

Abstract
To elucidate the role of thyroid hormone receptors (TRs) α1 and β in the development of hearing, cochlear functions have been investigated in mice lacking TRα1 or TRβ. TRs are ligand-dependent transcription factors expressed in the developing organ of Corti, and loss of TRβ is known to impair hearing in mice and in humans. Here, TRα1-deficient (TRα1−/−) mice are shown to display a normal auditory-evoked brainstem response, indicating that only TRβ, and not TRα1, is essential for hearing. Because cochlear morphology was normal in TRβ−/− mice, we postulated that TRβ regulates functional rather than morphological development of the cochlea. At the onset of hearing, inner hair cells (IHCs) in wild-type mice express a fast-activating potassium conductance, IK,f, that transforms the immature IHC from a regenerative, spiking pacemaker to a high-frequency signal transmitter. Expression of IK,f was significantly retarded in TRβ−/− mice, whereas the development of the endocochlear potential and other cochlear functions, including mechanoelectrical transduction in hair cells, progressed normally. TRα1−/− mice expressed IK,f normally, in accord with their normal auditory-evoked brainstem response. These results establish that the physiological differentiation of IHCs depends on a TRβ-mediated pathway. When defective, this may contribute to deafness in congenital thyroid diseases.
Thyroid hormone (triiodothyronine, T3) and its receptors are essential for the development of hearing. Congenital thyroid disorders impair hearing, and profound deafness is common when there is a prevalence of iodine deficiency (1–3). Also, hypothyroidism in mice and rats causes deformities in the organ of Corti and has indicated a critical window of development preceding the onset of hearing during which the hormone is required (4–6). Beyond these observations, however, little is understood of the mechanisms underlying T3 action in the auditory system.
T3 receptors (TRs) are ligand-dependent transcription factors encoded by the related TRα and TRβ genes (7, 8). The TRα gene encodes a T3-responsive receptor, TRα1, and a C-terminal splice variant, TRα2, of unknown function that does not bind T3 (9, 10). The TRβ gene encodes two N-terminal variants, TRβ1 and TRβ2, both of which function as T3 receptors (11, 12). Both TRα1 and TRβ are expressed during embryonic and postnatal development of the cochlea, indicating that the cochlea is a direct site of T3 action (13, 14). Deletion of TRβ by gene targeting in mice severely impairs the auditory-evoked brainstem response (ABR) (15), demonstrating that TRβ is essential for auditory development. Also, human resistance to thyroid hormone is associated with TRβ mutations and a proportion of cases of resistance to thyroid hormone exhibit deafness or mild hearing impairment (16, 17). However, the role of TRα1 in hearing remains unknown.
To determine the relative functions of TRα1 and TRβ in the auditory system, we have investigated ABR and cochlear physiology in mice lacking either TRα1 (18) or TRβ (19). Because TRβ-deficient (TRβ−/−) mice do not display histological defects in the cochlea (15), we have further tested the hypothesis that TRβ regulates functional, rather than morphological development of the cochlea. These studies have revealed a defect in a potassium current, IK,f, in inner hair cells (IHCs). IK,f has only recently been identified, and it normally appears immediately before the onset of hearing, preventing spiking behavior by the immature IHC and conferring a capability for high-frequency auditory signal transmission (20). Thus, these findings define a requirement for TRβ in cochlear development at the level of the physiological differentiation of IHCs.
MATERIALS AND METHODS
Mouse Strains.
Derivation of TRβ- and TRα1-deficient mice by using gene targeting has been described (15, 18). TRα1−/− mice specifically lack TRα1 but still express TRα2. TRβ−/− mice lack both TRβ1 and TRβ2 variants. The TRβ mutation was generated in W9.5 embryonic stem cells derived from a substrain of 129/Sv- + p + Tyr-cMgfSl-J/+ mice (strain JR0090, The Jackson Laboratory), which however, were wild type for the steel locus. The TRα1 mutation was generated in the E14 line of embryonic stem cells from the 129/OlaHsd mouse strain. Mutations were studied on mixed genetic backgrounds of 129/Sv × C57BL/6J (TRβ−/−) and 129/OlaHsd × BALB/c (TRα1−/−) mouse strains. Animal experiments followed approved institutional protocols at University of Tübingen, Mount Sinai School of Medicine, and University of Cincinnati.
ABR.
ABR was assessed as described (15, 21) on mice that had been anesthetized with Avertin (3.5 mg/10 g of body weight) in response to a click stimulus (band of 1–16 kHz) and pure-tone pips (8, 16, and 32 kHz). Thresholds were evoked by using 128–512 stimuli presented at a rate of 20/s. Mice were studied as young adults at 2–3 months of age to preclude complications from age-dependent hearing loss in C57BL/6J, BALB/c, and 129 strains (21). It was noted that some mice from occasional litters of both TRα1+/+ and TRα1−/− genotypes had elevated ABR thresholds, indicating that other genes in this strain background caused hearing loss. It is likely that substrain 129-derived genes account for this variability, because BALB/c mice do not show hearing loss until 1 year of age, whereas 129 substrains have at least one gene for earlier onset hearing loss (L.C.E., unpublished data).
Whole-Cell Recording.
Cochlear hair cells of TRβ−/−, TRα1−/−, and wild-type mice were studied either in Cell-Tak-mounted, organotypic cochlear cultures at postnatal-day (P) ages P1-P5 (ref. 22) or following acute dissection of the organ of Corti (P6–P60) from identified regions of the cochlea as described (20). Extracellular solution was composed of 144 mM NaCl, 0.7 mM NaH2PO4, 5.8 mM KCl, 1.3 mM CaCl2, 0.9 mM MgCl2, 5.6 mM d-glucose, and 10 mM Hepes⋅NaOH (pH 7.3) at room temperature. Vitamins and amino acids for Eagle’s minimal essential medium were added from concentrates (Gibco/BRL).
Membrane currents and voltages were studied at room temperature (20–25°C) by whole-cell patch-clamp using an Axopatch 200B amplifier. Patch pipettes were filled with intracellular solution containing 135 mM KCl, 0.1 mM CaCl2, 5 mM EGTA⋅KOH, 3.5 mM MgCl2, 2.5 mM Na2ATP, and 5 mM Hepes⋅KOH (pH 7.3) at room temperature. For measurements of Ca2+, Na+, and transduction currents, Cs+ was substituted for K+. Currents under voltage clamp are presented with capacitive transient and linear leak currents subtracted, and all voltages have been corrected for the voltage drop across the uncompensated series resistance, Rs (0.5–5 MΩ; mean ± SD = 1.3 ± 1.0 MΩ, n = 76), and for liquid-junction potentials (−4 mV) measured between intra- and extracellular solutions.
The fast potassium current, IK,f was measured at −25 mV between 2.4 and 3.6 ms from the onset of the depolarizing voltage steps as described (20). The fits of the developmental expression pattern are according to a sigmoidal logistic growth curve:
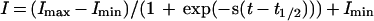 |
1 |
where I is current (nA), s is a slope factor (d−1), t is time (measured in days, d), and t1/2 is the time at which I is halfway between Imax and Imin. All statistical tests are two-tailed Student’s t tests.
Capacitance.
Linear membrane capacitances of cells were evaluated from current transients in response to −10 mV voltage steps from −84 mV.
Nonlinear capacitances of outer hair cells (OHCs) were measured by using a modified phase-tracking system. Charge movement by the OHC motor molecules was measured as a voltage-dependent capacitance by using a software lock-in technique (23, 24). Capacitance was monitored while slow voltage ramps were applied to the cell (1.8–7 s; −120–+70 mV). Phase angles were repeatedly adjusted between voltage sweeps. To exclude interference with large conductance changes induced by depolarization, potassium currents were blocked by substitution of 115 mM CsCl and 20 mM tetraethylammonium (TEA)⋅Cl for an equal amount of KCl in the pipette solution. Series resistances ranged from 3 to 9 MΩ in these measurements. Capacitance was plotted versus membrane voltage and fitted to the derivative of a two-state Boltzmann function:
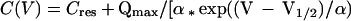 |
2 |
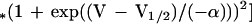 |
where Cres is a residual of the linear capacitance, Clin, not compensated for by the slow transient cancellation circuit of the amplifier. Qmax is the maximum whole-cell charge transferred by the motor molecules, slope factor α accounts for the voltage dependence of the charge translocation, and V1/2 is the voltage at half-maximum charge transfer. Maximum capacitance, Cmax, occurs at V1/2. Data are presented as voltage-dependent capacitance divided by the linear capacitance in fF/pF.
Endocochlear Potentials.
Surgical procedures followed described methods (25). Mice were anesthetized using 20% urethane at a dosage of 0.01 ml/g body weight. Potential measurements employed the Axopatch amplifier as a high-impedance voltmeter.
RESULTS
To investigate the relative functions of TRβ and TRα1 in the auditory system, ABR thresholds—a sensitive measure of auditory function—were determined for TRα1−/− mice (18). In contrast to the previously described deficiency in TRβ−/− mice (15), ABR thresholds were in the normal range in TRα1−/− mice (Fig. 1) for all frequency stimuli tested that span the most sensitive hearing range in mice. Thus, the roles of TRβ and TRα1 are not equivalent and TRα1 is nonessential for auditory function.
Figure 1.

Auditory-evoked brainstem responses in TRα1- and TRβ-deficient mice. Columns indicate mean ABR thresholds ± SEM in dB sound pressure level for click, 8-, 16-, and 32-kHz frequency stimuli. Thresholds of TRα1−/− mice were not significantly different from wt mice, whereas TRβ−/− mice have significantly elevated thresholds over TRβ+/+ or TRβ−/+ mice (∗∗, P < 0.01; ref. 15). Numbers of mice per group: n = 4, TRβ−/+; n = 9, TRβ−/−; n = 6, TRα1+/+; n = 8, TRα1−/−. TRβ−/− and TRα1−/− mice were compared with control mice (TRβ−/+ and TRα1+/+, respectively) of the corresponding genetic backgrounds (see Materials and Methods).
In the auditory system, TRβ is prominently expressed in the organ of Corti (13), suggesting that it has direct functions in cochlear development. Because previous analysis revealed that TRβ was not required for morphological development of the cochlea (15), we investigated instead several physiological functions in the cochlea, including the functions of individual hair cells.
Developmental expression of the fast-activating potassium conductance IK,f in IHCs was determined by whole-cell voltage- and current-clamp recordings made on hair cells from a large number of wild-type (wt), TRα1−/−, and TRβ−/− mice isolated from a range of developmental stages (Figs. 2 and 3). In wt mice of the same genetic background as TRβ−/− mice, IK,f was absent at P10 but was clearly expressed at P18 (Fig. 3A). Its expression followed a logistic growth function (Eq. 1) where half-maximal expression was reached on P17.5 with a slope factor s = 0.42/d (Fig. 3C), which compares closely with wt mice of the CD1 strain in which IK,f was recently described (20). In contrast, in TRβ−/− mice, IK,f was largely absent at P15–P18, corresponding to the period when their hearing impairment first becomes apparent (15). When studied over an extended time course of development, IK,f eventually appeared with a significant delay and reached half-maximal expression on P28.2 with s = 0.16/d. Fig. 3B shows that in adult TRβ−/− mice at P50, the characteristics of IK,f matched those of wt mice, indicating that it represented the same conductance. In accord with their normal ABR (Fig. 1), TRα1−/− mice displayed normal developmental expression of IK,f (Fig. 3 A and C), consistent with the proposed role of this conductance at the onset of hearing (20).
Figure 2.

Acutely isolated organ of Corti of a P18 wt mouse prepared for IHC recordings. (A) A section of the organ of Corti isolated from the basal region of the cochlea observed from the site of the stria vascularis. The short stereocilia of hair bundles of the IHCs are visible at the top. Pillar cells (PCs) rigidly couple IHCs to the OHCs, here seen at the level of their nuclei. (B) The same IHCs seen in A after OHCs and pillar cells have been removed mechanically with micropipettes. (C) IHC during the whole-cell voltage-clamp recording. A patch pipette has been sealed onto its cleaned basolateral surface while the cell remains inside the semiintact sensory epithelium of the organ of Corti.
Figure 3.

Whole-cell membrane currents of IHCs. (A) At P18, IHCs of wt and TRα1−/− mice expressed an additional, fast-activating K+ current, IK,f, that was absent in TRβ−/− mice. The very large membrane currents in the TRα1−/− cell at P18 caused a less effective voltage clamp of the membrane potential and a rounded-looking onset of the largest current traces shown. This very large current is not the result of a physiological difference, as it was not seen with smaller series resistances or smaller total membrane currents in other cells. The TRα1−/− recording denoted at P10 was derived from a cell at P13 not yet expressing the fast current component. (B) In adult animals, membrane currents are similar in IHCs of wt and TRβ−/− mice. (C) IK,f at −25 mV, measured at the points indicated by bars and arrows in A, as a function of the day of postnatal development. Curves represent IK,f in TRα1−/− and wt (solid line) and TRβ−/− (dotted line) mice. Individual points represent wt (♦), TRα1−/− (∗) and TRβ−/− mice (• and ○, apical and basal turn of cochlea, respectively). Fits are according to Eq. [1]. Imin (−208 pA) was determined by the IHCs’ Ca2+ currents. In mice older than about P40, Imax (4.26 and 3.14 nA in wt and TRβ−/− mice, respectively) was not significantly different (P > 0.05) in the two fits, based on the cells’ membrane capacitances, when data were expressed in current densities (nA/pF).
In TRβ−/− mice after P40, IK,f approached the magnitudes seen in wt mice. The curve fits of Fig. 3C suggest that final IK,f magnitude was less than in wt mice; however, the scatter of IK,f increased with age, and when normalized to membrane capacitances, the final magnitude of IK,f in TRβ−/− mice after P40 was not significantly different from IHCs from wt mice. When it was eventually expressed, IK,f of TRβ−/− mice was sensitive to charybdotoxin, with a dose-response curve at −25 mV showing a half-blocking concentration of 21 nM (n = 5 cells). This indicated that it was supported by the same conductance as described for wt CD1 mice (20).
Other hair cell membrane and transducer currents studied in TRβ−/− mice were normal, indicating that retarded expression of IK,f in IHCs was not caused by grossly impaired hair cell development. Before the onset of IK,f expression, membrane currents of IHCs and OHCs of wt, TRα1−/−, and TRβ−/− animals were indistinguishable. IHCs (n = 2) and OHCs (n = 5) of three TRβ−/− mice at P2–P4 were mechanically sensitive and responded with normal transducer currents (26) when their hair bundles were stimulated by a fluid jet (Fig. 4 A and B). At P10, IHCs had small Na+ and Ca2+ currents, and had the slowly activating potassium current, IK,neo (Fig. 3A; ref. 20), which on current injection produced slow action potentials (Fig. 4 C and D). IHCs of TRβ−/− mice at P15–P16 had Ca2+ currents typical of IHCs from wt mice before hearing onset.
Figure 4.

Normal physiology of immature cochlear hair cells in TRβ−/− mice. (A and B) When stimulated by a fluid jet at −84 mV, IHCs and OHCs of TRβ−/− mice responded with mechanoelectrical transducer currents, as found in wt cells. Currents were half-blocked by ≈3 μM d-tubocurarine as described for wt CD1 mice (22). (C and D) IHCs of TRβ−/− and wt mice responded similarly to current injections by forming slow Ca2+ action potentials. The TRβ−/− cell was slightly more depolarized and had a smaller input resistance than the wt cell (300 and 1,000 MΩ, respectively), effecting a faster membrane time constant and a more noisy-looking voltage response. For the same reason, the 20-pA current injection was less effective in the TRβ−/− cell in depolarizing the cell membrane, initiating only three action potentials. Other TRβ−/− cells had higher input resistances comparable to wt cells. (E and F) Voltage-dependent capacitance of OHCs in wt and TRβ−/− mice. Acutely isolated OHCs of apical turns at P8. Capacitances were normalized to accomodate differences in cell size by dividing by the cells’ linear, voltage-independent capacitances. We assume that observed differences in capacitance in TRβ−/− mice at P8 are not functionally significant.
OHCs of the organ of Corti are thought to contribute to an active amplification process of the cochlea (27) via the voltage-dependent motility of their cell bodies. Motility is assumed to depend on an integral membrane protein with piezoelectric properties. Although little is known of the normal development of OHC electromotility in the mouse, we compared OHCs from wt and TRβ−/− mice to investigate whether this function may be defective in TRβ−/− mice. We studied the electromotility indirectly by measuring the voltage-dependent capacitance it imposes on OHCs (refs. 24, 27; Fig. 4 E and F). Voltage-dependent capacitances are presented as normalized to the linear capacitances of the cells because according to their linear, voltage-independent capacitances, OHCs of TRβ−/− mice were smaller than those of wt mice (at P8, TRβ−/− = 4.0 ± 0.2 pF, n = 4; wt = 5.1 ± 0.5 pF, n = 6, P < 0.01).
Nonlinear capacitance could be demonstrated for OHCs isolated from both wt and TRβ−/− mice younger than P10. At P8, the nonlinear capacitance from six acutely isolated wt and four TRβ−/− OHCs was 290 ± 47 fF/pF and 150 ± 46 fF/pF, respectivley (Fig. 4 E and F). At P9, the nonlinear capacitance of six TRβ−/− cells was 271 ± 27 fF/pF. These results suggest that there is a developmental increase in voltage-dependent capacitances in mouse OHCs that may vary in progression between litters of pups. However, a complete developmental study that may be useful to address whether there is a significant difference in the development in wt versus TRβ−/− samples is difficult to perform, because OHCs die rapidly on isolation from mice older than P10. Nonetheless, the available results do not suggest an obvious defect in electromotility in TRβ−/− OHCs.
With the onset of hearing, the cochlea builds up an endocochlear potential (EP) in the scala media (25, 28) that contributes to the driving force for mechanoelectrical transduction. Na, K–ATPase subunits expressed in the stria vascularis that may contribute to generation of the EP have been suggested to be regulated by T3 (29). However, EPs of six TRβ−/− mice between P16 and P60 (85.2 ± 13.5 mV) were not significantly different (P > 0.05) from those of four wt mice (92.5 ± 13.5 mV). A delay in, as opposed to a permanent deficit in, EP formation was excluded because all three TRβ−/− mice studied at P16–P17 had normal EPs (25). Thus, hearing impairment in TRβ−/− mice is not the result of a defective EP, and retarded expression of IK,f cannot be ascribed to an indirect mechanism because of a defective EP. These findings support the conclusion that defective IK,f expression in TRβ−/− mice is not secondary to a general delay in cochlear development but results from the failure of a TRβ-dependent pathway that governs the functional maturation of IHCs.
DISCUSSION
In hearing, hair cells mediate the critical function of transducing acoustic stimuli into neural responses (30–32); however, the genetic controls that govern the specialized differentiation of these cells are poorly understood. The present results, identifying a defect in IHCs in TRβ−/− mice, indicate that a TRβ-mediated transcriptional pathway is required for the physiological differentiation of IHCs. In contrast to other deafness mutations that cause morphological defects in hair cells (33–35), TRβ is required at the level of the physiological maturation of IHC ionic conductances. Because TRβ is expressed in the greater epithelial ridge of the organ of Corti, where the immature IHCs reside (13), it could directly control expression of the presently unidentified genes encoding the ion channels that pass IK,f or other factors that control IK,f activity. However, more complex and indirect explanations cannot be excluded.
Given that basolateral potassium conductances help shape the IHC receptor potentials and accelerate IHC frequency performance (36), the deafness of TRβ−/− mice may be explained at least in part by retarded expression of IK,f. Neurons of the brainstem auditory pathway that extract timing information, such as the octopus or bushy cells of the ventral cochlear nucleus, have strongly differentiating temporal transfer characteristics (37) and may not respond properly if signal transmission by IHCs is retarded in the absence of IK,f. If IK,f is indeed critical for normal auditory function (20), it may be unexpected that adult TRβ−/− mice remain deaf with a permanently impaired ABR, even when IK,f eventually approaches normal magnitudes. This could be explained if early IK,f expression is required to facilitate the development of normal hearing in accord with the presence of a critical period for sensory inflow necessary for development of auditory function. This may be analogous to the visual system, in which studies of sensory deprivation have indicated a critical window for activity-dependent development of the ocular-dominance columns in the visual cortex (38). It also is not excluded that TRβ is required for other functions in hearing, for example, in synaptic transmission, although our results rule out major defects in hair cell transducer conductances, OHC electromotility, and endocochlear potentials.
TRα1 and TRβ bind to similar DNA sequences in vitro, and their expression overlaps in the organ of Corti (13, 14), suggesting that they have the potential to mediate common functions. However, our results indicate that only TRβ is essential for auditory function, perhaps reflecting that in vivo, each receptor can have distinct interactions with target genes or transcriptional cofactors. Further exploration of these possibilities must await the challenging task of identification and characterization of target genes for TRs in the cochlea.
Hypothyroidism in rodents causes malformation and loss of hair cells (4–6), indicating that T3 has a role in morphogenesis of the organ of Corti. However, loss of TRβ does not cause such deformities, nor can TRα1 be essential for morphogenesis, because TRα1 is not required for hearing. There may be several explanations for this lack of concordance between the defects that arise in the absence of hormone compared with those caused by the absence of T3 receptors. We previously suggested that TRβ mediates functional maturation, whereas TRα1 may mediate morphogenesis of the cochlea (15). This is still consistent with the present data if it is proposed that TRα1 may be substituted functionally by TRβ whereas TRβ cannot be substituted for by TRα1. Such partial redundancy between nuclear receptors may be similar to the situation for the retinoid receptor family (39). Alternatively, it has been shown in transfection assays that in the absence of T3, TRs can exert T3-independent repression on genes that are T3 inducible (40). Thus, hypothyroidism in vivo may result in abnormal and chronic repression by TRs that causes more severe developmental damage than loss of a receptor, where deleterious T3-independent repression by the TR could not occur.
Finally, these results implicate retarded expression of IK,f as a possible cause of hearing deficiency in the syndrome of resistance to thyroid hormone (16, 17). Failure to activate this TRβ-dependent function may also contribute to deafness in cases of congenital hypothyroidism. The present results also suggest that it is unlikely that TRα1 mutations will be found to underlie deafness in human disease.
Acknowledgments
We thank Prof. J. P. Ruppersberg, Department of Physiology, and Prof. H. P. Zenner, Ear, Nose, and Throat Hospital, University of Tübingen, for providing lab space and equipment. We also thank Dr. Karen Steel (University of Nottingham) for advice on measurement of endocochlear potentials in mice. This work was supported in part by a Sinsheimer Scholarship, the Deafness Research Foundation, the March of Dimes Birth Defects Foundation, the Human Frontiers Science Program, and National Institutes of Health Grant DC 03441 (D.F.) and Swedish Cancerfonden (B.V.).
ABBREVIATIONS
- T3
triiodothyronine
- TR
T3 receptor, TRα1, T3 receptor α1
- TRβ
T3 receptor β
- IK,f
fast-activating potassium conductance in cochlear inner hair cells
- IHC
inner hair cell
- OHC
outer hair cell
- ABR
auditory-evoked brainstem response
- EP
endocochlear potential
- P
postnatal day
References
- 1.Dussault J H, Ruel J. Annu Rev Physiol. 1987;49:321–324. doi: 10.1146/annurev.ph.49.030187.001541. [DOI] [PubMed] [Google Scholar]
- 2.DeLong, G. R. (1993) Am. J. Clin. Nutr. Suppl. 57, 286S–290S. [DOI] [PubMed]
- 3.Forrest D. J Clin Endocrinol Metab. 1996;81:2764–2767. doi: 10.1210/jcem.81.8.8768825. [DOI] [PubMed] [Google Scholar]
- 4.Deol M S. J Med Genet. 1973;10:235–242. doi: 10.1136/jmg.10.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uziel A. Acta Otolaryngol (Stockh) 1986;429:23–27. doi: 10.3109/00016488609122726. [DOI] [PubMed] [Google Scholar]
- 6.O’Malley B W, Li D, Turner D S. Hear Res. 1995;88:181–189. doi: 10.1016/0378-5955(95)00111-g. [DOI] [PubMed] [Google Scholar]
- 7.Sap J, Muñoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennström B. Nature (London) 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 8.Weinberger C, Thompson C C, Ong E S, Lebo R, Gruol D J, Evans R M. Nature (London) 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 9.Izumo S, Mahdavi V. Nature (London) 1988;334:539–542. doi: 10.1038/334539a0. [DOI] [PubMed] [Google Scholar]
- 10.Koenig R J, Lazar M A, Hodin R A, Brent G A, Larsen P R, Chin W W, Moore D D. Nature (London) 1989;337:659–661. doi: 10.1038/337659a0. [DOI] [PubMed] [Google Scholar]
- 11.Hodin R A, Lazar M A, Wintman B I, Darling D S, Koenig R J, Larsen P R, Moore D D, Chin W W. Science. 1989;244:76–78. doi: 10.1126/science.2539642. [DOI] [PubMed] [Google Scholar]
- 12.Sjöberg M, Vennström B, Forrest D. Development (Cambridge, UK) 1992;114:39–47. doi: 10.1242/dev.114.1.39. [DOI] [PubMed] [Google Scholar]
- 13.Bradley D J, Towle H C, Young W S., III Proc Natl Acad Sci USA. 1994;91:439–443. doi: 10.1073/pnas.91.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauterman J, ten Cate W J. Hear Res. 1997;107:23–28. doi: 10.1016/s0378-5955(97)00014-2. [DOI] [PubMed] [Google Scholar]
- 15.Forrest D, Erway L C, Ng L, Altschuler R, Curran T. Nat Genet. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 16.Refetoff S, DeWind L T, DeGroot L J. J Clin Endocrinol Metab. 1967;27:279–294. doi: 10.1210/jcem-27-2-279. [DOI] [PubMed] [Google Scholar]
- 17.Brucker-Davis F, Skarulis M C, Pikus A, Ishizawar D, Mastroianni M-A, Koby M, Weintraub B D. J Clin Endocrinol Metab. 1996;81:2768–2772. doi: 10.1210/jcem.81.8.8768826. [DOI] [PubMed] [Google Scholar]
- 18.Wikström L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennström B. EMBO J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forrest D, Hanebuth E, Smeyne R J, Everds N, Stewart C L, Wehner J M, Curran T. EMBO J. 1996;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- 20.Kros C, Ruppersberg J, Rüsch A. Nature (London) 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- 21.Erway L C, Willott J F, Archer J R, Harrison D E. Hear Res. 1993;65:125–132. doi: 10.1016/0378-5955(93)90207-h. [DOI] [PubMed] [Google Scholar]
- 22.Glowatzki E, Ruppersberg J, Zenner H, Rüsch A. Neuropharmacology. 1997;36:1269–1275. doi: 10.1016/s0028-3908(97)00108-1. [DOI] [PubMed] [Google Scholar]
- 23.Fidler N, Fernandez J. Biophys J. 1989;56:1153–1162. doi: 10.1016/S0006-3495(89)82762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gale J, Ashmore J. Pflügers Arch. 1997;434:267–271. doi: 10.1007/s004240050395. [DOI] [PubMed] [Google Scholar]
- 25.Steel K, Barkway C. Development (Cambridge, UK) 1989;107:453–463. doi: 10.1242/dev.107.3.453. [DOI] [PubMed] [Google Scholar]
- 26.Kros C, Rüsch A, Richardson G. Proc R Soc London Ser B. 1992;249:189–193. [Google Scholar]
- 27.Holley M. In: The Cochlea. Dallos P, Popper A, Fay R, editors. New York: Springer; 1996. pp. 386–434. [Google Scholar]
- 28.Rübsamen R, Lippe W R. In: Development of the Auditory System. Rubel E W, Popper A N, Fay R R, editors. New York: Springer; 1997. pp. 193–270. [Google Scholar]
- 29.Zuo J, Rarey K. Acta Otolaryngol (Stockh) 1996;116:422–428. doi: 10.3109/00016489609137867. [DOI] [PubMed] [Google Scholar]
- 30.Gillespie P G. Curr Opin Neurobiol. 1995;5:449–455. doi: 10.1016/0959-4388(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 31.Dallos P, Popper A, Fay R. The Cochlea. New York: Springer; 1996. [Google Scholar]
- 32.Hudspeth A. Neuron. 1997;19:947–950. doi: 10.1016/s0896-6273(00)80385-2. [DOI] [PubMed] [Google Scholar]
- 33.Steel K P, Brown S D M. Trends Genet. 1994;10:428–435. doi: 10.1016/0168-9525(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 34.Petit C. Nat Genet. 1996;14:385–391. doi: 10.1038/ng1296-385. [DOI] [PubMed] [Google Scholar]
- 35.Probst F, Fridell R, Raphael Y, Saunders T, Wang A, Liang Y, Morell R, Touchman J, Lyons R, Noben-Trauth K, et al. Science. 1998;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- 36.Kros C. In: The Cochlea. Dallos P, Popper A, Fay R, editors. New York: Springer; 1996. pp. 318–385. [Google Scholar]
- 37.Oertel D. Neuron. 1997;19:959–962. doi: 10.1016/s0896-6273(00)80388-8. [DOI] [PubMed] [Google Scholar]
- 38.Katz L, Shatz C. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 39.Kastner P, Mark M, Chambon P. Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 40.Damm K, Thompson C C, Evans R M. Nature (London) 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]