

Abstract
SEPT9_v1, the largest transcript of the septin gene family member, SEPT9, encodes a septin isoform implicated in the tumorigenic transformation of mammary epithelial cells. High levels of SEPT9_v1 expression also have been observed in both breast cancer cell lines, primary breast cancers as well as other solid tumor malignancies. We found a novel interaction between SEPT9_v1 and the c-Jun-N-terminal kinase (JNK), a mitogen- activated protein kinase important in cellular stress responses, cell proliferation, and cell survival. We found that up-regulation of SEPT9_v1 stabilizes JNK by delaying its degradation, thereby activating the JNK transcriptome. c-Jun kinase assays in mammary epithelial cells expressing SEPT9_v1, compared to controls, exhibited increased JNK/c-Jun transcriptional activity. This increase was associated with increased levels of cyclin D1, a critical component of the proliferative response required for progression through G1 of the cell cycle in many cell types. These findings demonstrate the first link between a septin protein and the JNK signaling pathway. Importantly, it suggests a novel functional role of SEPT9_v1 in driving cellular proliferation of mammary epithelial cells, a hallmark feature of oncogenesis that is directly relevant to breast cancer.
Keywords: JNK, septins, cell cycle regulation, cell proliferation, mammary epithelial cells, oncogenesis, cyclins
1. Introduction
SEPT9_v1 recently has been characterized as an important SEPT9 isoform that promotes cell proliferation and tumorigenesis in cell culture models of breast cancer as well as angiogenesis in prostate cancer [1, 2]. Several lines of evidence indicate functionally relevant crosstalk between mammalian septin family members and several signaling pathways implicated in oncogenesis, including HIF1α, Rho, and the DNA damage response [1, 3-6]. In addition, mammalian septins represent a network of polymers that are dynamically rearranged under the guidance of cellular signals. For example, Sheffield et al. demonstrated that a Cdc42 effector molecule, Borg3, regulates the formation of the SEPT2-SEPT6-SEPT7 septin complex [7]. Ito et al. demonstrated that constitutively active Rho disrupts the filament organization of SEPT9_v3, which co-localizes with actin stress fibers [3]. Finally, our previous findings suggested a strong affiliation of SEPT9_v1 with oncogenic phenotypes in breast cancer cells and with microtubule filaments where it seems to regulate microtubule stability [2]. However, the molecular mechanisms through which SEPT9_v1 promotes tumorigenesis and cellular transformation in mammary epithelial cells have not been elucidated.
SEPT9_v1 has also been functionally linked with angiogenesis through its activation of the hypoxia-inducible factor-1 (HIF-1) pathway, which promotes tumor progression in prostate cancer. Specifically, SEPT9_v1 overexpression was associated with activation of the HIF-1 transcriptome and increased cell proliferation rates [1]. Interestingly, JNK activation also had been previously implicated in the induction of HIF-1 [8]. In addition, the JNK/c-Jun pathway has been described as a critical component of the proliferative response and induction of cell cycle progression by increasing the transcriptional activity of c-Jun towards cyclin D1, a mechanism responsible for cell proliferation in breast cancer [9, 10].
c-Jun-N-terminal kinases are signal transduction proteins of mitogen-activated protein (MAP) kinases that are most strongly activated by stress stimuli including UV and ionizing radiation, heat shock, inflammatory cytokines, metabolic inhibitors, and osmotic or redox shock [11-14]. The JNK pathway also is involved in regulating many forms of stress-induced apoptosis and conflicting evidence indicates that JNK signaling may be either pro- or anti-apoptotic [12-14]. Evidence also is accumulating to suggest an important role for JNK in cell proliferation, cell transformation, tumor progression, and cell cycle regulation in G1 and M phases, occasionally due to signaling from upstream oncogenes [9, 15-20]. Strong evidence further supports the role of JNK in cell proliferation and cell cycle progression due to the changes that result from specific inhibition of basal JNK activity with SP600125 [11]. These diverse cellular functions are plausible because, similar to many other proteins, JNK operates in a manner that appears to be dependent on the cell type, stimulus, and context [17]. Typically, JNK mediates these numerous cellular responses by phosphorylating the N-terminal domain of c-Jun, thereby increasing its transactivation and upregulating AP-1-dependent transcription. AP-1-dependent transcription regulates the expression of many genes that are critically involved in cell proliferation, cell cycle, and survival [21, 22].
One of the target genes of JNK/c-Jun/AP-1 mediated transcription regulation is cyclin D1. The importance of cyclin D1 as a critical downstream target of JNK is suggested by the impaired transcription of cyclin D1-dependent kinases in c-Jun-/- mouse embryonic fibroblasts, resulting in a lack of cell proliferation and progression through the G1 phase of the cell cycle [23]. Cyclin D1 also has been found to be a transcriptional target of JNK and mediates its proliferative effects in hepatocytes [24]. Taken together, these findings emphasize that the cellular roles of JNK are not restricted to stress responses, but are also important for cellular proliferation and cell cycle regulation, and should facilitate future studies directed toward understanding how JNK can function in both pro-survival and pro-apoptotic signaling pathways.
Given the impact of SEPT9_v1 and JNK1,2 on cell proliferation, and the shared ability of SEPT9_v1 and JNK1,2 to regulate HIF-1, we elected to investigate the potential role of the interaction between SEPT9_v1 and JNK in cell proliferation. Since JNK activation resembled the effects of SEPT9_v1 over-expression, we hypothesized a crosstalk between the JNK signaling pathway and altered SEPT9_v1 expression, which could be implicated in accelerating cellular growth rates in mammary epithelial cells. We demonstrate here that JNK is, in fact, a novel SEPT9_v1 binding partner with the SEPT9 GTPase domain responsible for interaction. We also show that is functionally relevant to cell proliferation in mammary epithelial cells. JNK is also stabilized when SEPT9_v1 is ectopically expressed in mammary epithelial cells. In addition, we found functionally relevant correlations between the expression of SEPT9_v1, JNK activity, the up-regulation of the endogenous levels of cyclin D1, and accelerated proliferation in mammary epithelial cells. This implicates SEPT9_v1 as a modulator of phosphorylated c-Jun activity for the induction of CCND1 (cyclin D1) expression, presumably by regulating the endogenous level of JNK within the cells. Thus, in this report, we propose a novel mechanism by which SEPT9_v1 expression in human breast cancer causes activation of the JNK signaling pathway, contributing to its pro-proliferative effects that may promote tumor growth in mammary epithelial cells.
2. Materials and Methods
2.1 DNA constructs
Full length SEPT9_v1 gene was cloned into the pCMV-3Tag-1A expression vector. Deletion constructs were made as previously described in Makarova et al. by using site directed mutagenesis with one primer encompassing the desired deletion [25].
2.2 Cell Lines
A Human Papilloma Virus (HPV)-immortalized non-tumorigenic mammary epithelial cell line, HPV4-12, was developed and provided by S.P. Ethier while at the University of Michigan’s Comprehensive Cancer Center. Additional cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured under recommended conditions. Cells were transduced either with SEPT9_v1 or with SEPT9_v3 constructs in the pLPCX retroviral vector and stable clones expressing the constructs were selected using puromycin as previously described [2].
2.3 Immunoprecipitation
Cells were grown in 100mm dishes to 70% confluence before medium was removed, then the cells were washed twice with ice cold 1x PBS, and incubated for five minutes at 4°C in M-PER lysis buffer (Pierce Biotechnology, Inc. Rockford, IL). The cells were collected and lysates were cleared by centrifugation at 13 000 rpm for 30 minutes at 4°C. Approximately 400 μg of solubilized lysate was used for each immunoprecipitation using the Protein G Immunoprecipitation kit (Sigma Aldrich, St. Louis, MO) and immunoprecipitations were performed according to manufacturer’s instructions. The immunoprecipitated proteins were analyzed by SDS-PAGE and probed with the antibody described in the assay.
2.4 Western Blot
Western blotting was performed as previously described using 50 μg of whole cell lysates [2]. The following antibodies were used: a custom-made rabbit polyclonal anti-SEPT9_v1 antibody at a working dilution of 1:4000; SAPK/JNK rabbit monoclonal antibody; anti-cyclin D1 mouse monoclonal antibody; anti-phospho-cyclin D1 rabbit monoclonal antibody; and anti-phospho-c-Jun rabbit monoclonal antibody. All antibodies were purchased from Cell Signaling Technology (Beverly, MA) and used at working dilutions of 1:1000. In addition, anti-cyclin B1 mouse monoclonal, anti-cyclin E rabbit monoclonal and anti-JNK2 (D-2)HRP-linked antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and used at 1:200 dilution and 1: 500 respectively. An anti-Flag M2 monoclonal antibody used at 1:1000 dilution, a mouse ascites anti-β–actin antibody at a 1:10,000 dilution, a goat anti-rabbit:HRP secondary antibody (1:,000 dilution), and a goat anti-mouse:HRP (1:1000 dilution) antibody were all purchased from Sigma (St. Louis, MO). All antibodies were diluted in 5% non-fat dry milk and 0.05% Tween-20 in 1x TBS. The Super Signal West Pico Chemiluminescent kit (Pierce, Rockford, IL) was used for detection and blots were then exposed to Kodak XAR film. Relative to the loading control, semi-quantitative protein expression levels were determined by densitometry (Alpha Innotech IS-1000 Digital Imaging System, version 2.00).
2.5 GST Pull Downs
DH5α E. coli cells were transformed with plasmids for either pGEX-2T empty vector or pGEX-2T-SEPT9_v1-GST and were induced for four hours at 25°C with 1mM IPTG. Then, 1.0 mg of E. coli whole cell lysates were incubated for three hours at 4°C with glutathione sepharose 4B bead slurry (Amersham, Piscataway N.J.). Following washes, 1mg of whole cell lysates from HPV4-12-Flag:JNK1 stably transduced cells was added and incubated overnight at 4°C. After washing thoroughly with NTEN200 buffer (20mM Tris-HCl, 1mMEDTA, 0.5% NP40, 25μg/ml PMSF, 200mM NaCl), the beads were incubated for 15 minutes with 10 mM glutathione at room temperature. Samples were analyzed by Western blotting after boiling with Laemmli’s buffer.
2.6 Immunofluorescence
For immunofluorescence analysis, stable transductants and parental cell lines were grown on 17mm coverslips and fixed with 4% paraformaldehyde for 40 minutes at room temperature. Slides were then washed three times in 1x PBS for 10 minutes and blocked for one hour in blocking solution (5% dry milk, 1% BSA, 0.025% Tween-20 in 1x PBS). Cells were incubated overnight at 4°C in anti-SEPT9_v1 or anti-Flag-M2, and anti-SAPK/JNK antibody at a 1:30 dilution in blocking solution. Phalloidin conjugated to Alexafluor568 was used to identify F-Actin. Anti-rabbit:Alexafluor488 and anti-mouse:Alexafluor633 were used as secondary antibodies (Molecular Probes, Invitrogen Corporation, Carlsbad, CA) at a 1:500 dilution in blocking solution for one hour at room temperature. Cells were visualized using an Olympus FV-500 confocal microscope with a 100x objective.
2.7 Protein Stability Assay
Cells were plated in six-well plates and grown to 70% confluence. The cells were subjected to cycloheximide treatment as described [26]. Lysates were analyzed by Western blot at different timepoints. Quantification of relative endogenous JNK1,2 expression was performed by densitometry using β-Actin as the loading control (Alpha Innotech IS-1000 Digital Imaging System, version 2.00).
2.8 UV treatment
Sub-confluent cultures of the cells were irradiated with UV as described and incubated for one hour to recover [22]. Whole cell extracts were isolated as previous described and used for Western blotting [2].
2.9 Kinase Assay
In vitro
Whole cell extracts used for the evaluation of JNK were prepared by following the manufacturer’s directions (SAPK/JNK Assay Kit (non-radiactive), Cell Signaling Technology Beverly, MA). Briefly, cell extracts (250 μg of protein) were incubated with glutathione beads containing 1ug of GST-c-Jun (1-79) fusion protein, centrifuged, washed, and incubated in kinase buffer with 10 μM ATP for 30 minutes at 30°C. Phosphorylated substrate was detected by SDS-PAGE and immunoblotting using phospho-specific c-Jun antibody. Quantification of the JNK kinase activity relative to the parental and empty vector control was performed by densitometric scanning (Alpha Innotech IS-1000 Digital Imaging System, version 2.00).
In vivo
HPV4-12 human mammary epithelial cell lines, empty vector control (pLPCX), and SEPT9_v1 and SEPT9_v3 stable transductants were transiently transfected using FuGENE 6 according to manufacturer’s instructions (Roche, Nutley, NJ) [2]. Cell were plated in six-well culture plates and prepared following the manufacture’s instructions for the c-Jun TransLucent in vivo Kinase Assay Kit (Panomics Inc., Redwood City, CA). Briefly, the system is dependent upon the pTL-TAD expression vector, which contains the transcription activation domain (TAD) of cJun (amino acids 1-223), fused to the Gal4 DNA binding domain. The TAD-Gal4 fusion protein is constitutively expressed in cells and binds to the Gal4 binding domain, upstream of the luciferase gene in the pTL-Luc reporter plasmid. When TAD is phosphorylated by its specific kinase, JNK, it will activate transcription of luciferase. The cells were co-transfected with 0.5 μg of the pTL-c-Jun or the Translucent Control Vector (pTL-BD, which only has the Gal4 DNA binding domain), 1.0 μg of the pTL-Luc reporter plasmid, and 25 ng of pRL-TK (a plasmid containing Renilla luciferase reporter vector). Cells were lysed by adding lysis buffer from Promega Corporation (Madison, WI). Quantification of the JNK kinase activity relative to the parental and empty vector controls was performed by following the manufacture’s instructions in the Dual-Luciferase Reporter Assay System, (Promega Corporation, Madison, WI). Luciferase activity was assessed using a 1420 multi-label counter luminometer from PerkimElmer Life and Analytical Sciences (Oak Brook, IL).
2.10 JNK/c-Jun Transcriptome Activation Detection in vivo: Luciferase Assay
Whole cell extracts were treated following the manufacture’s instructions for the Pathway Profiling Luciferase System 3 kit for profiling cell proliferation and differentiation signaling pathways (Clontech Laboratories, Inc., Mountain View, CA). Briefly, HPV4-12 cells, the empty vectors controls (pLPCX), and SEPT9_v1 or SEPT9_v3 stable transductants were transiently transfected using FuGENE 6 (Roche, Nutley NJ) in a 6-well culture plate format. One microgram of either the pTAL-Luc vector used as a negative control or the pAP1-Luc vector (containing the cis-acting enhancer AP-1 element) and 25 ng of pRL-TK (a plasmid containing the Renilla Luciferase reporter vector) were used in this approach. The values obtained for the negative control, pTAL-Luc, were subtracted from the experimental results to normalize the data. Cells were lysed by adding lysis buffer from Promega Corporation (Madison, WI). Quantification of the relative fold of induction with the AP-1 enhancer to the parental and empty vector controls was performed by following the manufacturer’s instructions in the Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, WI). Luciferase activity was determined on a 1420 multi-label counter luminometer from PerkimElmer Life and Analytical Sciences (Oak Brook, IL)
2.12 Real-time Quantitative PCR
cDNA samples from IHMEC and BCCs were amplified in triplicate from the same starting material of total RNA following the manufacturer’s instructions (Omniscript RT kit, Qiagen, Valencia, CA). Samples were amplified using TaqMan MGB FAM dye-labeled probes from Applied Biosystems (Foster City, CA) in an ABI7900HT model Real-Time PCR machine. The following probes were used: Hs99999905_m1 (GAPDH) and Hs00277039_m1 (CCND1).
2.11 Cell synchronization
Cells were synchronized with a double thymidine block as described previously [27]. Briefly, cells were incubated in medium containing 2 mM thymidine for 12 hours, released into their normal medium for 8-10 hours, and then incubated for 12 hours in medium containing 2 mM thymidine. Samples were collected at regular intervals after releasing the cells from the thymidine block and the cell cycle distribution was detected by flow cytometry following staining with propidium iodide. Whole cell lysates were collected at the same timepoints and were analyzed by Western blotting to look at the SEPT9_v1 expression profile through the cell cycle.
3. Results
3.1 SEPT9_v1 interacts and co-localizes with JNK
To begin examining the role of SEPT9_v1 in breast cancer cells we first elected to elucidate SEPT9_v1 associated cellular mechanisms that are relevant to cell proliferation. We hypothesized that increased SEPT9_v1 expression may exert its effects on proliferation via association with the JNK signaling pathway. To test the hypothesis that altered SEPT9_v1 expression directly affects the JNK signaling pathway, we first studied potential JNK and SEPT9_v1 interactions by immunoprecipitation and immunofluorescence experiments. We used the immortalized human mammary epithelial cell line (IHMEC), HPV4-12, which develops tumorigenic properties after up-regulation of SEPT9_v1, and the breast cancer cell line Hs578T, which has significantly less endogenous SEPT9_v1 expression than HPV4-12, as we were specifically interested in mechanisms that may be relevant to malignant progression in breast cancer cells [2].
Whole cell lysates from HPV4-12 and Hs578T parental cells and their respective retroviral transductants stably expressing SEPT9_v1 were subjected to immunoprecipitation experiments using an anti-JNK1,2 antibody, which recognizes both JNK1 and JNK2. Immunoprecipitated proteins were then probed with an anti-SEPT9_v1 antibody. We found a strong specific signal corresponding to the SEPT9_v1 isoform in the whole cell lysates from cells expressing both exogenously and endogenously SEPT9_v1 that had been immunoprecipitated with anti-JNK1,2 antibodies (Fig 1A). These results suggested that SEPT9_v1 could interact with JNK proteins.
Figure 1. The SEPT9 isoforms SEPT9_v1 and SEPT9_v3 interact with JNK1,2.
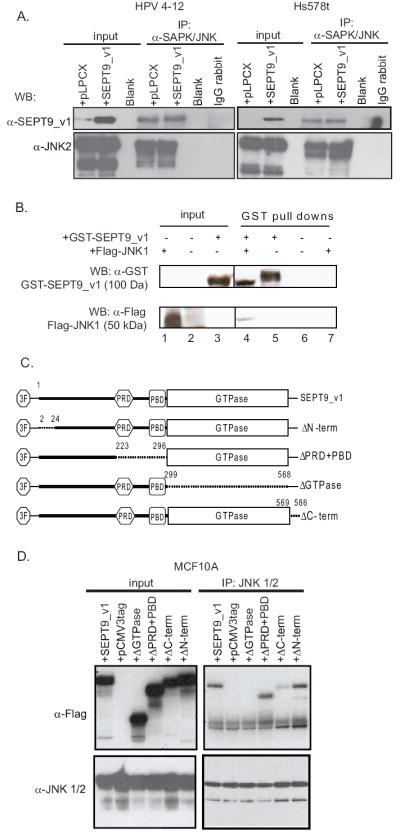
(A) Top panels: Co-immunoprecipitations with JNK1,2 antibody using whole cell lysates from HPV4-12 IHMEC cells (left side) and Hs578T breast cancer cells (right side) both empty vector cells and SEPT9_v1 retroviral transductants. Results show that SEPT9_v1 immunoprecipitates with JNK1,2 in both cell lines. Bottom panels: Western blotting using whole cell lysates and the indicated antibodies indicate the amount of starting material (“input”) used for the immunoprecipitation. (B) A GST pull-down using a GST-SEPT9_v1 fusion protein shows an interaction between SEPT9_v1 and ectopically expressed of Flag-tagged JNK1. Top panel: Western blotting using a GST antibody shows the presence of the GST-SEPT9_v1 fusion protein. Lane number 3 indicates GST-SEPT9_v1 fusion protein expression in the bacterial whole cell extract (WCE) as starting material. Bottom panel: Western blotting using an anti-Flag antibody to detect Flag-JNK1 shows the interaction between GST-SEPT9_v1 and JNK1 (lane 4). Lane number 1 indicates the expression of Flag-JNK1 in whole cells extracts (WCE) from transfected HPV4-12 cells, indicating the amount of JNK1 in the starting material for the GST pull-down. (C) Schematic representation of SEPT9_v1 and deletion mutants constructs. Every construct has three FLAGs as tag. Three FLAGs tag was denoted as 3F, PRD refers to proline rich domain, PBD refers to polybasic domain and GTPase – to GTPase binding domain. Numbers overlying the dotted lines refer to the amino acids positions flanking the deletion. (D) Co-Immunoprecipitation with JNK1/2 antibody of whole cell lysates of MCF10A transiently transfected with different constructs mentioned above.
To provide additional evidence of this interaction we performed GST pull-down experiments using a purified GST-SEPT9_v1 fusion protein and whole cell lysates from HPV4-12 cells transiently transfected with a construct for Flag-tagged JNK. The results shown in Figure 1B demonstrate that JNK and SEPT9_v1 also interact in vitro.
We next created Flag-tagged deletion constructs of SEPT9_v1 and performed co-immunoprecipitation experiments to determine which part of SEPT9_v1 interacted with JNK1,2 (Fig 1C). Whole cell extracts of MCF10A cells (IHMECs) transiently transfected with N-terminally Flag-tagged SEPT9_v1 and deletion mutants were co-immunoprecipitated with anti-JNK antibody and probed with the anti-FLAG antibody [2, 28]. We identified an interaction between ectopically expressed Flag-tagged SEPT9_v1 and JNK proteins, further indicating that SEPT9 isoforms can interact with JNK1 and JNK2. We found that all of the deletion mutants could interact with JNK1,2 except for the GTPase-domain deletion mutant (Fig. 1D).
We next compared the cellular localization of endogenous and ectopically expressed SEPT9_v1 with endogenous JNK in unstressed HPV4-12 cells. Cells were stained for immunofluorescence with anti-SEPT9_v1 and anti-JNK antibodies, phalloidin to label F-actin, and DAPI. As shown in panels depicting merged images, endogenous SEPT9_v1 and JNK co-localize at the nuclear periphery in the parental cells (Fig 2, subpanel e). When Flag-tagged SEPT9-v1 is overexpressed, it not only co-localizes with endogenous JNK at the nuclear periphery, but also inside the nucleus in interphase cells (Fig 2, subpanel j). Of interest, mitotic cells expressing SEPT9_v1 retroviral constructs showed co-localization of SEPT9_v1 and JNK at the midzone and the central spindle (Fig 2, subpanel o).
Figure 2. SEPT9_v1 co-localizes with JNK in interphase cells at the nuclear periphery and in mitotic cells at the midzone.
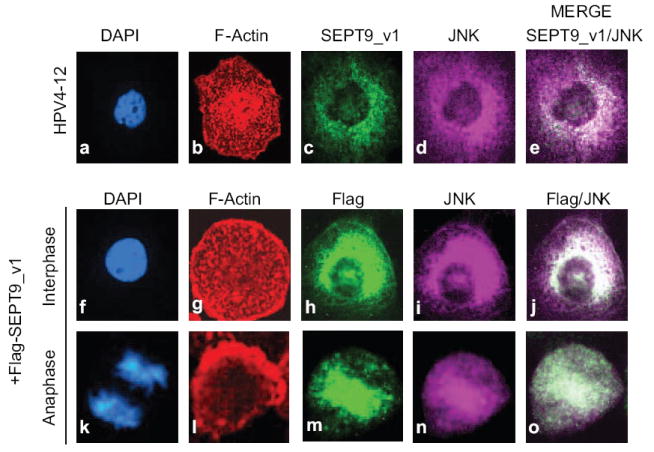
Top panel: Immunofluorescence of HPV4-12 cells in interphase shows the localization of F-Actin (b; red), endogenous SEPT9_v1 (c; green), JNK1,2 (d; purple), and the merged image (e) indicates the co-localization of SEPT9-v1 and JNK1,2 as indicated by the white color. Cells were counterstained with DAPI (subpanels a, f, and k; blue) to stain DNA. Middle panel (interphase cell) and bottom panel (anaphase cell): Immunofluorescence using HPV4-12 cells retrovirally transduced with Flag-tagged SEPT9_v1 constructs show localization of ectopic SEPT9_v1 in HPV4-12 cells, (subpanels h and m) and endogenous JNK1,2, (subpanels i and n). Co-localization between these proteins is indicated by the white color and the arrow in the merged image (subpanels j and o).
3.2 SEPT9_v1 stabilizes JNK in mammary epithelial cells
Next, we wanted to determine if SEPT9_v1 expression had any affect on JNK1,2 protein levels. As shown, increased SEPT9_v1 expression did not alter significantly alter the steady state JNK1,2 protein levels (Fig 1A). Therefore, we hypothesized that SEPT9_v1 modified JNK1 and/or JNK2 either post-transcriptionally or post-translationally. We studied the effects on JNK1,2 protein stability by using cycloheximide. Since cycloheximide inhibits new protein synthesis, JNK1,2 levels over time would predominantly reflect the degradation process. HPV4-12 clones stably expressing the retrovirally transduced pLPCX empty vector control or SEPT9_v1 constructs were exposed to cycloheximide across various time-points and JNK protein levels were subsequently analyzed by Western blot (Fig 3A and data not shown). Within 30 minutes of exposure to cycloheximide, JNK1,2 protein levels in control cells were decreased by 50%. However, in cells exogenously expressing SEPT9_v1, JNK1,2 protein levels did not decrease to 50% levels until 60 min of cycloheximide exposure. This data suggested that SEPT9_v1 is likely involved in cellular processes that regulate the stability and degradation of JNK1,2 proteins.
Figure 3. Up-regulation of SEPT9_v1 in immortalized human mammary epithelial cell culture models, cell line HPV4-12, stabilizes JNK expression and increases JNK kinase activity.
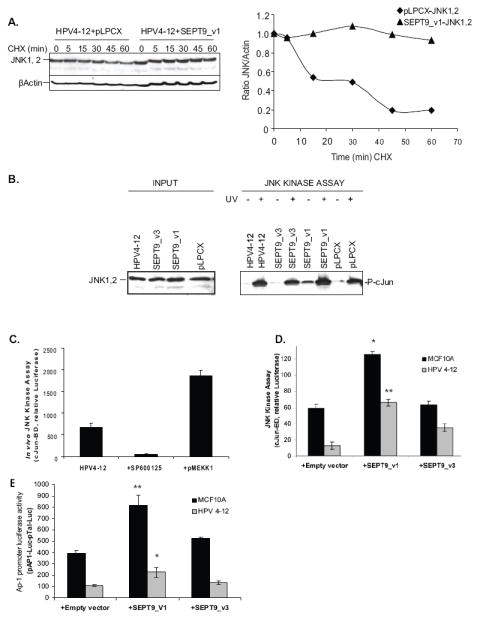
(A) Left panel: SEPT9_v1 stabilizes JNK1,2 protein levels. Western blotting for JNK1,2 protein levels at various timepoints during cycloheximide treatment in cells without (left) or with (right) stably expressed exogenous SEPT9_v1 and SEPT9_v3. β Actin was used as loading control. Right panel: A graphical representation of the data from the left panel, as determined by densitometry, highlights how SEPT9_v1 overexpression stabilized JNK1,2 within the cells. Within less than 30 minutes of exposure to cycloheximide, JNK1, 2 (♦) protein levels in HPV4-12+pPLCX cells were decreased by 50%. In cells transduced with SEPT9_v1, JNK1, 2 (▲) protein levels remained constant and did not decrease until 60 min of cycloheximide exposure (B) An in vitro kinase assay shows that SEPT9_v1 up-regulation increases JNK activity as indicated by increased levels of phosphorylated c-Jun in unstressed and UV-treated cells. Left panel: Whole cell extracts (input) show the initial endogenous JNK1,2 levels in the cells used for the kinase assay. Right panel: Cell extracts (250 μg of protein), were incubated with glutathione beads containing 1 μg of GST-c-Jun (1-79) fusion protein. Phosphorylated substrate was detected by SDS-PAGE and immunoblotting using phospho-specific c-Jun antibody. Quantification of the JNK1,2 kinase activity was performed by densitometric scanning showing that up-regulation of SEPT9_v1 caused a 7-fold increased induction of c-Jun phosphorylation in unstressed cells compared to parental and empty vector (pLPCX) controls. (C) The in vivo JNK kinase assay was validated for JNK specificity by transiently transfecting HPV4-12 cells with a well-characterized JNK inducer, pMEKK1 or by inhibiting JNK activity with the chemical inhibitor, SP600125. JNK kinase activity was induced by more than three fold with the pMEKK1 activator and the JNK inhibitor, SP600125, inhibited JNK kinase activity by 12 fold in HPV4-12 cells. (D) In vivo kinase assay shows SEPT9_v1 up-regulation increases JNK activity. Up-regulation of SEPT9_v1 in HPV4-12 and MCF10A IHMEC cells caused an induction of phosphorylated c-Jun-mediated reporter gene (Luciferase) expression primarily by the JNK pathway using the c-Jun Translucent in-vivo Kinase Assay Kit. The plot for this luciferase assay shows that SEPT9_v1 causes an increase of JNK1,2 kinase activity towards c-Jun by more than four-fold in HPV4-12 cells. SEPT9_v3 causes only a moderate increase in JNK1,2 kinase activity in vivo (ns: non significant *p<0.001, **p<0.0001; Student’s t-test). (E) SEPT9_v1 overexpression increases the expression of phosphorylated c-Jun target genes (the JNK/c-Jun signaling transcriptome) that have an AP-1 enhancer element. Cells were transiently transfected with the pTAL-Luc vector used as a negative control or the pAP1- Luc vector containing the cis-acting enhancer AP1 element and pRL-TK, a plasmid containing the Renilla Luciferase reporter. Compared to the empty vector control, up-regulation of SEPT9_v1 doubled the expression of the reporter construct when compared to HPV4-12 parental and empty vector controls. SEPT9_v3 up-regulation had an insignificant effect when compared to the empty vector control. In MCF10A cells, compared to empty vector control SEPT9_v1 up-regulation doubled the expression of the reporter construct or c-Jun activated transcription and SEPT9_v3 had a milder but significant effect (ns: non significant, *p<0.05, **p<0.01; Student’s t-test).
3.3 SEPT9_v1 up-regulation increases JNK kinase activity in vivo and in vitro
To examine further the effects of SEPT9_v1 up-regulation on JNK1,2 signaling in unstressed cells, we tested JNK1,2 kinase activity in HPV4-12 cells stably transduced with SEPT9_v1 or SEPT9_v3 isoforms, compared to the parental and empty vector controls. Strikingly, the ectopic expression of SEPT9_v1 caused an approximate sevenfold increase in c-Jun phosphorylation by JNK1,2 in unstressed cells (Fig 3B). Interestingly, even though SEPT9_v3 was also able to interact with JNK1,2 similar to SEPT9_v1 (data not shown) overexpression of the SEPT9_v3 isoform did not enhance JNK1,2 kinase activity (Fig. 3B). This suggests that SEPT9_v1’s extended unique amino terminus, which consists of 25 unique amino acids when compared to the otherwise identical SEPT9_v3 isoform, is likely involved in the SEPT9 associated activation of JNK1,2.
To quantify and further explore these findings, we used an in vivo JNK kinase assay where c-Jun binding sites are located in the promoter upstream of a Luciferase reporter. HPV4-12 cells stably expressing the empty vector, SEPT9_v1, or SEPT9_v3 isoforms were transiently co-transfected with the reporter luciferase plasmid and the in vivo kinase vector pTAD-cJun, which contains a transcriptional activator specific for the JNK signaling pathway. This in vivo kinase assay was validated by transiently transfecting HPV4-12 cells with pMEKK1, which induced the JNK kinase activity more than 3-fold, and also by treating the cells with a well characterized JNK inhibitor, SP600125, which inhibited JNK kinase activity by 12-fold (Fig 3C) [8, 24]. Quantitative data obtained with this dual reporter Luciferase system are shown in Figure 3D. In one immortalized human mammary epithelial cell line, MCF10A cells, SEPT9_v3 did not significantly increase reporter expression compared to controls although SEPT9_v1 did. Up-regulation of SEPT9_v1 and SEPT9_v3 in HPV4-12 cells significantly activated the kinase activity, as determined by the endpoint of Luciferase expression, by more than 5-fold and 3-fold, respectively, in HPV-12 parental cells when compared to the empty vector control (p<0.001; Student’s t-test). Interestingly, even though SEPT9_v1 can interact with JNK1,2 when it has a Flag-tag on the N-terminus, the presence of the tag abrogated the induction of JNK1,2 signaling (data not shown). This suggested that the three dimensional conformation of the N-terminus is important for JNK1,2 kinase activation and subsequent c-Jun phosphorylation even though the common C-terminal region encompassing the GTPase domain is required for the interaction between the proteins.
3.4 SEPT9_v1 Expression Amplifies JNK/c-Jun Signaling
The mechanism by which SEPT9_v1 mediates its tumorigenic properties via JNK1,2 activation, leading to c-Jun induction, may include the transcriptional up-regulation of c-Jun target gene expression. Transcriptional activity was measured by transiently co-transfecting cells with pRL-TK, expressing Renilla luciferase, and either a reporter plasmid containing the luciferase gene under a cis-acting enhancer AP-1 element (pAP1-Luc) or a negative control to determine the background signals associated with the culture medium and reporter activity (pTAL-Luc). The difference between the two reporters was used to determine differences in transcription activity. SEPT9_v3 up-regulation had an insignificant effect on the transcription activity when compared to empty vector controls in HPV 4-12. However, up-regulation of SEPT9_v1 increases the reporter gene expression or c-Jun activated transcription, more than two fold when compared to empty vector controls (Fig 3E, p=0.05; Student’s t-test). In MCF10A, up-regulation of SEPT9_v1 dramatically increases the c-Jun activated transcription but up-regulation of SEPT9_v3 showed a milder but significant effect (Fig3E, p<0.01; Student’s t-test). This data, combined with the lack of JNK kinase activation by SEPT9_v3 in Figure 3B, suggests that the unique N-termini of the two SEPT9 isoforms have different abilities to regulate the JNK signaling pathway. For this reason, we decided to focus our remaining studies on the effects of SEPT9_v1 on JNK signaling. We decided to include SEPT9_v3 in subsequent assays as a comparison to the effects of SEPT9_v1 up-regulation in this pathway.
3.5 Up-regulation of SEPT9_v1 Increases the Expression of Cyclin D1, a c-Jun Target Gene, at the Protein and mRNA Levels
The gene encoding cyclin D1 (CCND1) has emerged as an important target for the JNK/c-Jun pathway in driving cell proliferation [23, 24]. Ectopic expression of c-Jun induces expression of CCND1 through an AP-1 site in the promoter, which in turn is critical in driving G0 to G1 cell cycle progression [10, 29].
We therefore investigated whether the enhanced transcriptional activity of the JNK1,2 signaling pathway, as a result of SEPT9_v1 overexpression, increases Cyclin D1 levels in human mammary epithelial cells. Whole cell extracts of parental HPV4-12 cells, empty vector controls, and SEPT9_v1 overexpressing stable transductants were analyzed by Western blotting and probed with anti-Cyclin D1, anti-phospho-Cyclin D1 (Thr 286), and anti-Cyclin E antibodies. The same lysates used in the JNK Kinase assays and immunoprecipitation experiments were used for profiling the expression of various cyclin proteins to reproduce the testing conditions in unstressed cells. Asynchronous SEPT9_v1 expressing cells showed higher levels of Cyclin D1 expression and a 10-fold increase in phosphorylated Cyclin D1 when compared to the controls (Fig 4A). This finding agrees with the enhanced transcription from the luciferase reporter assay following SEPT9_v1 overexpression described in Figure 3E. Cyclin E expression was relatively unchanged in cells over-expressing SEPT9_v1. However, the high endogenous expression of cyclin E we observed in this human mammary epithelial cell line is consistent with previously described data showing that high levels of cyclin E expression is a consequence of the immortalization process, which precludes further up-regulation of this cyclin or our ability detect any subtle changes in cyclin E levels [30].
Figure 4. Increased SEPT9_v1 expression results in amplified expression of cyclin D1 and B1. SEPT9_v1 expression varies with the cell cycle in HPV4-12 IHMEC cells.
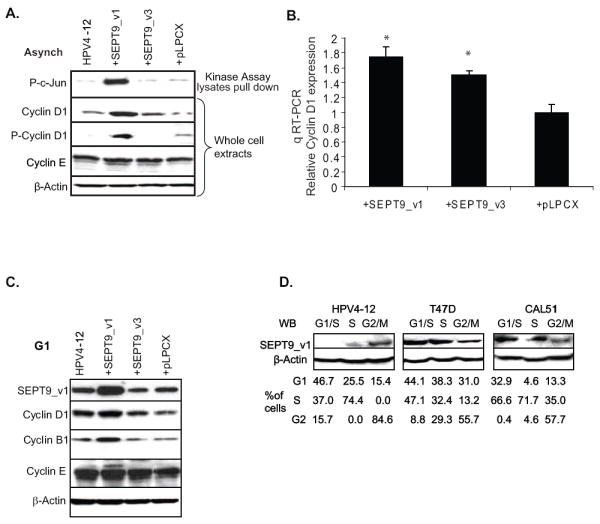
(A) Western blot shows that asynchronous HPV4-12 cells exogenously expressing SEPT9_v1 had higher levels of cyclin D1 expression and a 10-fold increase in phosphorylated cyclin D1 when compared to the controls. The top panel indicates the amount of phosphorylated c-Jun present in the cells, as previously shown in Figure 3B, while the remaining panels are whole cell lysates. β-Actin was used for a loading control. (B) A graphical representation of data from real-time quantitative RT-PCR shows that CCND1 (cyclinD1) expression is approximately doubled in HPV4-12 cells exogenously expressing SEPT9_v1 when compared to controls. Relative mRNA expression of CCND1 was determined against the GAPDH control (*p<0.05; Student’s t-test). (C) SEPT9_v1 over-expression results in elevated Cyclin D1 and Cyclin B1 levels in synchronized cells. Western blot analysis was performed for cyclin expression in whole cell lysates from cells synchronized at the G1/S boundary using a double thymidine block. HPV4-12 parental, empty vector control cells, and SEPT9_v1 stable transductants were analyzed for the expression profile of several cyclin proteins. SEPT9_v1 over-expressing cells had nearly double the amount of Cyclin D1 protein and a 2.5-fold increase in Cyclin B1 levels as determined by densitometry. β-Actin expression was used as a loading control. (D) SEPT9_v1 expression is deregulated in some breast cancer cell lines during the course of the cell cycle compared to HPV4-12 IHMEC cells. Whole cell lysates from three different breast cell lines (HPV4-12, and the cancer cell lines T47D, and CAL51) at various time points after releasing them from G1/S synchronization by a double thymidine block. In HPV4-12 IHMEC cells, SEPT9_v1 protein expression is highest in the G2/M phases whereas the breast cancer cell lines, T47D and CAL51, have altered patterns of SEPT9_v1 expression through the cell cycle such that expression is high in G1 and S phases but comparatively lower in cell populations enriched for the G2/M phases. β Actin was used as a loading control. The percentage of cells in the indicated phases of the cell cycle are listed below each lane and were determined by flow cytometry using propidium iodide staining for DNA content.
Confirmation of SEPT9_v1 activating transcription of the downstream targets of the JNK/c-Jun signaling pathway was achieved by determining the CCNDI mRNA relative expression compared to the GAPDH control, using quantitative RT-PCR. We found nearly a 2-fold increase in CCNDI expression in HPV4-12 cells expressing the SEPT9_v1 and SEPT9_v3 constructs (Fig 4B, p<0.05; Student’s t test).
3.6 SEPT9_v1 Up-regulation Correlates with Higher Cyclin Levels in the G1/S Phase of the Cell Cycle
Evidence to date indicates that up-regulation of cyclin D1 after the activating phosphorylation of c-Jun on residues Ser63 and Pro73 plays an important role in cell proliferation [9, 22]. Cyclin D1 serves as a key sensor and integrator of cellular signals of cells in early to mid-G1 phase [9, 31]. We studied the consequences of SEPT9_v1 up-regulation in G1/S cells synchronized using a double thymidine block method. Whole cell lysates of synchronized HPV4-12 parental, empty vector controls, SEPT9_v1 and SEPT9_v3 stable transductants were analyzed by Western blotting for the expression profile of several cyclin proteins. Cyclin E expression was the same in all of the cells regardless of the SEPT9 isoform expression or the cell stage as seen earlier in asynchronous cultures (Fig. 4A and 4C). Cells exogenously expressing SEPT9_v1 had nearly double the amount of Cyclin D1 protein and a 2.5-fold increase in Cyclin B1 levels (Fig 4C). Again, no significant changes were noted when SEPT9_v3 was expressed.
3.7 SEPT9_v1 Expression is De-regulated in some Breast Cancer Cell Lines
Considering that SEPT9_v1 altered the expression of the cell cycle protein cyclin D1 and we previously showed that SEPT9_v1 over-expressing cells have increased proliferation rates, we hypothesized that SEPT9_v1 expression may be cell cycle regulated in immortalized human mammary epithelial cells. However, breast cancer cells may have an aberrant pattern of expression at different stages of the cell cycle compared to IHMECs. We analyzed cultured cells at various time points after releasing them from G1/S synchronization by a double thymidine block. We used the IHMEC line, HPV4-12, to monitor normal SEPT9_v1 expression patterns and two breast cancer cell lines, T47D and CAL51, previously shown to express high endogenous levels of SEPT9_v1 [2]. As shown in Figure 4D, SEPT9_v1 protein expression was highest in the G2/M phases in HPV4-12 cells. T47D and CAL51 breast cancer cell lines showed altered patterns of expression. SEPT9_v1 levels were increased in the G1 and S phases of the cell cycle and still present in G2 but at a lesser extent in both cancer cell lines. These results suggested a different cell cycle-mediated regulation of SEPT9_v1 expression in breast cancers compared to non-tumorigenic cells.
3.8 Up-regulated Expression of SEPT9_v1 Contributes to JNK Pro-proliferative Effects in Mammary Epithelial Cells
We wanted to clarify the biological significance of the deregulation of SEPT9_v1 expression towards JNK kinase activity during the cell cycle. We hypothesized that increased SEPT9_v1 expression promoted cell cycle progression, possibly caused by the interaction with its binding partner, JNK1,2, thereby provoking the alteration in the JNK/c-Jun transcriptome. First, we studied the cell cycle histogram by FACS analysis of parental HPV4-12 and Hs578T cells, the empty vector negative controls (pLPCX), SEPT9_v1 and SEPT9_v3 transductants after releasing the cells from G1 synchronization. We found a promotion of the cell cycle progression when SEPT9_v1 is up-regulated, compared to parental and vector control (Fig 5A, 6A and data not shown). In both lines the cells progressed to the S phase (5 hours after release), entered into G2, (7 hours post-release), and entered the next cycle (15 hours after release) faster than the controls. A quantification of these data, as shown in Figures 5B and 6B, represents the percentage of cells in each phase of the cell cycle for each time point.
Figure 5. Up-regulated expression of SEPT9_v1 contributes to JNK1,2-mediated pro-proliferative effects in mammary epithelial cells.
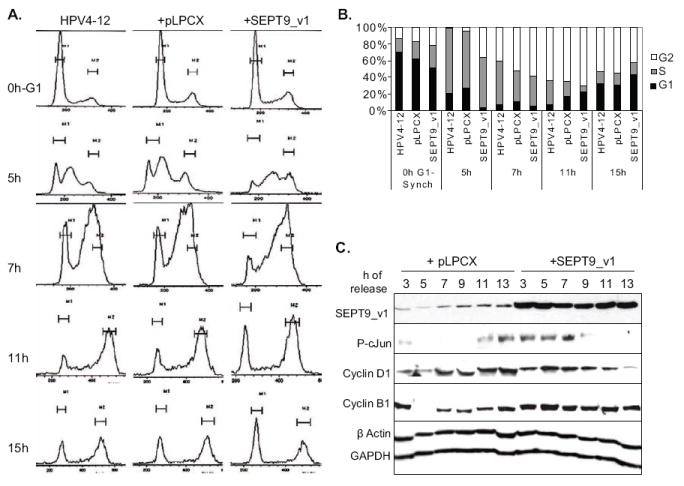
(A) Cell cycle histograms generated by FACS analysis of parental HPV4-12 cells, the empty vector negative controls (pLPCX), and SEPT9_v1 transductants after releasing the cells from G1/S synchronization via double thymidine block. (B) Quantification of the data in panel (A) represents the percentage of cells in each phase of the cell cycle for each time point for each of the cell lines. SEPT9_v1 promotes cell cycle progression when it is up-regulated, compared to parental and vector control. HPV4-12 cells exogenously expressing SEPT9_v1 progressed to the S phase (5 hours after release), entered into G2, (7 hour post-release), and entered the next cycle (15 hours after release) faster than the controls. (C) Up-regulation of SEPT9_v1 induced higher levels of phosphorylated c-Jun, Cyclin D1 and Cyclin B1 at earlier stages of the cells cycle. Western blot analysis was used to determine the levels of SEPT9_v1, phosphorylated c-Jun, cyclin D1 and cyclin B1 at different time points after release from double-thymidine synchronization for the empty vector controls and SEPT9_v1 transductants of HPV4-12 cells. The samples and time points studied here were collected at the same time as those used for subpanels (A) and (B). β Actin and GAPDH were used as loading controls.
Figure 6. High SEPT9_v1 expression promotes cell cycle progression in Hs578T cells.
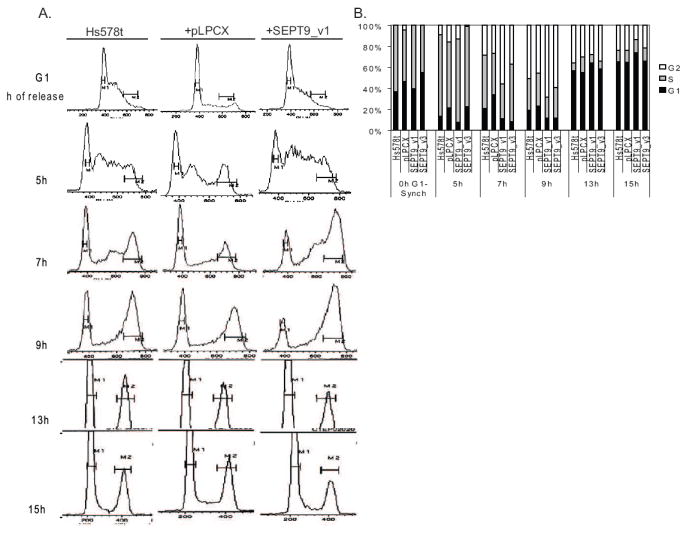
(A): Cell cycle histograms generated by FACS analysis of parental Hs578T cells, the empty vector negative control (pLPCX), and SEPT9_v1 transductants after releasing the cells from G1/S synchronization by double thymidine block. (B) A graphical representation of the data collected by FACS analysis, representing the percentage of cells in each phase of the cell cycle for each time point for each cell line. As shown at the 7 hour and 9 hour timepoints, HS578T cells exogenously expressing SEPT9_v1 enter mitosis sooner than the parental and empty vector controls.
In order to investigate the potential physiological relevance of SEPT9_v1 up-regulation in mammary epithelial cells at different cell cycle phases, we determined the levels of SEPT9_v1, phosphorylated c-Jun, cyclin D1 and cyclin B1 at different time points after release from synchronization for the empty vector controls and SEPT9_v1 transductants of HPV4-12 and Hs578T cells. Western blotting showed that up-regulation of SEPT9_v1 induced higher levels of phosphorylated c-Jun, cyclin D1 and cyclin B1 at earlier stages of the cells cycle (Fig 5C). The time points studied here correlated with the cell cycle histogram and the percentage of cells at each cell cycle stage mentioned above. Together, these data suggest that when SEPT9_v1 expression is up-regulated, as seen in breast cancer cell lines and primary breast cancers, SEPT9_v1 could hyper-activate the JNK signaling pathway, causing up-regulated expression of the downstream target genes, such as cyclin D1 [2]. This would ultimately push the cells through the cell cycle at a faster rate.
4. Discussion
In this study, we identified and characterized a novel interaction between the SEPT9 isoform, SEPT9_v1, and the JNK pathway in mammary epithelial cells. In an effort to understand the potential functional relevance of this to cell proliferation in breast cancer, we began exploring how this interaction may affect cell cycle progression in mammary epithelial cells. We found that unmodified JNK1,2 interacts with SEPT9_v1. This suggests that SEPT9_v1 can stimulate JNK signaling even when the cells are unstressed. Similar to our findings that SEPT9_v1 can promote the stabilization of JNK1,2, Amir et al. also found that SEPT9_v1 stabilized the interacting protein, HIF-1, thereby promoting cell proliferation [1]. Further studies are needed to investigate a possible mechanism that could explain the SEPT9_v1-mediated stabilization and regulation of these proteins.
We show that deletion of GTPase domain abolished the interaction between SEPT9_v1 and JNK. However it remains to be investigated whether GTPase activity per se is necessary for the binding.
Another interesting finding concerning the SEPT9_v1-JNK1,2 interaction is that both proteins colocalized to the midzone and central spindle late in mitosis (Fig. 2, subpanel o). This is particularly relevant because SEPT9 plays a major role in cleavage furrow formation in this region and JNK2 was previously shown to be involved in central spindle formation during anaphase [18]. Further studies focusing on the role of this protein-protein interaction could reveal crucial insights into the process of cytokinesis and may help elucidate the mechanism(s) by which SEPT9_v1 over-expressing cells become aneuploid and bi-nucleated [2].
A major finding presented here to support the interaction between SEPT9 and the JNK signaling pathway showed that retroviral expression of the SEPT9_v1 isoform consistently had stronger affects on JNK signaling and c-Jun transcription activation when compared to retroviral expression of the SEPT9_v3 isoform, a protein identical to SEPT9_v1 except for 25 amino acids at the N-terminus. This is in agreement with our previous report showing that retroviral expression of the full-length SEPT9_v1 variant resulted in the striking development of oncogenic phenotypes when expressed in breast cells as compared to the retroviral expression of SEPT9_v3 [2]. As previously mentioned, only 25 N-terminal amino acids differentiate these two isoforms; this suggests that these residues are crucial for the contrasting regulation of JNK1,2 signaling between these them. Interestingly, our work also indicates that shared domains or regions between these two proteins, particularly the GTPase domain, are necessary for the interaction between SEPT9 and JNK1,2. Importantly, this shared GTPase domain between SEPT9_v1, SEPT9_v3 and other septin isoforms might be responsible for the intermediate phenotypes seen in this study when SEPT9_v3 is up-regulated. Further studies are necessary to elucidate the dynamics of different SEPT9 isoforms and the activation of the JNK signaling pathway.
Our data correlate with previous reports in which the cyclin D1 gene, CCND1, has emerged as an important target of the JNK/c-Jun-AP1 pathway in driving proliferation [23, 24]. Previous studies associated this pathway with bona fide oncogenes, some of them mechanistically associated with Ras mutations [9]. Efforts to elucidate if SEPT9_v1 is responding to Ras mutations were performed by transiently transfecting the cells with HA-RAS but no significant alteration of endogenous SEPT9_v1 expression was observed (data not shown). This suggested that the up-regulation of SEPT9_v1 induced hyper-proliferation by a signaling mechanism involving JNK stabilization, but mediators or other components upstream of SEPT9_v1 are yet to be elucidated. However, we do propose that the JNK/c-Jun/AP-1 pathway and its downstream target genes are relevant physiological targets of SEPT9_v1 as their up-regulation gave a proliferative advantage to the cells by making them cycle faster through the G1 phase of the cell cycle and altered the expression pattern of regulatory cyclin proteins including Cyclin D1 and Cyclin B.
Taken together our data show a novel association of SEPT9_v1 with the JNK1,2 signaling pathway, an important signaling pathway that regulates cellular proliferation among other cellular roles. This further supports the hypothesized role of SEPT9 in cell signaling pathways that regulate cellular proliferation as previously suggested by studies of SEPT9_v1 and its association with HIF-1 alpha [1]. Here, we propose a mechanism by which increased SEPT9_v1 expression in breast cancer cells may impair cell cycle regulation, resulting in the increased transcription of key cyclins that drive progression through the cell cycle (Fig 7). Further characterization of the SEPT9_v1 domains important for this function and its interaction with JNK1,2 will be critical for both the understanding of normal cell proliferation control and its deregulation in cancer.
Figure 7. Proposed model for how increased SEPT9_v1 expression increases cell proliferation, a hallmark of cancer cells.

SEPT9_v1 stabilizes JNK1,2 kinase, allowing the JNK kinases to activate c-Jun by phosphorylation. c-Jun activity thus is not properly regulated and induces AP-1 mediated transcription of cyclin D1, and other AP-1 controlled target genes, to cause increased proliferation and “faster” movement through the cell cycle. In an unidentified, or related, mechanism, cyclin B1 expression is also increased, causing the cells to prematurely enter mitosis.
5. Conclusions
In this study, we show that up-regulation of SEPT9_v1 expression throughout the cell cycle, particularly in the G1 phase, significantly alters cell cycle progression via the JNK signaling pathway. We found a novel interaction between SEPT9_v1 and JNK1,2 kinases, which controls the activation of the transcription factor c-Jun by phosphorylation thereby resulting in the transcription of target genes, such as CCND1 (cyclin D1), with AP-1 sites in their promoters. Through its interaction with (and stabilization of) JNK1,2, SEPT9_v1 causes cells to progress at a faster rate through the cell cycle due to the deregulated expression of cyclin proteins, particularly cyclin D1 and cyclin B1, which are responsible for cell cycle control. These findings shed new light on possible mechanisms by which SEPT9_v1 alters cellular proliferation and contributes to malignant progression in breast cancers.
Acknowledgments
This work was supported by NIH NCI grant RO1CA072877 to EMP, the Department of Defense Breast Cancer Research Pre-doctoral Fellowship award to LMP #BC050310, the NIH National Research Service Award #5-T32-GM07544 from the National Institute of General Medicine Sciences to LMP and EAP and by the NIH National Research Service Award F31 to Promote Diversity in Health Related Research #5-F31-CA123639-02 to EAP. We thank Amy Silvers for helpful data analysis and discussion, and Dorraya El-Ashry for critical review of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amir S, Wang R, Matzkin H, Simons JW, Mabjeesh NJ. Cancer Res. 2006;66(2):856–866. doi: 10.1158/0008-5472.CAN-05-2738. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez ME, Peterson EA, Privette LM, Loffreda-Wren JL, Kalikin LM, Petty EM. Cancer Res. 2007;67(18):8554–8564. doi: 10.1158/0008-5472.CAN-07-1474. [DOI] [PubMed] [Google Scholar]
- 3.Ito H, Iwamoto I, Morishita R, Nozawa Y, Narumiya S, Asano T, Nagata K. Oncogene. 2005;24(47):7064–7072. doi: 10.1038/sj.onc.1208862. [DOI] [PubMed] [Google Scholar]
- 4.Kremer BE, Adang LA, Macara IG. Cell. 2007;130(5):837–850. doi: 10.1016/j.cell.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagata K, Inagaki M. Oncogene. 2005;24(1):65–76. doi: 10.1038/sj.onc.1208101. [DOI] [PubMed] [Google Scholar]
- 6.Sudo K, Ito H, Iwamoto I, Morishita R, Asano T, Nagata K. Hum Mutat. 2007;28(10):1005–1013. doi: 10.1002/humu.20554. [DOI] [PubMed] [Google Scholar]
- 7.Sheffield PJ, Oliver CJ, Kremer BE, Sheng S, Shao Z, Macara IG. J Biol Chem. 2003;278(5):3483–3488. doi: 10.1074/jbc.M209701200. [DOI] [PubMed] [Google Scholar]
- 8.Comerford KM, Cummins EP, Taylor CT. Cancer Res. 2004;64(24):9057–9061. doi: 10.1158/0008-5472.CAN-04-1919. [DOI] [PubMed] [Google Scholar]
- 9.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Embo J. 2001;20(13):3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. J Biol Chem. 1995;270(40):23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 11.Du L, Lyle CS, Obey TB, Gaarde WA, Muir JA, Bennett BL, Chambers TC. J Biol Chem. 2004;279(12):11957–11966. doi: 10.1074/jbc.M304935200. [DOI] [PubMed] [Google Scholar]
- 12.Lin A. Bioessays. 2003;25(1):17–24. doi: 10.1002/bies.10204. [DOI] [PubMed] [Google Scholar]
- 13.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Science. 2000;288(5467):870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 14.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Science. 1995;270(5240):1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 15.Rennefahrt U, Illert B, Greiner A, Rapp UR, Troppmair J. Cancer Lett. 2004;215(1):113–124. doi: 10.1016/j.canlet.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Liu HT. Cell Res. 2002;12(1):9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Lin A. Cell Res. 2005;15(1):36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 18.MacCorkle RA, Tan TH. J Biol Chem. 2004;279(38):40112–40121. doi: 10.1074/jbc.M405481200. [DOI] [PubMed] [Google Scholar]
- 19.Oktay M, Wary KK, Dans M, Birge RB, Giancotti FG. J Cell Biol. 1999;145(7):1461–1469. doi: 10.1083/jcb.145.7.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mingo-Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL. Oncogene. 2004;23(2):596–604. doi: 10.1038/sj.onc.1207147. [DOI] [PubMed] [Google Scholar]
- 21.Shaulian E, Karin M. Oncogene. 2001;20(19):2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 22.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. Cell. 1994;76(6):1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 23.Wisdom R, Johnson RS, Moore C. Embo J. 1999;18(1):188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. Hepatology. 2003;37(4):824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 25.Makarova O, Kamberov E, Margolis B. Biotechniques. 2000;29(5):970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- 26.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. Cancer Cell. 2003;3(4):363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 27.Fan M, Du L, Stone AA, Gilbert KM, Chambers TC. Cancer Res. 2000;60(22):6403–6407. [PubMed] [Google Scholar]
- 28.Kalikin LM, Sims HL, Petty EM. Genomics. 2000;63(2):165–172. doi: 10.1006/geno.1999.6077. [DOI] [PubMed] [Google Scholar]
- 29.Nelsen CJ, Rickheim DG, Timchenko NA, Stanley MW, Albrecht JH. Cancer Res. 2001;61(23):8564–8568. [PubMed] [Google Scholar]
- 30.Martin LG, Demers GW, Galloway DA. J Virol. 1998;72(2):975–985. doi: 10.1128/jvi.72.2.975-985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Endocrinology. 2004;145(12):5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]