF (BD Bioscience, San Jose, CA, USA), and scanned on a FACSan flow cytometer (BD Bioscience) using cellquest software as previously described (Thorpe et al, 1994; Emery et al, 2002). Protein analysis was performed on an automated high-performance liquid chromatography (HPLC) analyser (Variant II β-thalassaemia short program; Bio-Rad Laboratories, Hercules, CA, USA).
Haematological analysis
For reticulocyte analysis, 1 μl of peripheral blood was incubated in Hank’s balanced salt solution with 0.01 μg/ml of thiazole orange (Sigma-Aldrich, St Louis, MO, USA), 2 mmol/l of EDTA and 0.02% sodium azide for 1–2 h at room temperature as previously described (Davis et al, 1995). The stained cells were analysed on a FACSan flow cytometer using cellquest software. All other blood cell analyses were performed on a Cell-DYN 3500 hematology analyzer (Abbott Diagnostics, Santa Clara, CA, USA).
FLC transplantation
Donor fetal livers were harvested from embryonic day 15.5 embryos obtained by intercrossing either Hbbth–3/+ mice or Hbbth–3/+ mice homozygous for the γlo or γhi transgene alleles. Fetuses were diagnosed by colour phenotype and PCR so as to identify Hbbth–3/Hbbth–3 homozygotes. These fetal liver preparations were mixed in various ratios and transplanted at a dose of 3 × 106 total cells per recipient into congenic C57BL/6J mice previously treated with 1100 cGy myeloablating total body irradiation. Transplant recipients were monitored routinely for signs of anaemia and were killed when moribund or profoundly anaemic with haematocrits ≤15%.
Results
Generation and expression analysis of β-thalassaemic/γ-globin transgenic lines
The β-globin knockout mouse line Hbbth–3 (Yang et al, 1995), and two γ-globin transgenic mouse lines, line 1279 (γlo), which expresses low levels of γ-globin (Enver et al, 1989; Constantoulakis et al, 1991), and line 135/4-2 (γhi), which expresses high levels of γ-globin (Stamatoyannopoulos et al, 1993), were backcrossed to C57BL/6J and then crossed to generate a series of β-thalassaemia/γ-transgene hybrids. As shown in Fig 1 and discussed more fully in the Materials and methods section, these two transgenic lines express different levels of human γ-globin as a result of differences in 5′-promoter regions and transgene copy numbers. For further analysis, we chose to focus on three γ-globin transgene allele combinations: heterozygous γlo (γlo/−), homozygous γlo (γlo/γlo), and homozygous γhi (γhi/γhi). The amount of γ-globin expressed from these three transgene allele combinations against both a WT C57BL/6J background and on the heterozygous Hbbth–3/+β-thalassaemia background were determined at the level of RNA (by RNase protection) and at the level of protein (by HPLC).
In the WT background, γ-globin RNA expression levels ranged from 7.0 ± 0.8% (versus total mouse α-globin) for the γlo/− transgene allele, 13.9 ± 1.3% for the γlo/γlo transgene combination, and 27.2 ± 5.9% for the γhi/γhi transgene combination (Fig 2, left panel). These levels were consistent with previously reported expression levels for these transgenic lines (Enver et al, 1989; Constantoulakis et al, 1991; Stamatoyannopoulos et al, 1993) and were significantly different from one another (P ≤ 0.001). In these same lines, protein expression levels ranged from 2.7 ± 0.4% (HbF versus total Hb) for the γlo/− transgene allele, 5.2 ± 0.7% for the γlo/γlo transgene combination, and 12.0 ± 2.7% for the γhi/γhi transgene combination. These levels were also significantly different from one another (P ≪ 0.001), but represented an average 60% reduction compared with RNA levels. This discrepancy between RNA and protein levels may reflect a modest competitive advantage of mouse β-globin over human γ-globin in associating with mouse α-globin or possibly a modest discrepancy in the relative efficiency of translation.
Fig 2.

γ-Globin expression in wild type and β-thalassaemia intermedia mice. Peripheral blood was collected at 10–20 weeks of age from wild type and Hbbth–3/+ individuals containing one or two copies of the γlo transgene alleles (γlo/− and γlo/γlo respectively) or two copies of the γhi transgene allele (γhi/γhi). Samples were analysed for the amount of γ-globin RNA (as a percentage of total mouse α-globin, open bars) by RNase protection, and for the amount of γ-globin protein (in the form of hybrid mouse/human HbF as a percentage of total Hb, grey bars) by high-performance liquid chromatography. Results are presented as mean ± SD for a total of five to 12 individuals for each group for the RNA analysis and seven to 29 individuals for each group for the protein analysis. *P < 0.001 vs. RNA; P < 0.001 vs. wild type.
Evidence for selective advantage of murine thalassaemic red cells containing fetal Hb
RNA levels remained essentially unchanged when these same transgene allele combinations were crossed onto the Hbbth–3/+β-thalassaemia background, ranging from 6.8 ± 1.5% for the γlo/− transgene allele, 15.8 ± 1.6% for the γlo/γlo transgene combination, and 27.1 ± 4.2% for the γhi/γhi transgene combination (Fig 2, left). This suggests that: (i) γ-globin transgene expression fails to convey a selective survival advantage at the early stages of erythrocyte development when RNA is still present; and (ii) transcription of the γ-globin transgene is not effected when there is deficient β-globin transcription in trans. However, protein analysis provided strong evidence of a selective survival advantage for mature RBC expressing the γ-globin transgene, as shown by an increase in protein expression levels ranging from 13.7 ± 1.2% for the γlo/− transgene allele, 25.4 ± 3.5% for the γlo/γlo transgene combination, and 40.0 ± 4.7% for the γhi/γhi transgene combination. This represented a highly significant increase in the levels of γ-globin protein compared to the levels in WT mice, with an average increase of 5.1-fold for the γlo/− combination, 4.9-fold for the γlo/γlo transgene combination, and 3.3-fold for the γhi/γhi transgene combination.
To investigate further this apparent selective survival advantage for RBC expressing the γ-globin transgene, immunofluorescent staining and flow cytometry was used to assess the γlo/− transgene expression pattern in both WT and β-thalassaemia backgrounds. As seen in the example presented in Fig 3A and summarised in Fig 3B, this transgene allele generated a clearly heterocellular pattern of expression in the WT background, with an average 64 ± 7% of RBC expressing HbF. This was consistent with the initial characterisation of this line, where the fraction of F cells was estimated at 25% by a less sensitive staining method (Constantoulakis et al, 1991). In contrast, this same allele generated a nearly pancellular expression pattern in the β-thalassaemia background, with an average 93 ± 3% of RBC expressing HbF. As seen in Fig 3A and summarised in Fig 3C, this shift was also accompanied by an increase in the absolute level of γ-globin expression within the population of HbF(+) RBC. In the WT background, HbF(+) RBC stained with an average intensity of 53 ± 10 fluorescent units, while in the β-thalassaemia background the average intensity was 195 ± 70 units. Combining this 3.7-fold increase in the level of expression within HbF(+) cells with the 1.4-fold increase in the overall frequency of HbF(+) cells fully accounts for the 5.1-fold increase in total HbF expression determined by HPLC analysis. Taken together, these results provide clear evidence that human γ-globin can compensate for the diminished levels of endogenous β-globin in this murine model of β-thalassaemia intermedia, and that this compensation results in the selective survival and expansion of HbF(+) RBC. This in turn results in a post-transcriptional increase in both the fraction of RBC expressing HbF, as well as the overall level of HbF expressed within these RBC.
Fig 3.

In vivo selection of red blood cell (RBC) expression γ-globin. Peripheral blood was collected from non-transgenic wild-type (WT) mice, WT mice containing a single copy of the γlo transgene alleles (γlo/−), and Hbbth–3/+ thalassaemia mice containing a single copy of the γlo transgene allele, fixed and stained with a phycoerythrin-conjugated monoclonal antibody to HbF, and analysed on a flow cytometer. (A) Typical analysis showing the lack of staining in a non-transgenic control (no Tg; filled histogram), as well as the shift from a heterocellular pattern of γ-globin expression in a transgenic WT mouse (γlo/− WT; heavy line) to a pancellular and significantly elevated pattern of expression in a Hbbth–3/+ mouse (γlo/− Thal; light line). The marker used to define HbF(+) is indicated. (B) Percentage of HbF(+) RBC in WT versus Hbbth–3/+ mice containing a single copy of the γlo transgene allele showing a selective expansion of these cells in the thalassaemia background. Data represents mean ± SD for at least six individuals. (C) Mean fluorescence of HbF(+) RBC from panel (B), showing a concomitant increase in the intensity of staining in the thalassaemia background. *P < 0.01 vs. WT.
Human γ-globin production fully corrects the phenotype of murine β-thalassaemia intermedia
We assessed multiple haematological parameters for the three Hbbth–3/+ × γ-globin transgenic hybrids described above, as well as non-transgenic Hbbth–3/+ and WT controls. As seen in the series of panels to the left of Fig 4, non-transgenic Hbbth–3/+ mice exhibited a phenotype of β-thalassaemia intermedia, with a decrease in RBC (10.4 ± 0.4 ×1012/l vs. 11.7 ± 0.6 × 1012/l for WT), total Hb (12.8 ± 0.7 g/dl vs. 17.5 ± 1.1 g/dl for WT), and haematocrit (45.2 ± 1.9% vs. 57.8 ± 2.6% for WT), and a notable increase in reticulocytes (22.1 ± 4.0% vs. 5.5 ± 2.0% for WT). Addition of a single γ-globin transgene allele (γlo/−) increased RBC counts to levels that were indistinguishable from WT (11.6 ± 0.8 × 1012/l), and generated a modest improvement in Hb (15.2 ± 1.2 g/dl), haematocrit (50.5 ± 3.4%), and reticulocytes (10.1 ± 3.7%). Addition of two copies of this γ-globin transgenic allele (γlo/γlo) lead to the full normalisation of all four of these parameters, including RBC counts (11.9 ± 0.8 × 1012/l), Hb (17.6 ± 0.9 g/dl), haematocrit (58.9 ± 2.7%), and reticulocytes (5.5 ± 1.0%). This was also the case for two copies of the γhi transgene allele (γhi/γhi), including RBC counts (11.6 ± 1.1 × 1012/l), Hb (17.1 ± 0.9 g/dl), haematocrit (57.2 ± 1.1%), and reticulocytes (3.8 ± 1.9%). Taken together, these results demonstrate that the deficits in overall Hb and RBC production in this model of murine β-thalassaemia intermedia can be fully normalised by expression of hybrid mouse/human HbF at about 25% (4.5 g/dl) HbF, and that this can be achieved by expression of γ-globin RNA at about 15% of total mouse α-globin.
Fig 4.

Effects of γ-globin expression on haemopoietic and red blood cell (RBC) indices. Peripheral blood was collected at 10–20 weeks of age from none-transgenic Hbbth–3/+ (Thal) and wild-type (WT) individuals, as well as from Hbbth–3/+ individuals containing one or two copies of the γlo transgene alleles (γlo/− and γlo/γlo respectively) or two copies of the γhi transgene allele (γhi/γhi). Samples were analysed for total RBC counts, total haemoglobin, haematocrit, the percentage of reticulocytes, as well as mean corpuscular volume, mean corpuscular haemoglobin, mean corpuscular haemoglobin concentration and red cell distribution width. Results represent mean ± SD for three to 11 individuals for each group. *P < 0.05 vs. the Thal controls; P < 0.05 vs. the WT control.
The higher levels of γ-globin expression were necessary to fully normalise multiple indices used to characterise mature RBC (Fig 4). RBC from Hbbth–3/+ mice exhibited a decrease in mean corpuscular volume (MCV; 41.5 ± 1.7 fl vs. 50.3 ± 1.2 fl for WT) and mean corpuscular Hb (MCH; 12.3 ± 0.5 pg vs. 15.5 ± 0.4 pg for WT), and a notable increase in red cell distribution width (RDW; 35.6 ± 2.2% vs. 15.8 ± 1.6% for WT). Addition of either one or two copies of the γlo transgene allele (γlo/− or γlo/γlo respectively) improved all of these indices relative to the Hbbth–3/+ controls, but failed to fully normalise them relative to the WT controls. This included an increase in MCV (43.5 ± 1.4 and 47.3 ± 0.6 fl respectively) and MCH (13.1 ± 0.5 and 14.1 ± 0.3 pg), and a decrease in RDW (26.4 ± 2.8% and 20.6.8 ± 1.7%). It was not until two copies of the γhi transgene allele (γhi/γhi) were included that all these indices normalised, including MCV (48.6 ± 0.4 fl), MCH (14.8 ± 0.8) and RDW (17.6 ± 1.1%). As indicated in Fig 4, there were no significant differences in mean corpuscular Hb concentration between the various lines tested. These differences in RBC morphology were confirmed by histological analysis (Fig 5). In this case, Hbbth–3/+ (Thal) RBC showed a significant level of dismorphic features, including hypochromic microcytosis, target cells, basophilic stippling, and poikilocytes, as well as acanthocytes, spherocytes, burr cells, and schistocytes. There was also evidence of Howell–Jolly bodies, polychromatophilia, and a low level of nucleated RBCs indicative of a modest level of RBC regeneration. These features were progressively diminished in γlo/+ and γlo/γlo RBC, and were completely normalised in γhi/γhi RBC compared with RBC from a normal C57BL/6J (WT) control.
Fig 5.
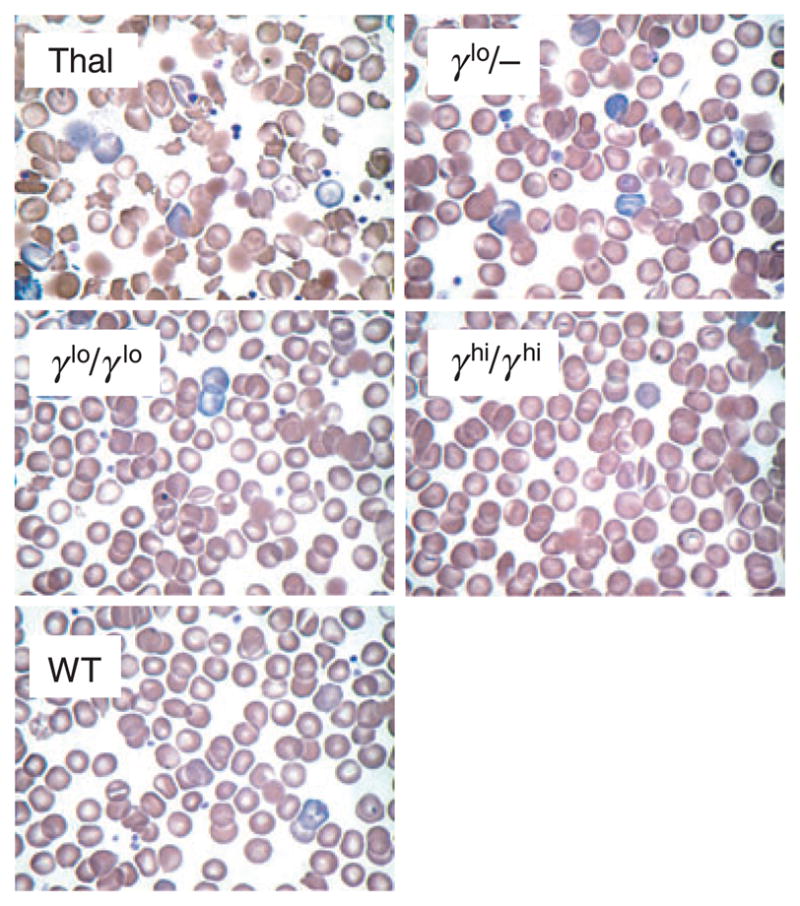
Effects of γ-globin expression on red blood cell histology. Wright–Giemsa stained peripheral blood smears from individual animals are presented, including wild-type C57BL/6J, non-transgenic β-thalassaemia intermedia Hbbth–3/+ (Thal) or Hbbth–3/+ individuals containing one copy of the γlo transgene allele (γlo/−), two copies of the γlo transgene allele (γlo/γlo) or two copies of the γhi transgene allele (γhi/γhi) (original magnification ×60).
Taken together, these results demonstrate that the deficits in multiple RBC indices observed in this murine model of β-thalassaemia intermedia can be fully normalised by hybrid mouse/human HbF, but that this requires expression at about 40% (6.8 g/dl) HbF, which in turn can be achieved by expression of γ-globin RNA at about 27% of total mouse α-globin.
Hybrid mouse α/human γ-Hb is unable to rescue from lethality the murine model of β-thalassaemia major characterised by complete β-chain deficiency
We next sought to determine the degree to which human γ-globin expression could mitigate the lethal phenotype in the homozygous Hbbth–3/Hbbth–3 model of murine β-thalassaemia major. Crosses were produced in which the mice were homozygous for a complete β-globin chain deficiency and in which the only none α-globin chains produced were human γ-globin. These mice were expected to have a very large excess of α-globin chains and the question was whether, and to what level, the synthesis of human γ-globin chains could rescue these animals from embryonic lethality. As detailed in Table I, crosses between Hbbth–3/+ individuals containing no γ-globin transgenes (γ−/γ−) and two copies of the line γlo transgene allele (γlo/γlo), as well as intercrosses between Hbbth–3/+ individuals containing two copies of the γlo allele (γlo/γlo), failed to generate viable offspring homozygous for the Hbbth–3 knockout (0 of 35 for the γlo/− offspring and 0 of 68 for the γlo/γlo offspring, with an expectation rate of 1 in 4). This is consistent with the results of Person et al (2001), who, by similar crosses, found 0 of 49 viable β0-thalassaemia homozygotes containing a transgene expressing human γ-globin at 13%, with an expectation rate of 1 in 16.
Table I.
Breeding for β-null mice.
| Parents | Target genotype | Target frequency of offspring (1/4 expected) |
|---|---|---|
| Hbbth–3/+, γlo/γlo × Hbbth–3/+, γ−/γ− | Hbbth–3/Hbbth–3, γlo/− | 0 of 35 |
| Hbbth–3/+, γlo/γlo × Hbbth–3/+, γlo/γlo | Hbbth–3/Hbbth–3, γlo/γlo | 0 of 68 |
| Hbbth–3/+, γhi/γhi × Hbbth–3/+, γhi/γhi | Hbbth–3/Hbbth–3, γhi/γhi | 1 of 29* |
Died at 2 months of age.
We also performed intercrosses between Hbbth–3/+ individuals containing two copies of the γhi transgene allele (γhi/γhi), which was shown to produce γ-globin protein at c. 40% in the thalassaemia intermedia mice described above. Surprisingly, these crosses produced only one offspring of 29 live births that was homozygous for the Hbbth–3 knockout allele, while the expected number was 7.5 (1/4). This animal had a total Hb level of 11.5 g/dl and haematocrit of 37%. This level of anaemia is compatible with survival in mice, and is equivalent to the haematological phenotype of W/Wv mice (Antonchuck et al, 2004). However, even this animal failed to survive past 2 months of age. These results indicate that the failure of even high levels of human γ-globin to rescue the Hbbth–3/Hbbth–3 mice resides in the functional properties of the mouse/human hybrid Hb. This in turn suggests that the murine model of homozygous β-thalassaemia is not an appropriate animal model for testing the therapeutic potential of elevated levels of fetal Hb.
Studies in a murine β-thalassaemia major stem cell transplantation model
We next turned to the transplantation model for murine β-thalassaemia major, first described by Rivella et al (2003). This involved collecting liver cells from day 15.5 Hbbth–3/Hbbth–3 fetuses that were also homozygous for the γlo or γhi transgene alleles, mixing these in different ratios with Hbbth–3/Hbbth–3 FLCs containing no γ-globin transgenes, and transplanting the mixtures of cells into myeloablated congenic C57BL/6J recipients. Recipients of unmodified Hbbth–3/Hbbth–3 FLCs were expected to die around 50 d post-transplant due to profound anaemia that was coincidental with the loss of normal recipient RBCs. If γ-globin gene expression could compensate for the complete β-globin chain deficiency and hybrid mouse α-globin/human γ-globin tetramers could functionally replace native mouse HbA, we would expect the transplant recipients to be corrected haematologically and to survive.
As seen in Fig 6, in the absence of γ-globin transgenes, transplant recipients in our studies survived a median of 48 d, eventually succumbing to profound anaemia coincident with the eventual loss of normal recipient RBC. Animals transplanted with FLCs from WT C57BL/6 donors or congenic Hbbth–3/+ donors typically survived long-term (data not shown). The use of γlo/γlo FLCs in ratios of 10%, 25%, 50% and even 100% failed to significantly increase survival in this model, with a median survival of 63 d at the highest ratio (Fig 6A). Likewise, the use of γhi/γhi FLCs at a ratio of 10% failed to significantly increase survival, with a median survival of 52 d. In contrast, the use of γhi/γhi FLCs at 25%, 50% and 100% did significantly increase survival, to a median of 77, 75 and 84 d respectively. Analysis at near 40 d post-transplant (latest time tested) indicated a clear dose–response in multiple haematological indices. This included an overall increase in haematocrit, ranging from 14 ± 9% for the 0% controls, 24 ± 3% for 100% γlo/γlo, and 31 ± 9% for 100% γhi/γhi, as well as in total Hb, ranging from 4.8 ± 3.8 g/dl for the 0% controls, 8.6 ± 1.1 g/dl for 100% γlo/γlo, and 9.4 ± 1.1 g/dl for 100% γhi/γhi. Taking into account the fraction of RBC expressing HbF (40 ± 14%) and the fraction of total Hb arising from HbF (37 ± 15%) in the 100% γhi/γhi recipients at this time point, it was estimated that the γhi/γhi transgenes resulted in the production of HbF at 7.7 ± 1.9 g/dl. This is slightly higher than the level observed in Hbbth–3/+ recipients containing this same transgene combination (6.8 g/dl), but well below the 11.5 g/dl observed in the one live-born individual with this same genetic constitution from the breeding studies described above. Immunofluorescent analysis at 83 d post-transplant of surviving γhi/γhi transplant recipients demonstrated that essentially 100% of peripheral RBC consisted of HbF(+) donor cells.
Fig 6.

Effects of γ-globin expression on survival of mice transplanted with Hbbth–3/Hbbth–3 fetal liver cells (FLCs). FLCs were collected from day 15.5 homozygous Hbbth–3/Hbbth–3 fetuses containing either no transgenes or two copies of either the γlo of γhi transgene alleles (γlo/γlo or γhi/γhi respectively). These cells were mixed in different ratios (shown as the percentage of transgenic FLCs), and transplanted into myeloablated congenic C57BL/6J recipients. Transplant recipients were monitored closely and killed when moribund or severely anemic with hematocrits <15%. (A) Survival of recipients transplanted with mixtures of γlo/γlo FLCs. (B) Survival of recipients transplanted with mixtures of γhi/γhi FLCs. *P ≤ 0.01 vs. 0% controls.
This level of γ-globin gene expression was, however, clearly insufficient to support the long-term survival in this transplantation model of murine homozygous β-thalassaemia. This is in contrast to the findings of Rivella et al (2003), who reported that expression of a recombinant virus vector for human β-globin at levels resulting in production of mouse/human hybrid HbA at an average 6.5 ± 2.9 g/dl could support long-term survival in this same model. This discrepancy shows that human γ-globin may provide an even poorer substitute for mouse β-globin than does human β-globin in this and other murine models of β-thalassaemia, and implicates the function of the hybrid mouse α/human γ-Hb, rather than the amount of this hybrid Hb, in the failure of γ-globin to rescue the lethal phenotype in this model.
Discussion
The gene therapy approaches that are currently being pursued for the treatment of the β-chain haemoglobinopathies rely on the use of recombinant virus vectors to stably transfer expression cassettes for either human β-globin or γ-globin into reconstituting haematopoietic stem cells of thalassaemia patients. Vectors for human γ-globin have been pursued due to the potent anti-sickling properties of γ-globin for the treatment of sickle cell disease, as well as the therapeutic potential of γ-globin for the treatment of the β-thalassaemic patients. Several lines of evidence suggest that γ-globin gene transfer should prove curative for the β-thalassaemic patients. For example, compound heterozygotes for β-thalassaemia and deletional HPFH exhibit a very mild phenotype (Weatherall, 2001), compound heterozygotes for β-thalassaemia and a non-deletional HPFH allele exhibit a β-thalassaemia intermedia phenotype (Fessas & Stamatoyannopoulos, 1964; Sofroniadou et al, 1975; Tate et al, 1986), and compound δβ-thalassaemia/β-thalassaemia heterozygotes exhibit a β-thalassaemia intermedia to mild phenotypes (Stamatoyannopoulos et al, 1969). Homozygous β0-thalassaemia individuals who have also coin-herited an unlinked gene for heterocellular HPFH have higher levels of HbF and mild clinical course (Prchal & Stamatoyannopoulos, 1981). Homozygotes for δβ-thalassaemia have only fetal Hb in their blood and a phenotype of intermediate thalassaemia (Weatherall, 2001), while homozygotes for deletional HPFH have only fetal Hb in their blood and are clinically normal, although they have elevated haematocrits and dismorphic RBC indices, presumably secondary to the increased oxygen affinity of fetal Hb (Wheeler & Krevans, 1961; Charache et al, 1976). Collectively, the experience of a very large number of clinical studies over 40–50 years strongly indicated that a gene therapy approach based on γ-globin should prove effective for the treatment of β-thalassaemia.
As a prerequisite to gene therapy clinical trials, it is essential that the efficacy and safety of candidate globin gene transfer vectors be tested in animal models of β-thalassaemia. Several studies to date have used murine models of relatively mild β-thalassaemia intermedia (May et al, 2000, 2002; Imren et al, 2002; Person et al, 2003; Hanawa et al, 2004), and have shown that β-globin gene transfer with lentivirus-based vectors can fully correct the thalassaemic phenotype in this animal model. The studies reported here demonstrated that γ-globin gene transfer can fully correct the phenotype of murine β-thalassaemia intermedia, indicating that Hbbth–3/+ mice represents a good model for testing candidate vectors for human γ-globin. In particular, we documented a clear dose-dependent improvement in multiple haematopoietic indices, culminating in the complete normalisation of the β-thalassaemia phenotype at the highest levels of γ-globin expression tested. We also documented the selective survival of RBC expressing threshold levels of γ-globin, a property predicted to be critical for the therapeutic success of globin gene therapy. These studies also establish the level of γ-globin expression necessary to fully normalise the phenotypic sequelae in this model (27.1 ± 4.2% of mouse α-globin RNA, correlating to expression of hybrid HbF at 40.0 ± 4.7% or about 6.8 g/dl).
Given these results, it was a surprise to find that expression of human γ-globin at even higher levels (giving rise to total Hb levels in the 7.7–11.5 g/dl range) failed to support long-term survival of Hbbth–3/Hbbth–3 homozygous mice that manifest a phenotype of embryonic lethal β-thalassaemia major. This was especially true in light of studies with virus vectors for human β-globin in this model where long-term survival was achieved at total Hb levels of 6.5 g/dl or less (Rivella et al, 2003). Likewise, clinical studies indicate that humans can survive, albeit with serious disease, with all or nearly all Hb consisting of HbF at levels of 6 g/dl or even lower (Weatherall & Clegg, 1981). Thus, it would appear that, in contrast to the heterozygous Hbbth–3/+ mice with β-thalassaemia intermedia, the homozygous Hbbth–3/Hbbth–3 mice constitute an inappropriate model for testing the efficacy of γ-globin gene transfer vectors.
We can propose two possible explanations for the failure of γ-globin to rescue the lethal phenotype in Hbbth–3/Hbbth–3 mice. First, previous studies have demonstrated that γ-globin has a strong propensity for forming Hb Bart’s (γ4 homotetramers) in transgenic mice (Arcasoy et al, 1997). Although the underlying mechanisms for this propensity are unclear, it suggests a basic inefficiency of hybrid mouse α-globin/human γ-globin heterotetramer formation. The formation of Hb Bart’s would further decrease the already limited pool of binding partners for excess α-globin chains, resulting in increased α-chain precipitation and proerythrocyte destruction. Secondly, previous studies have also demonstrated that hybrid mouse/human HbF, as well as Hb Bart’s, have decreased oxygen affinity (Arcasoy et al, 1997; Shear et al, 1998), which, in combination, would diminish the ability of surviving RBC in these mice to efficiently deliver oxygen.
In summary, human γ-globin can fully correct the heterozygous Hbbth–3/+ mouse model of β-thalassaemia intermedia in a dose-dependent manner and in association with an increased survival of RBC-containing hybrid mouse/human HbF. As such, Hbbth–3/+ mice constitute a good model for the pre-clinical assessment of gene transfer vectors for human γ-globin. However, even high levels of human γ-globin cannot rescue the lethal phenotype of the homozygous Hbbth–3/Hbbth–3 mouse model of severe β-thalassaemia major, and as such this model is not appropriate for testing therapeutic interventions based on human γ-globin gene expression.
Acknowledgments
We wish to thank Qiliang Li for probes used in the RNase protection analysis and Betty Mastropaolo for help with photomicrographs. This work was supported by grants from the National Heart, Lung, and Blood Institute, National Institutes of Health.
References
- Antonchuck J, Hyland CD, Hilton DJ, Alexander WS. Synergistic effects of erythropoiesis, thrombopoiesis, and stem cell competitiveness in mice deficient in thrombopoietin and steel factor receptors. Blood. 2004;104:1306–1313. doi: 10.1182/blood-2004-04-1522. [DOI] [PubMed] [Google Scholar]
- Arcasoy MO, Romana M, Fabry ME, Skarpidi E, Nagel RL, Forget BG. High levels of human γ-globin gene expression in adult mice carrying a transgene of deletional-type hereditary persistence of fetal hemoglobin. Molecular and Cellular Biology. 1997;17:2076–2089. doi: 10.1128/mcb.17.4.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Clegg JB, Weatherall DJ. The negro variety of hereditary persistence of fetal haemoglobin is a mild form of thalassaemia. British Journal of Haematology. 1976;34:527–534. doi: 10.1111/j.1365-2141.1976.tb03599.x. [DOI] [PubMed] [Google Scholar]
- Constantoulakis P, Josephson B, Mangahas L, Papayannopoulou T, Enver T, Costantini F, Stamatoyannopoulos G. Locus control region – a gamma transgenic mice: a new model for studying the induction of fetal hemoglobin in the adult. Blood. 1991;77:1326–1333. [PubMed] [Google Scholar]
- Davis BH, Ornvold K, Bigelow NC. Flow cytometric reticulocyte maturity index: a useful laboratory parameter of erythropoietic activity in anemia. Cytometry. 1995;22:35–39. doi: 10.1002/cyto.990220107. [DOI] [PubMed] [Google Scholar]
- Emery DW, Yannaki E, Tubb J, Nishino T, Li Q, Stamatoyannopoulos G. Development of virus vectors for gene therapy of β chain hemoglobinopathies: flanking with a chromatin insulator reduces γ-globin gene silencing in vivo. Blood. 2002;100:2012–2021. doi: 10.1182/blood-2002-01-0219. [DOI] [PubMed] [Google Scholar]
- Enver T, Ebens AJ, Forrester WC, Stamatoyannopoulos G. The human beta-globin locus activation region alters the developmental fate of a human fetal globin gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:7033–7037. doi: 10.1073/pnas.86.18.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessas P, Stamatoyannopoulos G. Hereditary persistence of fetal hemoglobin in Greece. A study and a comparison. Blood. 1964;24:223–240. [PubMed] [Google Scholar]
- Hanawa H, Hargrove PW, Kepes S, Srivastava DK, Nienhuis AW, Persons DA. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassaemia. Blood. 2004;104:2281–2290. doi: 10.1182/blood-2004-03-0863. [DOI] [PubMed] [Google Scholar]
- Imren S, Payen E, Westerman KA, Pawliuk R, Fabry ME, Eaves CJ, Cavilla B, Wadsworth LD, Beuzard Y, Bouhassira EE, Russell R, London IM, Nagel RL, Leboulch P, Humphries RK. Permanent and panerythroid correction of murine beta thalassaemia by multiple lentiviral integration in hematopoietic stem cells. Proceedings of the National Academy of Science of the United States of America. 2002;99:14380–14385. doi: 10.1073/pnas.212507099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, Sadelain M. Therapeutic haemoglobin synthesis in β-thalassaemic mice expressing lentivirus-encoded human β-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- May C, Rivella S, Chadburn A, Sadelain M. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99:1902–1908. doi: 10.1182/blood.v99.6.1902. [DOI] [PubMed] [Google Scholar]
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, Humphries RK, Beuzard Y, Nagel RL, Leboulch P. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- Person DA, Allay ER, Sabatino DE, Kelly P, Bodine DM, Nienhuis AW. Functional requirements for phenotypic correction of murine beta-thalassemia: implications for human gene therapy. Blood. 2001;97:3275–3282. doi: 10.1182/blood.v97.10.3275. [DOI] [PubMed] [Google Scholar]
- Person DA, Hargrove PW, Allay ER, Hanawa H, Nienhuis AW. The degree of phenotypic correction of murine beta-thalassemia intermedia following lentiviral-mediated transfer of a human gamma-globin gene is influenced by chromosomal position effects and vector copy number. Blood. 2003;101:2175–2183. doi: 10.1182/blood-2002-07-2211. [DOI] [PubMed] [Google Scholar]
- Prchal J, Stamatoyannopoulos G. Two siblings with unusually mild homozygous beta-thalassemia: a didactic example of the effect of a nonallelic modifier gene on the expressivity of a monogenic disorder. American Journal of Medical Genetics. 1981;10:291–300. doi: 10.1002/ajmg.1320100312. [DOI] [PubMed] [Google Scholar]
- Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Rivella S, Lisowski L, Samakoglu S, Riviere I. Globin gene transfer for treatment of the beta-thalassemias and sickle cell disease. Best Practice and Research Clinical Haematology. 2004;17:517–534. doi: 10.1016/j.beha.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Shear HL, Grinberg L, Gilman J, Fabry ME, Stamatoyannopoulos G, Goldberg DE, Nagel RL. Transgenic mice expressing human fetal globin are protected from malaria by a novel mechanism. Blood. 1998;92:2520–2526. [PubMed] [Google Scholar]
- Sofroniadou K, Wood WG, Nute PE, Stamatoyannopoulos G. Globin chain synthesis in the Greek type (A gamma) of hereditary persistence of fetal hemoglobin. British Journal of Haematology. 1975;29:137–148. doi: 10.1111/j.1365-2141.1975.tb01807.x. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos G, Fessas P, Papayannopoulou T. F-thalassemia. A study of thirty-one families with simple heterozygotes and combinations of F-thalassemia with A2-thalassemia. American Journal of Medicine. 1969;47:194–208. doi: 10.1016/0002-9343(69)90146-6. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos G, Josephson B, Zhang JW, Li Q. Developmental regulation of human gamma-globin genes in transgenic mice. Molecular and Cellular Biology. 1993;13:7636–7644. doi: 10.1128/mcb.13.12.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate VE, Wood WG, Weatherall DJ. The British form of hereditary persistence of fetal hemoglobin results from a single base mutation adjacent to an S1 hypersensitive site 5′ of the A gamma globin gene. Blood. 1986;68:1389–1393. [PubMed] [Google Scholar]
- Thorpe SJ, Thein SL, Sampietro M, Craig JE, Mahon B, Huehns ER. Immunochemical estimation of haemoglobin types in red blood cells by FACS analysis. British Journal of Haematology. 1994;87:125–132. doi: 10.1111/j.1365-2141.1994.tb04881.x. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ. The thalassemias. In: Stamatoyannopoulos G, Majerus PW, Perlmutter RM, Varmus H, editors. The Molecular Basis of Blood Diseases. 3. W.B. Sounders Co; Philadelphia, PA: 2001. pp. 183–226. [Google Scholar]
- Weatherall DJ, Clegg JB. The Thalassemia Syndromes. Blackwell Scientific Publications; Oxford: 1981. [Google Scholar]
- Wheeler JT, Krevans JR. The homozygous state of persistent fetal hemoglobin and the interaction of persistent fetal hemoglobin with thalassemia. Bulletin of the Johns Hopkins Hospital. 1961;109:217–233. [PubMed] [Google Scholar]
- Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for beta 0-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]