

Abstract
The ATP binding cassette (ABC)-transporters are energy dependent efflux pumps which regulate the pharmacokinetics of both anti-cancer chemotherapeutic agents, e.g. taxol, and of HIV-1 protease inhibitors (HPIs), e.g. saquinavir. Increased expression of several ABC-transporters, especially P-gp and MRP2, are observed in multidrug resistant (MDR) tumor cells and on HIV-1 infected lymphocytes. In addition, due to their apical expression on vascular endothelial barriers, both P-gp and MRP2 are of crucial importance towards dictating drug access into sequestered tissues. However, although a number of P-gp inhibitors are currently in clinical trials, possible inhibitors of MRP2 are not being thoroughly investigated. The experimental leukotriene receptor antagonist (LTRA), MK-571 is known to be a potent inhibitor of MRP transporters. Using the MRP2 over-expressing cell line, MDCKII-MRP2, we evaluated whether the clinically approved LTRAs, e.g. montelukast (Singulair™) and zafirlukast (Accolate™), can similarly suppress MRP2-mediated efflux. We compared the efficacy of increasing concentrations (20-100 μM) of MK-571, montelukast, and zafirlukast, in suppressing the efflux of calcein-AM, a fluorescent MRP substrate, and the radiolabeled [3H-] drugs, taxol and saquinavir. Montelukast was the most potent inhibitor (p<0.01) of MRP2-mediated efflux of all three substrates. Montelukast also increased (p<0.01) the duration of intracellular retention of both taxol and saquinavir. More than 50% of the drugs were retained in cells even after 90 mins post removal of montelukast from the medium. Our findings implicate that montelukast, a relatively safe anti-asthmatic agent, may be used as an adjunct therapy to suppress the efflux of taxol and saquinavir from MRP2 overexpressing cells.
Keywords: ABC transporter, Drug efflux, Inhibition, Montelukast, Taxol, Saquinavir
Introduction
By transporting drugs against their concentration gradients, the ATP-binding cassette (ABC) transporters can significantly alter the bioavailability and tissue distribution of numerous therapeutic agents 1-5). Since a number of these ABC-transporters can confer multidrug resistant (MDR) phenotype to tumor cells 6, 7) strategies to suppress chemotherapy efflux from MDR-tumors are of crucial significance 8, 9). Several anti-retroviral drugs, especially the HIV-1 protease inhibitors (HPIs) are also substrates for ABC-transporters 5, 10-12). The efflux of HPIs, from both HIV-1 infected cells and from subendothelial viral reservoirs, can severely compromise the antiviral efficacy of HPIs and facilitate the selection of drug resistant viruses. Hence, strategies to inhibit anti-retroviral drug efflux would also be very important. In an attempt to inhibit these drug-efflux mechanisms, several approaches have been utilized in the past 9, 13-16). However, due to their crucial physiological significance, especially in keeping toxic xenobiotics out of important tissues such as the central nervous system (CNS) 17), development of effective ABC-transporter inhibitors without systemic toxicities, has been a formidable challenge. A frequently used approach has been the use of competitive inhibitors of ABC-transporters 18), and several such inhibitors had been screened by their structure-activity-relationships (SAR) and in vitro inhibitory potencies 16, 19). However, despite efficacy in preclinical trials, their in vivo toxicities have hampered the clinical approval of these experimental compounds. A second approaches been the use of clinically approved drugs which are known to be competitive inhibitors of specific ABC-transporters, and is currently showing significant promise in the advancement specific ABC-transporter inhibition.
The best characterized ABC-transporter is P-glycoprotein (P-gp); also known as ABCB1 19). P-gp plays a crucial role in chemoresistance of a variety of different tumors and in regulating drug transport across the blood-brain-barrier (BBB) since it displays a broad substrate spectrum comprising of both neutral and cationic organic compounds. Another ABC-transporter, with substrate specificities similar to that of P-gp, is breast cancer resistant protein (BCRP), a.k.a. ABCG2. Increased BCRP expression has been shown in both placental barriers and cancer stem cells 20). Hence, most of the ABC-transporter inhibitors currently under development are targeted towards P-gp and BCRP 21). Even though the newly discovered MDR-associated proteins (MRPs), a.k.a. the ABCC family of transporters, are also known to transport a variety of therapeutic agents 22, 23), there appears to be only a handful of studies to screen for inhibitors of different MRPs.
The MRP transporter family consists of at least nine identified members, each having slightly different substrate specificities, tissue distribution, and transport kinetics 23). A number of chemotherapeutics are substrates for MRPs 24-28) and several MRPs can decrease intracellular levels of HPIs 10-12). Different tumors are known to overexpress different MRPs and a specific pattern is observed on HIV-1 infected lymphocytes 29, 30). Previous studies by us 5) as well as others 4, 31, 32) showed that vascular endothelial cells express functional MRPs, as well. Furthermore, unlike P-gp and BCRP, which are both expressed on apical surfaces of membranes, specific patterns of expression, on either apical or basal surfaces of membranes, are seen with the MRPs. These findings implicated the importance of MRPs in regulating sub-endothelial drug concentrations and suggested that inhibition of specific MRPs may be possible without manifesting systemic side effects.
Both MRP1 and MRP2 are frequently associated with drug resistance, and transport hydrophilic anionic compounds, large molecules and peptidomimetics 14, 33). Both MRP1 and MRP2 have similar substrate specificities for anti-cancer 34-36) and anti-HIV agents 10, 11, 29, 30). The anti-microtubule agent, taxol and its derivatives, widely used in the treatment of breast and ovarian cancers, are substrates of both MRP1 and MRP2 36). Similarly, several HPIs, e.g. saquinavir and lopinavir, often used in highly active antiretroviral therapy (HAART) combinations in HIV-positive patients, are also substrates of both MRP1 and MRP2 5, 11). However, the inhibition of MRP2 would be critical significance since it is expressed on the apical surfaces of membranes, unlike MRP1 which is mostly expressed on the basal surfaces of membrane barriers 17, 27, 37). Hence, the identification of clinically approved drugs which can be used as MRP2 inhibitors would be of profound advantage.
Since MRPs are known to co-transport drugs with glucuronide, glutathione or sulphate, investigators have tried to develop MRP inhibitors using chemically modified glucuronides or glutathiones 13, 14, 33), however, their in vitro toxicities were significant deterrents to their advancement to the clinic. The clinically approved drug used to treat gout, probenecid (Benuryl™), had shown some promise as an MRP inhibitor 38, 39). However, probenecid's side effects and its inconsistent inhibitory potential 36, 38), decreased its utility as an MRP inhibitor. Since MRPs are also known to transport endogenous substances such as leukotrienes (LTs) 22), several studies had monitored whether leukotriene receptor antagonists (LTRA) can inhibit MRPs. Indeed, the experimental LTRA, MK-571 was found to be a potent MRP inhibitor 40). However, although several clinically approved LTRAs are currently available 41), their MRP inhibitory potencies had not been previously investigated.
In the present study, we monitored the efficacy of the clinically approved LTRAs, montelukast (Singulair™) and zafirlukast (Accolate™), in suppressing the efflux of taxol and saquinavir, from cells stably transfected with MRP2. Our findings showed that montelukast is a potent, specific and durable inhibitor of MRP2-mediated efflux of both taxol and saquinavir.
Materials and methods
Reagents
MK571 was purchased from Biomolecules International (Philadelphia, PA) and dissolved in distilled water to a final concentration of 9.3 mM. Probenecid was obtained from Sigma-Aldrich (USA) and dissolved in ethanol to a final concentration of 7.0 mM. After reconstitution, both drugs were aliquoted and stored at -70°C. Montelukast (Singulair ™; Merck, Whitehouse Station, NJ) and Zafirlukast (Accolate™; AstraZeneca, Wilmington, DE)), were obtained from Tulane University Medical Center (TUMC) pharmacy. Before every experiment, fresh stock solutions of montelukast (16.4 mM) and zafirlukast (17.3 mM) were prepared in distilled water. The calcein-AM efflux assay kit was purchased from Molecular Probes (Invitrogen Life Science, USA). The radiolabeled [3H-] drugs, saquinavir and taxol, were obtained from Moravek Radiochemicals (Brea, CA). Antibodies to human MRP2 were purchased from Abcam (Cambridge, MA) and Alexa Fluor-488 conjugated goat anti-mouse antibody was obtained from Invitrogen (Carlsbad, CA). The PCR primers were synthesized from Midland certified reagent company (Midland, TX) and reagents for reverse transcription (RT) and polymerase chain reaction (PCR) were obtained from Promega (Madison, WI).
Cell culture
The Madin-Darby canine kidney cell line, MDCK-II and the MRP2 overexpressing cell line, MDCKII-MRP2, were kind gifts from Professor P. Borst, Netherlands Cancer Research Institute, The Netherlands 42). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Media tech; Hernden,VA) supplemented with 10% FBS (Alanta Biologicals; Norcross, GA) and 100 μg/ml of the antibiotics, penicillin and streptomycin (Sigma). Cells were cultured in a 37°C incubator with 5% CO2 and were passaged every 3-4 days by trypsinization and by subculturing at a 1:5 dilution. Cells were cryopreserved in liquid-N2, in complete media containing 10% dimethyl sulfoxide (DMSO). Before carrying out experiments, newly thawed cells were checked for MRP2 expression and MRP-mediated efflux.
RT-PCR assay
Total RNA was isolated from cells using the Trizol RNA isolation reagent (Invitrogen; Carlsbad, CA) according to the manufacturer's instructions. The RNAs were quantified using a spectrophotometer and aliquots were frozen at -80°C for later use in reverse transcription (RT) and polymerase chain reaction (PCR) assays. The cDNAs were generated from RNA (0.5μg) by using the murine leukemia virus (MuLV) RT enzyme (0.5 U), Oligo-dT (2.4μg/ml) and dNTPs (100 mM) by incubation at 42°C for 60 min. The RT products (cDNAs) were aliquoted and stored at -20°C. For each PCR amplication reaction, 10 μl of the RT solution was used. The PCR reactions were carried out by using a thermal cycler; Model 9600 (Perkin-elmer; Boston, MA) in the presence of Taq DNA-polymerase (0.5 U), in RED-Taq PCR buffer; containing KCl (500 mM), MgCl2 (11 mM), dNTPs (200 μM) and the gene specific primers. The RT-PCR primer sequences and amplification protocols for human MRP2 and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were according to our previous publications 5). The PCR products were electrophoresed on a 2.0 % agarose gel containing ethidium bromide (10 μg/ml). Band intensities for PCR-products were determined from scanned gel pictures, by using a GS-700 imaging densitometer (Bio-Rad). The intensity of amplified MRP2 mRNAs were normalized to the values obtained with GAPDH mRNA levels in the respective samples.
Immunofluorescence microscopy
Immunofluorescence microscopy (IFM) was carried out to measure MRP-2 expression in cells. The MDCKII and MDCKII-MRP2 cells were grown on collagen-I coated chamber slides (BD Biosciences) and processed as described previously 43). The 4′, 6-diamidino-2-phenylindole hydrochloride (DAPI) (Molecular Probes, Eugene, OR) stain was used to visualize the cell nuclei. The primary antibodies were mouse antibodies generated against human MRP2 protein and the secondary antibody used was a fluorochrome (Alexa Fluor-488) conjugated goat anti-mouse antibody. The primary antibodies were used at a dilution of 1:20 and the secondary antibody used at a 1:200 dilution. Images were acquired with a Zeiss Axioplan II microscope (Carl Zeiss, Thornwood, NY). Series of horizontal optical sections (0.3 μm each) were collected and subsequently deconvolved using Slidebook 4.1 software (Intelligent Imaging Innovations, Denver, CO). Post-collection processing of the images was performed using Adobe PhotoShop.
Calcein-AM efflux assay
Calcein acetoxymethyl ester (Calcein-AM), a lipid soluble fluorescent dye, is a known substrate for both P-gp and MRP. The calcein-AM drug-efflux assay was used to monitor MRP function, according to our previous publication 5). Briefly, cells were cultured in 24-well opaque plates until 90% confluent. Prior to the addition of calcein-AM, cells were pre-exposed to increasing concentrations of the MRP-inhibitors (20-100 μM), MK-571, montelukast, zafirlukast or probenecid, for 15 minutes. Cells were then incubated with calcein-AM (0.25μM) in the dark for 30 minutes at 37°C and 5% CO2. Cells were then washed 3 times in ice-cold PBS and calcein-retentions were determined by using an FLx 800 fluorimeter (Bio-Tek instruments). The absorption maximum was set at 494 nm and emission maximum at 517 nm, and readings were taken from the bottom of the wells. Subsequently, cells were lysed and extracted proteins were quantified by using the BCA protein estimation kit (Pierce; Rockford, IL). The average fluorescence in each well was normalized to the protein contents in the respective samples, and are represented as Mean Fluorescence Units (MFU)/μg of protein.
Drug retention assay
Radiolabeled [3H-] taxol {370 GBq (10.0 Ci/mM)} and saquinavir {37 GBq (1.0 Ci/mM)} were used to determine intracellular accumulation and temporal efflux rates of these MRP2 substrates, similar to our previous studies 5). The MDCKII-MRP2 cells (1×105 cells/well) were seeded onto 24-well clear plates and cultured until 90% confluency. Cells were first incubated with either montelukast (0 -100 μM) or MK-571 (0 - 100 μM) for 15–20 min, followed by a 2.0 hr exposure to either 3H-taxol (74 Bq) or 3H-saquinavir (18.5 Bq). At the end of the 2 hr incubation period, cells were washed with cold PBS and cell extracts were obtained for determination of ‘peak loading’ or total drug accumulation. In order to measure ‘temporal efflux’ or efflux rates of the radiolabeled drugs, slight modifications in the experimental protocol was done. Following the 2.0 hr of loading (0-time point) cells were washed with PBS (at 37° C) and incubated with fresh medium in the absence of the MRP inhibitors. Cells were harvested at 15 min intervals (15, 30, 45, 60 and 90 min) and the intracellular retention of taxol and saquinavir were monitored. In both ‘peak loading’ and ‘temporal efflux’ experiments, cell extracts were obtained by lysing cells with 1.0 ml of NH4OH (1.0 M) and incubation for 5 min at room temperature. Approximately 100 μl of extracts were transferred to scintillation vials containing 10 ml of Ecolite scintillation cocktail (MP Biomedicals; Irvine, CA) and another 100 μl of the extract was used to measure protein levels. The counts per minute (CPM) values were determined by using a Tri-Carb 2800TR Liquid Scintillation counter (Perkin Elmer; Boston, MA) and radioactivities were normalized to the protein contents in respective in cell extracts, and represented as CPM/μg of protein.
Statistical analysis
All statistical analyses were carried out using the INSTAT Graph Pad-2 software (Graph Pad, San Diego, CA). Data are the mean values obtained from triplicate or quadruplicate wells within the same experiment. Each experiment was performed at least three to five times to obtain the ± SEM of values. Significant changes from control values were determined by using a two-tailed Student t test and comparison between three or more groups were carried by two-way analysis of variance (ANOVA). Results obtained were considered to be significant when P was less than 0.05 (P < 0.05).
Results
MRP2 gene expression and protein levels in MDCKII and MDCKII-MRP2 cells
The canine MDCKII cells were stably transfected with human MRP2 cDNA to generate the MDCKII-MRP2 cells 49). We first wanted to confirm that the MDCKII-MRP2 cells used in our experiments over-express MRP2 mRNA and protein. Using established RT-PCR primers and protocols 5) we first compared human MRP2 mRNA levels in parental (MDCKII) cells and in MRP2 over-expressing (MDCKII-MRP2) cells (Fig.1A). Furthermore, by IFM analysis using an anti-MRP2 primary antibody, we monitored MRP2 protein levels in both cell types (Fig.1B). The human MRP2 gene was specifically expressed in MDCKII-MRP2 cells, but no detectable band was seen in MDCKII cells. Although, faintly detectable MRP2 staining was seen in the parental cells, the MDCKII-MRP2 cells showed strong peripheral staining with MRP2 (green stain) which were evident around the Dapi stained nuclei (blue stain). These studies demonstrated that the MDCKII-MRP2 cells express a significantly higher level of MRP2, as compared to the parental cells.
Figure-1.
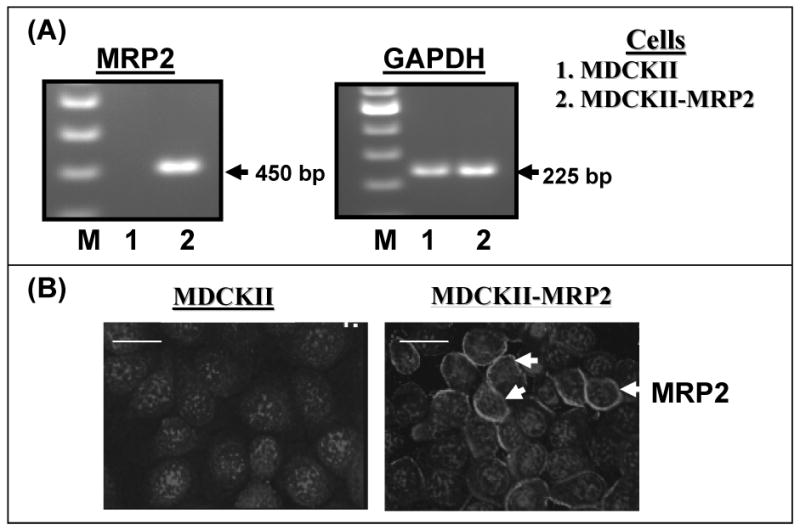
MRP2 gene expression and protein levels in MDCKII and MDCKII-MRP2 cells. In (A), RT-PCR amplification of mRNAs isolated from MDCKII and MDCKII-MRP2 cells were carried out. The PCR products were separated in an agarose gel containing ethidium bromide. A representative gel picture (n=4) showing the molecular weight markers (M), and bands for human MRP2 (450 bp) and GAPDH (225 bp) products, are depicted. In (B), immunofluorescence microscopy (IFM) of human MRP2 was carried out in both MDCKII and MDCKII-MRP2 cells by using an Alexa Fluor-tagged anti-MRP2. A representative photograph (n=3) showing MRP2 localization is depicted. Dapi stained nuclei are shown in blue and MRP2 staining is shown in green. The white bars represent 10 micro meter, and the red arrows are depicting perinuclear staining of MRP2. The MDCKII-MRP2 cells show constitutively higher expression of MRP-2 as compared to the parental MDCKII cells.
Maximal suppression of MRP2-mediated calcein efflux is achieved with montelukast coexposure
Both the parental and MRP2 overexpressing cells were used to study MRP2 specific drug-efflux function, in order to determine the potency of different MRP-inhibitors, e.g. MK-571, montelukast, zafirlukast or probenecid, in inhibiting the efflux of calcein (Fig. 2). Intracellular calcein levels (MFU/ μg of protein) were much lower in the MRP2 over-expressing cells (B), as compared to the parental cells (A), suggesting a significantly (p<0.01) higher rate of efflux. Interestingly, MK-571 was able to suppress calcein efflux in both cell types, however, montelukast was more potent in inhibiting calcein-efflux from the MRP2 overexpressing cells. In the MDCKII-MRP2 cells, pre-exposure to MK-571 showed a 3-fold increase in calcein retention, whereas montelukast coexposure showed almost a 5-fold increase. Neither zafirlukast nor probenecid significantly blocked calcein efflux from the parental cells and had minimal effects on calcein efflux in the MRP2 overexpressing cells. These findings suggested that the higher rate of efflux in MDCKII-MRP2 cells is primarily due to MRP2. As compared to MK-571, montelukast was found to be a more potent inhibitor of MRP2-mediated calcein efflux.
Figure-2.
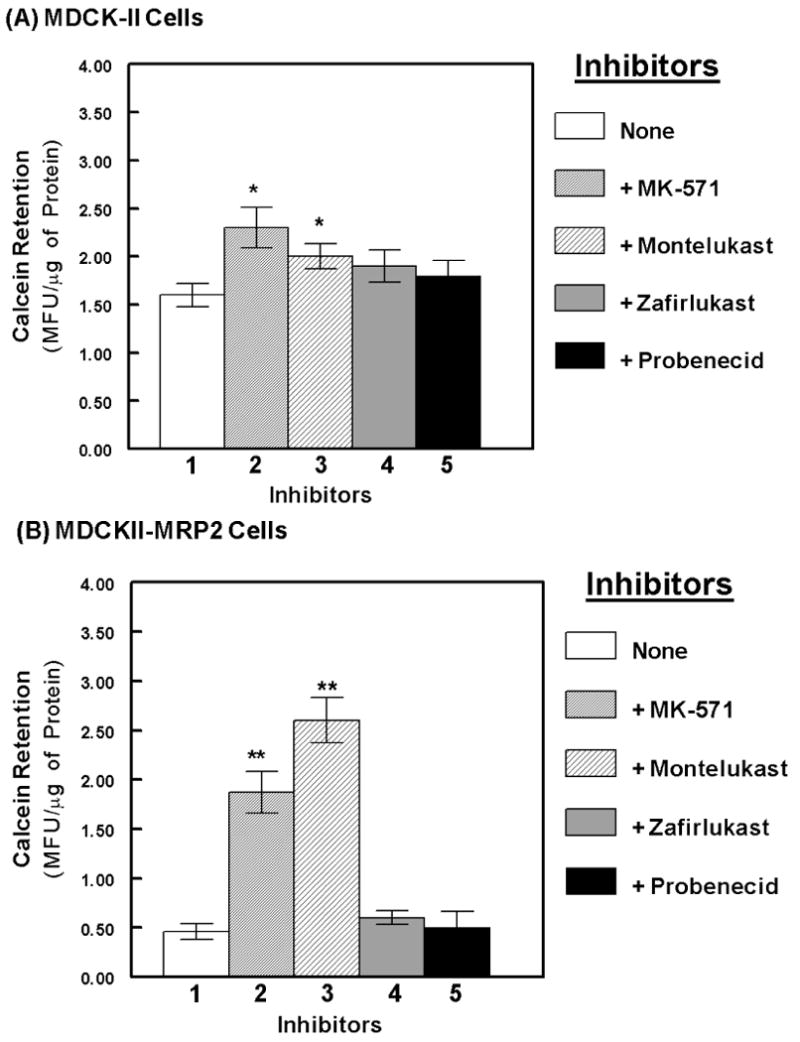
Suppression of MRP2-mediated calcein-AM efflux by the MRP inhibitors. The fluorescent MRP substrate, calcein-AM was used to measure efflux function in both MDCKII and MDCKII-MRP2 cell lines. Intracellular calcein retention is presented as mean fluorescent units (MFU) per μg of protein in cell extracts. Changes in intracellular fluorescence, in the absence (lane-1) or presence of the inhibitors (100 μM each), MK-571 (lane-2), montelukast (lane-3), zafirlukast (lane-4) or probenecid (lane-5), is shown. Bar graphs represent data from 4-5 separate experiments carried out in quadruplicate cultures, and error bars depict the standard error of means (±SEM). The symbols, * represent P<0.05 and ** represent P<0.01. Montelukast showed the highest MRP2 inhibitory potential.
Similar calcein-AM retention studies were also carried out in a P-gp over-expressing breast cancer cell line, NCI/ADR 5). Although verapamil, a P-gp inhibitor, was able to block calcein-efflux, neither MK-571 nor montelukast showed any significant increase in intracellular calcein levels (data not shown). These observations further indicated that montelukast is a specific inhibitor of MRP2, but not of P-gp.
Montelukast is a potent suppressor of MRP2-mediated efflux of both taxol and saquinavir
The anti-cancer agent, taxol 36) and the anti-HIV drug, saquinavir 11), are known to be MRP2 substrates. Hence, in the following studies we wanted to determine whether montelukast can suppress the efflux of both taxol and saquinavir from the MDCKII-MRP2 cells (Fig.3). We determined the intracellular retention of 3H-taxol (A) and 3H-saquinavir (B). After 2 hrs of incubation of the radiolabeled drugs, in the presence or absence of the four different MRP inhibitors, total cellular accumulations (loading concentrations) were determined and represented as CPM/ μg of protein. Similar to the calcein-efflux studies (Fig.2), neither zafirlukast nor probenecid had a significant effect on intracellular accumulation of either of these drugs. Coexposure to MK-571 suppressed taxol efflux by 1.7 fold and saquinavir efflux by almost 2.5 fold. However, montelukast coexposure suppressed taxol efflux by almost 3.0 fold and saquinavir efflux by more than 5.0 fold. These studies demonstrated that both taxol and saquinavir are substrates for MRP2 and montelukast possesses the highest inhibitory potency against MRP2-mediated efflux of both taxol and saquinavir.
Figure-3.
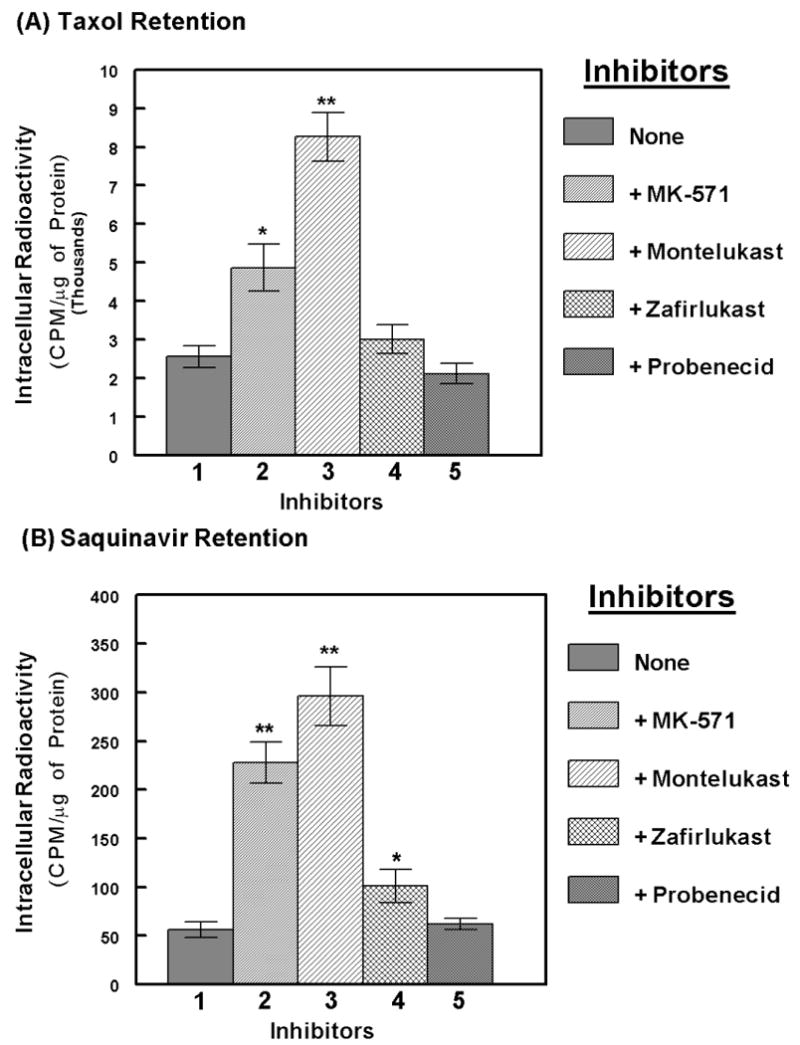
Suppression of MRP2-mediated taxol and saquinavir efflux by the MRP inhibitors. Intracellular accumulation of 3H-taxol (A) or 3H-saquinavir (B) were measured, in the absence (lane-1) or presence of MK-571 (lane-2), montelukast (lane-3), zafirlukast (lane-4), or probenecid (lane-5). Cells were pre-exposed to the inhibitors (100 μM) for 15 mins followed by addition of the substrates and further incubation for 2 hrs. Plates were washed and cell extracts were obtained to monitor intracellular taxol or saquinavir retention, represented as radioactivity (CPM) per μg of protein. In each panel, bar graphs represent data from 3-4 independent experiments carried out in triplicates and error bars depict ±SEM. The symbols, * and ** represent P<0.05 and P<0.01, respectively. Montelukast was found to inhibit the MRP2-mediated efflux of both taxol and saquinavir.
Both taxol and saquinavir efflux studies were also carried out in the parental MDCKII cells (data not shown). Similar to our findings with the calcein-assays (Fig.2) no significant efflux of either of these two substrates was seen in the MDCKII cells, and neither MK-571 nor montelukast showed a significant increase in their intracellular levels. These observations indicated that the significant efflux of taxol and saquinavir observed in the MDCKII-MRP2 cells (Fig. 3) is primarily a function of MRP2, and not due to any of the other endogenous MRPs.
Concentration dependent effects of montelukast and MK-571 on MRP2-mediated efflux
Although at the highest concentration, i.e.100 μM, both montelukast and MK-571 was able to suppress MRP2-mediated efflux of all three substrates (Fig. 2 & Fig. 3), we wanted to compare the inhibitory potencies of these two inhibitors, by exposing cells to a range of lower concentrations, i.e. 20, 40 and 80 μM. The relative efficacies of MRP2 blockade were assayed using calcein-AM (Fig. 4A), 3H-taxol (Fig. 4B) or 3H-saquinavir (Fig. 4C). Even with the lower concentrations, intracellular levels of all three substrates were consistently higher with montelukast, as compared to that observed with MK-571. Although at 20 μM, MK-571 did not show a significant increase in drug accumulations, montelukast's inhibitory effects were consistently observed even at this lowest concentration. At both 40 μM and 80 μM of montelukast, a 2-4 fold higher intracellular accumulation of all three substrates were also clearly evident. Interestingly, the inhibitory effect of montelukast on the efflux of both taxol and calcein were more pronounced, than that observed with MK-571. However, at all concentrations studied, both montelukast and MK-571 showed a similar level of inhibition of saquinavir efflux. This suggested that a substrate specific inhibition of MRP2 mediated efflux may occur via different MRP2 inhibitors.
Figure-4.
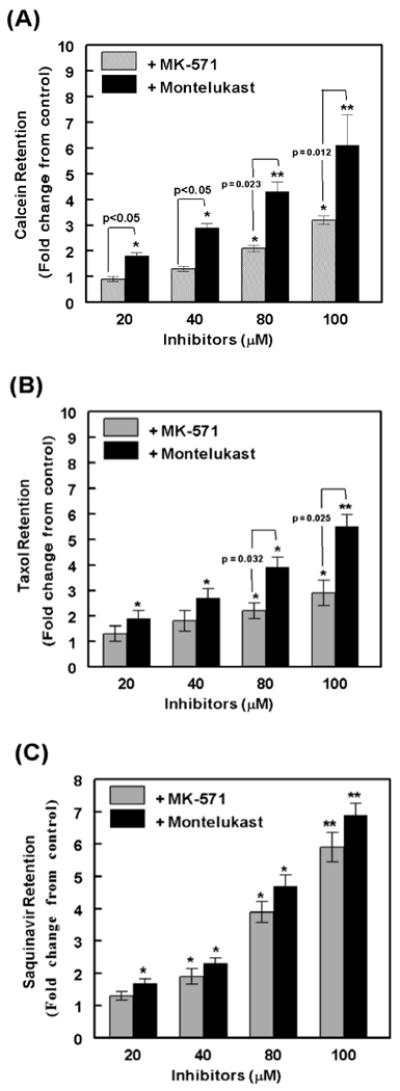
Comparing the MRP2 inhibitory potencies of MK-571 and montelukast. The concentration dependent effect of MK-571 or montelukast (20-100 μM) on inhibition of calcein (A), taxol (B) or saquinavir (C) efflux from the MDCKII-MRP2 cells was assessed. Data obtained from three independent experiments (n=3) are shown. In each bar graph, data is presented as fold change as compared to controls (no inhibitor added). Error bars depict ±SEM and symbols, * and ** represent P<0.05 and P<0.01, respectively. At equimolar concentrations of each inhibitor, significant differences between MK-571 and montelukast are shown as P-values above the bar graphs. Montelukast is a more potent inhibitor of MRP2-mediated efflux of taxol than MK-571, and is similar in potency to MK-571 with respect to the inhibition of saquinavir efflux.
Montelukast mediated inhibition of MRP2 is sustained for an extended period of time
Ideally, competitive inhibitors having a longer duration of binding, possibly due to an increased affinity towards the transporter, may augment intracellular drug concentrations and would be a more efficacious MDR-modulator when used in adjunct therapy. In the following experiments, we monitored the duration of MRP2 inhibition by both montelukast and MK-571 (Fig. 5). Similar to previous experiments, cells were first preloaded with either 3H-taxol (Fig. 5A) or 3H-saquinavir (Fig. 5B) in presence or absence of each of the inhibitors, montelukast or MK-571 (100 μM each). Cultures were washed and replaced with medium without inhibitors and cell extracts were obtained at different time points (15-90 min) post wash, in order to determine the duration of intracellular retention of each substrate. Results showed that coexposure to montelulkast increased peak loading concentration (0-time point) of taxol by almost 4-fold and of saquinavir by almost 3-fold. Within 15 min post-wash, a significant decrease in intracellular concentrations of both taxol and saquinavir were seen in both control cells and in those exposed to the inhibitors. However, a clear difference in this gradual decrease was evident post removal of different inhibitors. In cells exposed to MK-571, the intracellular levels of both taxol and saquinavir decreased close to the basal levels seen in control cells within 30 min. However, in cells exposed to montelukast, even after 90 min post-wash both taxol and saquinavir were retained at almost fifty percent of its loading concentrations. Thus, as compared to MK-571, a significantly (p< 0.01) lower MRP2-mediated efflux and a significantly (p< 0.05) longer intracellular retention of both taxol and saquinavir were clearly evident when cells were coexposed to montelukast.
Figure-5.
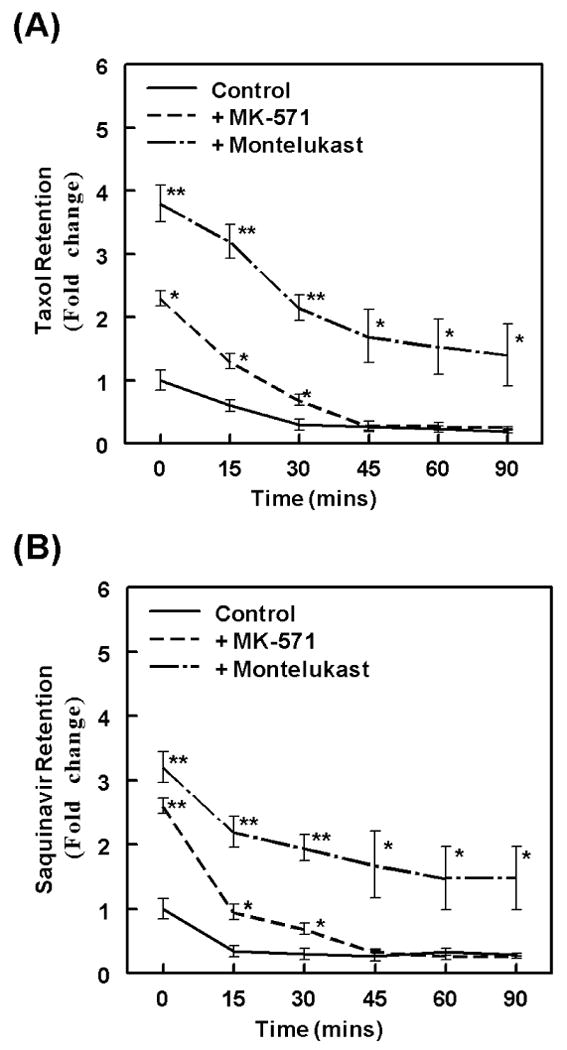
Montelukast is a durable inhibitor of MRP2-mediated taxol and saquinavir efflux. We wanted to measure the duration of MRP2 inhibition by MK571 or montelukast. The MDCKII-MRP2 cells were exposed to the inhibitors (100 μM each) for 30 mins prior to the incubation of cells with taxol (A) or saquinavir (B) for 2 hrs. Following the loading step in the presence of the inhibitors, cells were thoroughly washed and cultured in the absence of inhibitors. Intracellular radioactivities (CPM/μg protein) were determined at different time points (15-90 mins) post wash. Data presented in the line graphs depict fold change in radioactivity, as compared to the control values obtained at the 0-time point. These studies showed the peak-loading and temporal-efflux of taxol and saquinavir. Error bars depict the ±SEM of values, and significant differences are depicted as P-values (*, P<0.05 and **, P<0.01).
Discussion
Clinical approval of a new drug is a long, arduous, and extremely costly endeavor, starting with target identification in vitro, validation of their effectiveness in pre-clinical studies in vivo, and eventually their safety and efficacy determinations in human clinical trials. Only a few of the investigational new drug (IND) candidates finally make it through to the clinic, primarily due to their poor pharmacokinetics and toxicity profiles in patients. However, if certain clinically approved drugs can be used towards different therapeutic indications, this long and expensive process may be circumvented. In this respect, since a number of available drugs are inhibitors of ABC-transporters, in vitro evidence showing their utility when used in combination with anti-cancer or anti-HIV agents, will enhance their translational value as adjunct therapy. Our in vitro findings demonstrated that the anti-asthmatic agent montelukast is a potent and durable inhibitor of MRP2 mediated efflux.
The MRP transporters can modulate absorption, distribution, metabolism, and excretion properties of drugs primarily due to their polarized expression on membrane barriers. Thus, MRP inhibitors may be able to significantly increase tissue levels of drugs and decrease the persistence of both tumor-cells and HIV-infected lymphocytes, sequestered within subvascular reservoirs. Our studies primarily focused on the identification of MRP2 inhibitors; because in addition to their expression on tumor cells 34, 35) and HIV-1 infected lymphocytes 29, 44), the MRP2 transporters are known to be expressed on apical surfaces of vascular barriers 17) and may play crucial roles in regulating the directional transport of therapeutic agents. Although a number of pan-inhibitors of MRPs had been previously documented 16, 17, 19, 37, 48), most of these agents have significant side effects in vivo. Furthermore, previous studies using either probenecid or sulfinpyrazone had shown that their MRP inhibitory effects are inconsistent, and in certain cell types, they may stimulate rather than inhibit the efflux function 36, 38, 45). Indeed, in our experiments, probenecid coexposure slightly decreased calcein-efflux, however, it had no effect on saquinavir-efflux and slightly increased taxol-efflux (Fig. 2 & Fig. 3).
Amongst the four MRP inhibitors investigated in the current study, montelukast was found to be the most potent and consistently inhibited the efflux of all three MRP substrates. Another major advantage of using montelukast maybe its longer duration of binding to MRP2 which would be an important characteristic during adjunct therapy, since under physiological conditions the endothelial cells are exposed to peak plasma levels of the inhibitors for only a short period of time. On vascular barriers, the inhibitory effects should persist long enough to enable a durable MRP2 inhibition, enabling a higher rate of entry rather than efflux. Thus, montelukast mediated inhibition of MRP2 should augment drug entry via vascular barriers, and facilitate drug entry into cells sequestered within the underlying tissues 46).
In the clinical setting, it is known that montelukast (Singulair) is rapidly absorbed following oral administration and mean oral bioavailability is approximately 60-70% 47). After administration of the 10-mg tablet, the mean peak plasma concentration (Cmax) was found to be 1-2 μM which is achieved within 3 to 4 hrs. The mean area under the plasma concentration (AUC) of montelukast is approximately 2.5 μg·h/mL and the half-life (t1/2) ranges from 2.7 to 5.5 hrs. Although the Cmax, achieved with the 10 mg dose, is much lower than that used in our studies (20-100 μM) to enable a significant level of MRP2 suppression, previous studies had used the 50 mg dose of montelukast in humans without significant short-term toxicities. Hence, a higher level of montelukast may be used for blockade of MRP2 during short term chemotherapy, but this may not be as feasible with a long-term antiretroviral regimen. Since accumulation of toxic levels of montelukast has not been indicated even with the higher doses, the high AUC and long plasma t1/2 of montelukast may implicate its utility in the clinic as an adjunct to anti-cancer or anti-HIV agents.
Interestingly, the intracellular accumulation of saquinavir was found to be almost 50-fold lower than that observed with taxol (Fig. 3). Indeed, the HPIs (e.g. saquinavir) are known to have very low membrane permeability and drug efflux mechanisms are significant problems with these peptidomimetic agents 46). Hence, a high rate of efflux may primarily dictate the concentrations of HPIs achieved in HIV-infected cells. A number of strategies, such as phospholipid encapsulation 47) and pro-drug synthesis 48) are being examined to increase intracellular levels of HPIs. Our findings showed that coexposure to montelukast can increase the intracellular saquinavir retention by more than 6-fold. In addition, montelukast was found to be as potent as MK-571, in inhibiting saquinavir efflux (Fig. 4C). In addition, montelukast coexposure enabled a significantly (p<0.01) longer intracellular retention of saquinavir than MK-571 (Fig. 5B). We envision that using other currently available and/or experimental LTRAs, it would be possible to screen for MRP2 inhibitors in vitro. All of the known leukotrienes are transported by the MRP transporters, hence, the LTRAs, which are leukotriene analogs are also substrates for MRPs 40, 49). However, different leukotrienes and LTRAs may be transported at distinctly different rates by individual MRPs. In this respect, montelukast may be more effective in both competitive blockade and duration of binding to MRP2 due to specific modifications in its structure. Thus, it is likely that the structures of LTRAs may predict those which would be better MRP inhibitors 50).
The cysteinyl leukotrienes: LTC4, LTD4, LTE4, have two types of receptors; cysLT1 and cysLT2. All clinically available LTRAs have high-affinity binding to the cysLT1 receptor, and the rank order of affinities of leukotrienes towards the CysLT1 receptor is, LTD4 ≫ LTE4 = LTC4 ≫ LTB451). Since LTD4 is the primarily secreted form, analogs of LTD4 were first synthesized containing added aliphatic acid structures and aryl groups, which formed the basic structures of many LTRAs (Fig. 6) 49, 51). Since both MK-571 and montelukast have close structural homology, their potency towards blocking MRP2 may be higher than that observed with zafirlukast. Although previous studies 52), have shown that the flavonoid myrcetin is a potent inhibitor of MRP2-mediated vincristine efflux, our findings demonstrated that an approved drug montelukast has inhibitory effects at similar concentrations. Our observations also indicated that substrate specific differences in inhibition may occur with different LTRAs. Indeed, previous findings by Nagayama S. et al., (1998), had shown the ability of pranlukast (ONO-1078) to chemosensitize MDR-tumor cells towards vincristine 53). Although we did not use this newly approved drug, pranlukast (Ultair®), it is possible that its lipid backbone may enable it to inhibit MRPs for a longer duration, especially at the plasma membrane.
Figure-6.
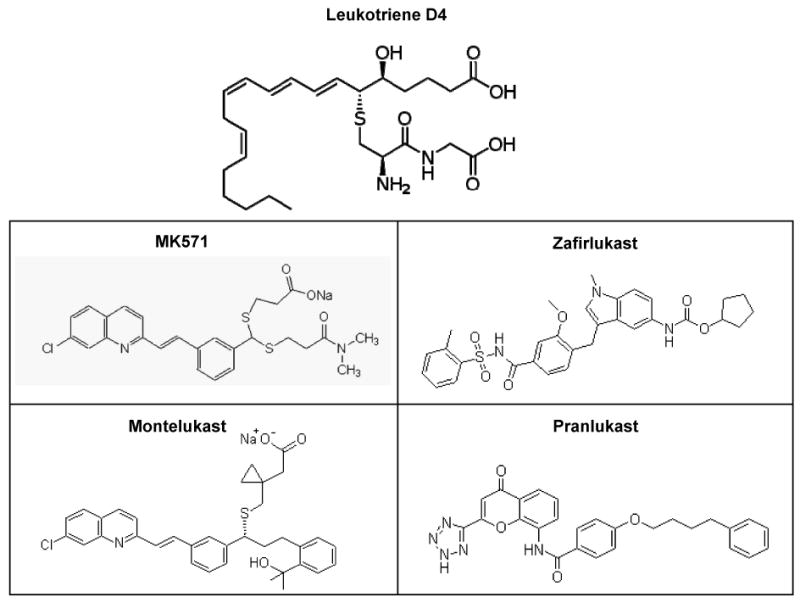
Structures of LTRAs, MK571, montelukast, zafirlukast and pranlukast. The structure of leukotriene D4 (LTD4) is compared to the synthetic LTRAs. The first LTRA, MK-571 was developed by a dithioacetal linkage incorporation and replacement of carboxylic acids by an amide moiety. A quinolone-containing structure generated from MK-571 was developed to first yield verlukast (MK679) and the enantiomers were resolved to generate montelukast (Singulair®). Initial SAR analyses led to the fabrication of an indole-containing compound to yield zafirlukast and lastly, by introducing a tetrazole and a lipid backbone, the newly approved LTRA, pranlukast (ONO-1078) (Ultair®) was generated.
Although our studies in MDCKII and MDCKII-MRP2 cells suggest that montelukast is a potent inhibitor of MRP2, it is possible that montelukast is blocking multiple MRP transporters in these cells. It has been previously reported that MDCKII cells express low levels of other endogenous (canine) MRPs 42). However, these investigators had also shown that MDCKII-MRP2 cells express 25-fold more MRP2 than the parental MDCKII cells. Our RT-PCR studies had shown higher expression of MRP2 in these stably transfected cells (Fig. 1). Furthermore, in Fig. 1(B), we clearly see a much higher MRP2 staining intensity in the MDCKII-MRP2 cells by immuno-fluorescence microscopy. In addition, drug-efflux assays no significant MRP mediated efflux in the parental cells, however, a significantly lower calcein retention and significant inhibition of efflux function by both montelukast (p<0.01) and MK-571 (p<0.05) (Fig. 2B) clearly confirmed that MRP2 is the main drug-efflux mechanism in the MDCKII-MRP2 cells. Hence, the montelukast-mediated inhibition in drug-efflux is primarily due to the inhibition of MRP2. Although, our findings may also suggest that the differences in the inhibition of drug-efflux by the other two LTRAs, MK-571 and zafirlukast, may be due to their ability to affect other MRPs, although at a lower level than that observed with montelukast. Our findings suggest the need to improve the potency of monterukast, possibly by chemical modifications, to develop novel compounds that display higher potency for MRP2 inhibition than that observed for its antagonistic activity on LT receptors so that the side effects can be minimized.
Since montelukast is generally a safe drug with minimal side effects, and is indicated in both adults and pediatric patients (12 months of age and older) 54, 55), our observations on its potent and durable inhibitory effects on MRP2 mediated efflux of taxol and saquinavir, may be of significant therapeutic importance. However, it is unclear at this time how the relatively high concentrations of montelukast, required to transiently inhibit the MRP2 mediated efflux function, will ultimately affect the hepatobiliary eliminations and regulate the directional transport of other drugs and regulate drug-drug interactions 56). Accordingly, caution should be exercised in extrapolating our results towards the in vivo efficacy of montelukast as an adjunct therapy to taxol or saquinavir.
Acknowledgments
The authors thank Prof. P. Borst from the National Cancer Institute (Amsterdam, The Netherlands) who kindly provided the MDCKII and MDCKII-MRP2 cell lines. This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) grant R21-AI064048 and by Louisiana Cancer Research Consortium (LCRC).
References
- 1.Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4(7):923–939. doi: 10.1517/17425255.4.7.923. [DOI] [PubMed] [Google Scholar]
- 2.Colabufo NA, Berardi F, Contino M, Niso M, Perrone R. ABC pumps and their role in active drug transport. Curr Top Med Chem. 2009;9(2):119–129. doi: 10.2174/156802609787521553. [DOI] [PubMed] [Google Scholar]
- 3.Girardin F. Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci. 2006;8(3):311–321. doi: 10.31887/DCNS.2006.8.3/fgirardin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 5.Eilers M, Roy U, Mondal D. MRP (ABCC) transporters-mediated efflux of anti-HIV drugs, saquinavir and zidovudine, from human endothelial cells. Exp Biol Med (Maywood) 2008;233:1149–1160. doi: 10.3181/0802-RM-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Figueiredo-Pontes LL, Pintão MC, Oliveira LC, Dalmazzo LF, Jácomo RH, Garcia AB, Falcão RP, Rego EM. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin Cytom. 2008;74(3):163–168. doi: 10.1002/cyto.b.20403. [DOI] [PubMed] [Google Scholar]
- 7.Kuo MT. Roles of multidrug resistance genes in breast cancer chemoresistance. Adv Exp Med Biol. 2007;608:23–30. doi: 10.1007/978-0-387-74039-3_2. [DOI] [PubMed] [Google Scholar]
- 8.Annereau JP, Szakács G, Tucker CJ, Arciello A, Cardarelli C, Collins J, Grissom S, Zeeberg BR, Reinhold W, Weinstein JN, Pommier Y, Paules RS, Gottesman MM. Analysis of ATP-binding cassette transporter expression in drug-selected cell lines by a microarray dedicated to multidrug resistance. Mol Pharmacol. 2004;66:1397–1405. doi: 10.1124/mol.104.005009. [DOI] [PubMed] [Google Scholar]
- 9.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal S, Pal D, Mitra AK. Both P-gp and MRP2 mediate transport of Lopinavir, a protease inhibitor. Int J Pharm. 2007;339:139–147. doi: 10.1016/j.ijpharm.2007.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huisman MT, Smit JW, Crommentuyn KM, Zelcer N, Wiltshire HR, Beijnen JH, Schinkel AH. Multidrug resistance protein 2 (MRP2) transports HIV protease inhibitors, and transport can be enhanced by other drugs. AIDS. 2002;16:2295–2301. doi: 10.1097/00002030-200211220-00009. [DOI] [PubMed] [Google Scholar]
- 12.Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood-CNS interfaces: Summary of current knowledge and recommendations for further research. Antiviral Res. 2009;82(2):A99–109. doi: 10.1016/j.antiviral.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen ZS, Furukawa T, Sumizawa T, Ono K, Ueda K, Seto K, Akiyama SI. ATP-Dependent efflux of CPT-11 and SN-38 by the multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol Pharmacol. 1999;55:921–928. [PubMed] [Google Scholar]
- 14.Williamson G, Aeberli I, Miguet L, Zhang Z, Sanchez MB, Crespy V, Barron D, Needs P, Kroon PA, Glavinas H, Krajcsi P, Grigorov M. Interaction of positional isomers of quercetin glucuronides with the transporter ABCC2 (cMOAT, MRP2) Drug Metab Dispos. 2007;35:1262–1268. doi: 10.1124/dmd.106.014241. [DOI] [PubMed] [Google Scholar]
- 15.Yuan H, Li X, Wu J, Li J, Qu X, Xu W, Tang W. Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. Curr Med Chem. 2008;15(5):470–476. doi: 10.2174/092986708783503258. [DOI] [PubMed] [Google Scholar]
- 16.Zhou SF, Wang LL, Di YM, Xue CC, Duan W, Li CG, Li Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15(20):1981–2039. doi: 10.2174/092986708785132870. [DOI] [PubMed] [Google Scholar]
- 17.Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7(7):861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 19.Zhou SF. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica. 2008;38(78):802–832. doi: 10.1080/00498250701867889. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed-Belkacem A, Pozza A, Macalou S, Pérez-Victoria JM, Boumendjel A, Di Pietro A. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2) Anticancer Drugs. 2006;17(3):239–243. doi: 10.1097/00001813-200603000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Kuhnle M, Egger M, Muller C, Mahringer A, Bernhardt G, Fricker G, Ko□nig B, Buschauer A. Potent and Selective Inhibitors of Breast Cancer Resistance Protein (ABCG2) Derived from the p-Glycoprotein (ABCB1) Modulator Tariquidar. J Med Chem. 2009;52(4):1190–1197. doi: 10.1021/jm8013822. [DOI] [PubMed] [Google Scholar]
- 22.Loe DW, Deeley RG, Cole SP. Biology of the multidrug resistance-associated protein, MRP. Eur J Cancer. 1996;32A:945–957. doi: 10.1016/0959-8049(96)00046-9. [DOI] [PubMed] [Google Scholar]
- 23.Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22:7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 24.Aneja R, Liu M, Yates C, Gao J, Dong X, Zhou B, Vangapandu SN, Zhou J, Joshi HC. Multidrug resistance-associated protein-overexpressing teniposide-resistant human lymphomas undergo apoptosis by a tubulin-binding agent. Cancer Res. 2008;68(5):1495–1503. doi: 10.1158/0008-5472.CAN-07-1874. [DOI] [PubMed] [Google Scholar]
- 25.Damiani D, Tiribelli M, Raspadori D, Michelutti A, Gozzetti A, Calistri E, Candoni A, Chiarvesio A, Lenoci M, Russo D, Fanin R. The role of MDR-related proteins in the prognosis of adult acute myeloid leukaemia (AML) with normal karyotype. Hematol Oncol. 2007;25(1):38–43. doi: 10.1002/hon.806. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto Y, Tamiya T, Nagao S. Resistance to topoisomerase II inhibitors in human glioma cell lines overexpressing multidrug resistance associated protein (MRP) 2. Med Invest. 2005;52(12):41–48. doi: 10.2152/jmi.52.41. [DOI] [PubMed] [Google Scholar]
- 27.Kamazawa S, Kigawa J, Kanamori Y, Itamochi H, Sato S, Iba T, Terakawa N. Multidrug resistance gene-1 is a useful predictor of Paclitaxel-based chemotherapy for patients with ovarian cancer. Gynecol Oncol. 2002;86(2):171–176. doi: 10.1006/gyno.2002.6738. [DOI] [PubMed] [Google Scholar]
- 28.Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 29.Janneh O, Jones E, Chandler B, Owen A, Khoo SH. Inhibition of P-glycoprotein and multidrug resistance-associated proteins modulates the intracellular concentration of lopinavir in cultured CD4 T cells and primary human lymphocytes. J Antimicrob Chemother. 2007;60(5):987–993. doi: 10.1093/jac/dkm353. [DOI] [PubMed] [Google Scholar]
- 30.Janneh O, Hartkoorn RC, Jones E, Owen A, Ward SA, Davey R, Back DJ, Khoo SH. Cultured CD4T cells and primary human lymphocytes express hOATPs: intracellular accumulation of saquinavir and lopinavir. Br J Pharmacol. 2008;155(6):875–883. doi: 10.1038/bjp.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26(8):1052–1065. doi: 10.1038/sj.jcbfm.9600254. [DOI] [PubMed] [Google Scholar]
- 32.Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2(1):86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slot AJ, Wise DD, Deeley RG, Monks TJ, Cole SP. Modulation of human multidrug resistance protein (MRP) 1 (ABCC1) and MRP2 (ABCC2) transport activities by endogenous and exogenous glutathione-conjugated catechol metabolites. Drug Metab Dispos. 2008;36(3):552–560. doi: 10.1124/dmd.107.019661. [DOI] [PubMed] [Google Scholar]
- 34.Lagas JS, Vlaming ML, van Tellingen O, Wagenaar E, Jansen RS, Rosing H, Beijnen JH, Schinkel AH. Multidrug resistance protein 2 is an important determinant of paclitaxel pharmacokinetics. Clin Cancer Res. 2006;12:6125–6132. doi: 10.1158/1078-0432.CCR-06-1352. [DOI] [PubMed] [Google Scholar]
- 35.Liedert B, Materna V, Schadendorf D, Thomale J, Lage H. Overexpression of MOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J Invest Dermatol. 2003;121:172–176. doi: 10.1046/j.1523-1747.2003.12313.x. [DOI] [PubMed] [Google Scholar]
- 36.Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer. 2005;116:824–829. doi: 10.1002/ijc.21013. [DOI] [PubMed] [Google Scholar]
- 37.Jedlitschky G, Hoffmann U, Kroemer HK. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin Drug Metab Toxicol. 2006;2(3):351–366. doi: 10.1517/17425255.2.3.351. [DOI] [PubMed] [Google Scholar]
- 38.Kim HS, Min YD, Choi CH. Double-edged sword of chemosensitizer: increase of multidrug resistance protein (MRP) in leukemic cells by an MRP inhibitor probenecid. Biochem Biophys Res Commun. 2001;283(1):64–71. doi: 10.1006/bbrc.2001.4746. [DOI] [PubMed] [Google Scholar]
- 39.Horikawa M, Kato Y, Tyson CA, Sugiyama Y. The potential for an interaction between MRP2 (ABCC2) and various therapeutic agents: probenecid as a candidate inhibitor of the biliary excretion of irinotecan metabolites. Drug Metab Pharmacokinet. 2002;17:23–33. doi: 10.2133/dmpk.17.23. [DOI] [PubMed] [Google Scholar]
- 40.Gekeler V, Ise W, Sanders KH, Ulrich WR, Beck J. The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem Biophys Res Commun. 1995;208:345–352. doi: 10.1006/bbrc.1995.1344. [DOI] [PubMed] [Google Scholar]
- 41.Buccellati C, Fumagalli F, Viappiani S, Folco G. Leukotriene modifiers: novel therapeutic opportunities in asthma. Farmaco. 2002;57(3):235–242. doi: 10.1016/s0014-827x(02)01209-0. [DOI] [PubMed] [Google Scholar]
- 42.Cui Y, Konig J, Buchholz JK, Spring H, Leier I, Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- 43.Höner zu Bentrup K, Ramamurthy R, Ott CM, Emami K, Nelman-Gonzalez M, Wilson JW, Richter EG, Goodwin TJ, Alexander JS, Pierson DL, Pellis N, Buchanan KL, Nickerson CA. Three-dimensional organotypic models of human colonic epithelium to study the early stages of enteric salmonellosis. Microbes Infect. 2006;8(7):1813–1825. doi: 10.1016/j.micinf.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 44.Owen A, Hartkoorn RC, Khoo S, Back D. Expression of P-glycoprotein, multidrug-resistance proteins 1 and 2 in CEM, CEM(VBL), CEM(E1000), MDCKII(MRP1) and MDCKII(MRP2) cell lines. AIDS. 2003;17(15):2276–2278. doi: 10.1097/01.aids.0000088212.77946.65. [DOI] [PubMed] [Google Scholar]
- 45.Evers R, de Haas M, Sparidans R, Beijnen J, Wielinga PR, Lankelma J, Borst P. Vinblastine and sulfinpyrazone export by the multidrug resistance protein MRP2 is associated with glutathione export. Br J Cancer. 2000;83:375–383. doi: 10.1054/bjoc.2000.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowman MC, Archin NM, Margolis DM. Pharmaceutical approaches to eradication of persistent HIV infection. Expert Rev Mol Med. 2009;11:e6. doi: 10.1017/S1462399409000970. [DOI] [PubMed] [Google Scholar]
- 47.Kapitza SB, Michel BR, van Hoogevest P, Leigh ML, Imanidis G. Absorption of poorly water soluble drugs subject to apical efflux using phospholipids as solubilizers in the Caco-2 cell model. Eur J Pharm Biopharm. 2007;66(1):146–158. doi: 10.1016/j.ejpb.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 48.Rouquayrol M, Gaucher B, Roche D, Greiner J, Vierling P. Transepithelial transport of prodrugs of the HIV protease inhibitors saquinavir, indinavir, and nelfinavir across Caco-2 cell monolayers. Pharm Res. 2002;19(11):1704–1712. doi: 10.1023/a:1020913631309. [DOI] [PubMed] [Google Scholar]
- 49.Bernstein PR. Chemistry and structure--activity relationships of leukotriene receptor antagonists. Am J Respir Crit Care Med. 1998;157:S220–S225. [PubMed] [Google Scholar]
- 50.Zelcer N, Huisman MT, Reid G, Wielinga P, Breedveld P, Kuil A, Knipscheer P, Schellens JH, Schinkel AH, Borst P. Evidence for two interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2) J Biol Chem. 2003;278:3538–3544. doi: 10.1074/jbc.M303504200. [DOI] [PubMed] [Google Scholar]
- 51.Montuschi P. Leukotrienes, antileukotrienes and asthma. Mini Rev Med Chem. 2008;8(7):647–656. doi: 10.2174/138955708784567395. [DOI] [PubMed] [Google Scholar]
- 52.van Zanden JJ, de Mul A, Wortelboer HM, Usta M, van Bladeren PJ, Rietjens IM, Cnubben NH. Reversal of in vitro cellular MRP1 and MRP2 mediated vincristine resistance by the flavonoid myricetin. Biochem Pharmacol. 2005;69(11):1657–1665. doi: 10.1016/j.bcp.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Nagayama S, Chen ZS, Kitazono M, Takebayashi Y, Niwa K, Yamada K, Tani A, Haraguchi M, Sumizawa T, Furukawa T, Aikou T, Akiyama S. Increased sensitivity to vincristine of MDR cells by the leukotriene D4 receptor antagonist, ONO-1078. Cancer Lett. 1998;130(12):175–182. doi: 10.1016/s0304-3835(98)00132-3. [DOI] [PubMed] [Google Scholar]
- 54.Hung GR. Principles of managing children with asthma in the emergency department. Paediatr Child Health. 2007;12:479–481. doi: 10.1093/pch/12.6.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knorr B, Holland S, Rogers JD, Nguyen HH, Reiss TF. Montelukast adult (10-mg film-coated tablet) and pediatric (5-mg chewable tablet) dose selections. J Allergy Clin Immunol. 2000;106(3 Suppl):S171–S178. doi: 10.1067/mai.2000.109424. [DOI] [PubMed] [Google Scholar]
- 56.Bodó A, Bakos E, Szeri F, Váradi A, Sarkadi B. The role of multidrug transporters in drug availability, metabolism and toxicity. Toxicol Lett. 2003;140-141:133–143. doi: 10.1016/s0378-4274(02)00497-6. [DOI] [PubMed] [Google Scholar]