

Abstract
Objectives/Hypothesis
Identify correlations among SLC26A4 genotype, cochlear structural anomalies, and hearing loss associated with enlargement of the vestibular aqueduct (EVA).
Study Design
Prospective cohort survey, National Institutes of Health, Clinical Center, a federal biomedical research facility.
Methods
83 individuals, 11 months to 59 years of age, with EVA in at least one ear. Correlations among pure-tone hearing thresholds, number of mutant SLC26A4 alleles, and the presence of cochlear anomalies detected by computed tomography or magnetic resonance imaging.
Results
Linear mixed-effect model indicates significantly poorer hearing in ears with EVA from individuals with two mutant alleles of SLC26A4 than in those with EVA and a single mutant allele (p = .012) or no mutant alleles (p = .007) in this gene. There was no detectable relationship between degree of hearing loss and the presence of structural cochlear anomalies.
Conclusions
The number of mutant alleles of SLC26A4, but not the presence of cochlear anomalies, has a significant association with severity of hearing loss in ears with EVA. This information will be useful for prognostic counseling of patients and families with EVA.
Keywords: enlarged vestibular aqueduct, SLC26A4, hearing
Introduction
Enlargement of the vestibular aqueduct (EVA) is a common radiological malformation of the inner ear in children with early-onset sensorineural hearing loss (SNHL). It is identified by computed tomography (CT) or magnetic resonance imaging (MRI) 1–6. EVA can be unilateral or bilateral and either syndromic or nonsyndromic. It is the most penetrant feature of Pendred syndrome (PDS), an autosomal recessive disorder caused by biallelic mutations of the SLC26A4 gene 7 (OMIM605646). PDS was originally defined as goiter and profound congenital SNHL, but is now known to comprise variable thyroid and auditory phenotypes 8. Nonsyndromic EVA can also be associated with SLC26A4 mutations 9, 10. Approximately 25% of EVA patients have only one detectable mutant allele of SLC26A4 and up to 50% of EVA patients have no mutations that we can detect in this gene 11, 12, further contributing to nosologic confusion.
The auditory phenotype of EVA can include fluctuations, progressive changes in hearing, or both 4, 13. The pathophysiological mechanism of the hearing loss is unknown. There is no correlation of either the degree of hearing loss or the occurrence of sudden hearing loss and the degree of enlargement of the vestibular aqueduct or its contents, the endolymphatic duct 3, 4. There are contradictory reports on the effect of the number of SLC26A4 mutations on hearing 11, 14–18 and on inner ear radiologic findings. One study 11 suggested that biallelic mutations are associated with Mondini dysplasia (incomplete cochlear partition with a scala communis) more frequently than with EVA while another 15 reported a correlation of number of SLC26A4 mutant alleles with the width of the vestibular aqueduct. However, other studies failed to demonstrate a significant relationship between the number of mutant SLC26A4 alleles with the severity of the radiological phenotype 19, 5. The purpose of our study was to identify and characterize potential correlations among SLC26A4 genotype, inner ear radiologic findings, and hearing in a cohort of patients with EVA.
Materials and Methods
Subjects
This study was approved by the Combined Neuroscience Institutional Review Board, National Institutes of Health (NIH), Bethesda, MD. We obtained written, informed consent from adult subjects and parents of minor subjects. Audiological data were available for 83 of 86 subjects with nonsyndromic EVA or PDS who were previously described in reports on SLC26A4 genotype and thyroid phenotype 12, 20, 21. Participants self-described race and ethnicity in accordance with our institutional designations (Table 1). All subjects had unilateral or bilateral EVA, defined as a diameter >1.5 mm at the midpoint of the course of the vestibular aqueduct between the posterior cranial fossa and the vestibule of the inner ear, or an otherwise grossly malformed morphology of the vestibular aqueduct 12.
Table I.
Eighty-three EVA subjects analyzed in this study.
| Variable | Classification | n (% of cohort) |
|---|---|---|
| Gender | Male | 38 (46%) |
| Female | 45 (54%) | |
| Ethnicity | White, non-Hispanic | 72 (87%) |
| Black, non-Hispanic | 2 (2%) | |
| Hispanic | 1 (1%) | |
| Other/unknown | 8 (10%) | |
| Number of mutant alleles of SLC26A4 | 0 (M0) | 47 (57%) |
| 1 (M1) | 16 (19%) | |
| 2 (M2) | 20 (24%) | |
| EVA laterality | Bilateral | 63 (76%) |
| Unilateral | 20 (24%) |
SLC26A4 genotype
SLC26A4 genotypes for the subjects have been previously reported 12, 20. We categorized subjects according to number of detected mutant alleles of SLC26A4: zero (M0), one (M1), or two (M2). There are six hypofunctional variants (c.-60A>G, c.-3-2A>G, p.F335L, p.C565Y, p.L597S, and p.M775T) of uncertain pathogenicity within the cohort 20. Our preliminary analysis revealed no statistically significant effect of how we classified these variants (pathogenic versus nonpathogenic) upon comparisons of hearing (0.5/1/2/4-kHz pure-tone air-conduction threshold average) among M0, M1, and M2 subjects. Therefore, these hypofunctional variants were considered nonpathogenic for subsequent analyses, as in Madeo et al 21. Twenty (24%) subjects were classified as M2, 16 (19%) as M1, and 47 (57%) as M0.
Radiologic phenotype
We performed MRI of the inner ear, whenever possible, with fast induction employing steady state excitation (FIESTA), T2-weighted fast spin echo, or both on a 1.5-Tesla system at the NIH Clinical Center 12. Seventy of 83 subjects also had previous imaging of the inner ear either by CT (57), MRI (4), or both (9) available for review. All imaging studies were reviewed together at the completion of this study by a Neuroradiologist (J.A.B.) and an Otolaryngologist-Head and Neck Surgeon (A.J.G). We classified 146 ears with EVA into two groups: EVA, if no cochlear anomaly was identified, or EVA-plus-cochlear-anomaly, if an anomaly was identified. Potential cochlear anomalies included Michel deformity, cochlear aplasia or hypoplasia, common cavity, and incomplete cochlear partition. We did not classify hypoplastic modiolus as a cochlear anomaly due to its extremely high prevalence in ears with EVA 22. The cochlear structure of four ears was considered indeterminate due to insufficient technical quality of the images and these ears were excluded from further analysis involving radiologic data.
Audiologic phenotype
Three ears with cochlear implants were excluded from analysis. Thus, audiometric data for 143 ears with EVA from 83 subjects were evaluated (Table 1; age range = 11 months to 59 y, x̄ = 11.3 y, SD = 12.0 y). Sixty-one subjects had an audiological evaluation at the NIH Clinical Center and 22 subjects provided outside audiological records for review.
Audiological evaluations at the NIH included, when possible, pure-tone air- (250 to 8000 Hz) and bone-conduction (250 to 4000 Hz) threshold tests using clinical audiometers in double-walled sound suites in accordance with ANSI standards 23, 24. Middle ear function was assessed by tympanometry (226-Hz probe tone). We attempted to obtain previous and subsequent audiograms performed at other facilities, which were included for analysis only if the reliability had been rated as good by the examining audiologist. For each subject, we defined the reference audiogram as the most recent and complete audiological evaluation conducted at the NIH, or elsewhere if the NIH evaluation was incomplete.
Type of hearing loss was based upon 0.5/1/2-kHz pure-tone averages for air- and bone-conduction thresholds and degree of hearing loss was classified according to the 0.5/1/2/4-kHz pure-tone air-conduction threshold average as described previously 25. We also calculated low-(0.25 to 0.5 kHz), mid- (1 to 2 kHz), and high-frequency (4 to 8 kHz) pure-tone air-conduction threshold averages. The air-bone gap (ABG) was calculated for each octave frequency from 0.25 to 4 kHz and as an average for at 0.5, 1, and 2 kHz.
Seventy of 83 subjects had serial audiograms spanning at least one month for comparison to their earliest available (baseline) evaluation. We adapted the method of Madden et al 2 to categorize longitudinal pure-tone thresholds as stable, progressive, fluctuating, improving, fluctuating/improving, or fluctuating/progressive according to definitions in Table 2.
Table II.
Longitudinal categorization of pure-tone threshold data.
| Category | Definition |
|---|---|
| Stable | No significant fluctuation, improvement or progression as defined below |
| Improving | 10-dB improvement at any three frequencies |
| or 15-dB improvement at any two frequencies | |
| or ≥ 20-dB improvement at any one frequency | |
| Progressive | 10-dB decline at any three frequencies |
| or 15-dB decline at any two frequencies | |
| or ≥ 20-dB decline at any one frequency | |
| Fluctuating* | Interim audiogram shows 10-dB change at any three frequencies |
| or 15-dB change at any two frequencies | |
| or ≥ 20-dB change at any one frequency | |
| and final audiogram does not show significant improvement or progression from baseline | |
| Fluctuating/Improving* | Interim audiogram shows 10-dB change at any three frequencies |
| or 15-dB change at any two frequencies | |
| or ≥ 20-dB change at any one frequency | |
| and final audiogram shows significant improvement from baseline | |
| Fluctuating/Progressive* | Interim audiogram shows 10-dB change at any three frequencies |
| or 15-dB change at any two frequencies | |
| or ≥ 20-dB change at any one frequency | |
| and final audiogram shows significant progression from baseline |
≥ 3 audiograms required to assess.
Statistical Analyses
Data from ears with evidence (i.e., abnormal tympanometric results) of middle ear pathology were removed from ABG and longitudinal analyses. We used one-way analysis of variance (ANOVA) to investigate possible relationships between the magnitude of the ABG and number of mutant alleles of SLC26A4. An independent-samples t-test was performed to search for associations of ABG size with cochlear radiological phenotype (EVA versus EVA-plus-cochlear-anomaly). We calculated Pearson’s correlation coefficient for ABG magnitude and the vestibular aqueduct diameter on CT scans. We performed a one-way ANOVA to search for relationships between the size of the vestibular aqueduct measured by CT scan and the number of mutant alleles of SLC264.
Fisher’s exact test was used to evaluate for associations between degree of hearing loss, number of mutant alleles of SLC26A4 (M0, M1, M2), and cochlear radiological phenotype (EVA, EVA-plus-cochlear-anomaly). In order to avoid confounding effects between ear-specific data (degree of hearing loss and radiological phenotype) and person-specific data (genotype), we excluded 24 individuals (28 ears with EVA) for whom data from only one ear was available.
We analyzed the 143 ears with EVA using a linear mixed-effects model, which accounts for correlation structure due to inclusion of two ears from the same individual. We evaluated relationships between hearing (0.5/1/2/4-Hz, low-, mid- or high-frequency pure-tone threshold averages) and the following independent variables: age, gender, genotype, presence/absence of cochlear anomalies, and thyroid phenotype (goiter versus normal). We performed a similar mixed-effects analysis to model the interval change in 0.5/1/2/4-kHz pure-tone threshold average from baseline to final audiogram. In order to avoid ceiling effects, we did not include individuals with severe or profound hearing loss at baseline in this latter analysis.
All exploratory and modeling analyses were performed using SPSS software for Macintosh version 15 (SPSS Inc, Chicago, Illinois).
Results
Radiologic phenotype
Thirty-two (22.5%) of 142 ears with EVA with radiological data also had a cochlear anomaly. Thirty (21.1%) ears had an incomplete cochlear partition (“Mondini deformity”), one (0.7%) ear had a common cavity, and one (0.7%) ear had cochlear hypoplasia.
Audiologic phenotype
Ninety ears had sufficient data to classify the type of hearing loss. Sixty (67%) ears had SNHL, four (4%) had conductive loss, and 25 (28%) had mixed loss. One (1%) ear was classified as normal based upon the 0.5/1/2-kHz pure-tone threshold average, although SNHL was present at 6000 Hz and 8000 Hz.
Degree of hearing loss could be defined in 139 ears (range = 10 to 125 dB HL, x̄ = 70.63 dB HL, SD = 29.2 dB). It was mild in 18 (13%) ears, moderate in 43 (31%), severe in 36 (26%), and profound in 36 (26%). Six (4%) ears, all in subjects less than 14 years of age, were classified as having normal hearing based on the 0.5/1/2/4-kHz pure-tone air-conduction threshold average, although all six had hearing loss (>20 dB HL) for at least one frequency. We did not observe normal hearing in either ear of any subject with two mutant alleles of SLC26A4, although one subject (#1627 in Pryor et al 12) with unilateral hearing loss and EVA was classified as M1 in our analysis but would be M2 if p.F335L is pathogenic 20.
Fifty-seven ears had reference audiograms with both air- and bone-conduction threshold data and normal tympanometry. We detected air-bone gaps (ABGs) of 15 to 55 dB for at least one stimulus frequency from 0.25 to 4 kHz in 49 (86%) ears and for at least two frequencies in 37 (65%) ears. There was no significant (α = .05) relationship between the magnitude of the ABG at any frequency with the number of mutant alleles of SLC26A4, the presence or absence of a cochlear-anomaly, or the diameter of the vestibular aqueduct.
Relationships of hearing with genotype and cochlear structure
Figure 1a shows that mean air-conduction thresholds are greater in M2 ears than in M0 or M1 ears. Although there was a trend for poorer hearing in ears with EVA and a coinciding cochlear anomaly, a significant difference was not observed (Figure 1b).
Figure 1.

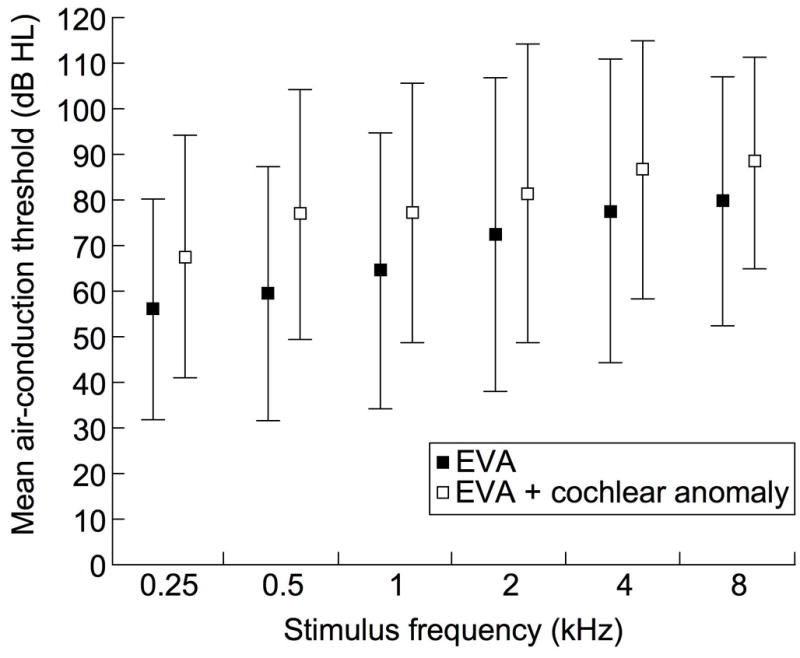
Mean (+/− 1SD) pure-tone air-conduction thresholds in ears with EVA grouped according to (a) number of mutant alleles of SLC26A4 and (b) presence or absence of cochlear anomalies.
There was a significant association of the number of mutant alleles of SLC26A4 with the degree of hearing loss (Fisher’s exact test, p = .002), and an association of the number of mutant alleles of SLC26A4 with the presence of a cochlear anomaly that approached significance (p = .058). There was no significant association between degree of hearing loss and the presence of a cochlear anomaly (p = .82).
We sought to further explore these relationships and the potential influences of age, gender, and thyroid phenotype with a linear mixed-effects model of hearing loss on the number of mutant alleles of SLC26A4 and the presence/absence of cochlear anomalies, including these factors as covariates. According to this model, hearing in the M2 group was 21.3 dB (p = .007) poorer than in the M0 group and 21.9 dB (p = .012) poorer than in the M1 group. Hearing in the M1 group was 5.9 dB poorer than in the M0 group, but this was not statistically significant (p = .50). Hearing in ears with cochlear anomalies was not significantly different (0.05 dB, p = .99) from ears without a cochlear anomaly. The model identified no effect of age on hearing, no significant difference in hearing between males and females, and no effect of thyroid phenotype. There was no significant (α = .05) relationship between the size of the vestibular aqueduct and the number of mutant SLC26A4 alleles (p = .35).
Longitudinal hearing analyses
Twenty-eight (26%) ears had stable hearing over an average duration of 3.7 years of follow-up (Table 3). Thirty-three (30%) ears had progressive hearing loss when followed, on average, for 4.9 years, and eight (7%) ears had improvement in hearing when followed for an average of 2.7 years. The remaining three categories, all characterized by fluctuations (Table 2), included 40 (37%) ears. Twenty-three (58%) of 40 ears with fluctuations showed progressive loss of hearing.
Table III.
Longitudinal hearing descriptive statistics.
| Stable | Progressive | Fluctuating- progressive | Fluctuating | Fluctuating-improving | Improving | |
|---|---|---|---|---|---|---|
| Number of ears |
||||||
| Total | 28 | 33 | 23 | 12 | 5 | 8 |
| No mutations+ | 13 | 12 | 13 | 9 | 2 | 7 |
| 1 mutations+ | 6 | 9 | 6 | 2 | 1 | 1 |
| 2 mutations+ | 9 | 12 | 4 | 1 | 2 | 0 |
| (−) Cochlear anomaly | 20 | 27 | 19 | 5 | 5 | 8 |
| (+) Cochlear anomaly | 8 | 5 | 4 | 6 | 0 | 0 |
| No data | 0 | 1 | 0 | 1 | 0 | 0 |
| x̄ (SD) |
||||||
| Duration of follow-up (y) | 3.7 (4.2) | 4.9 (4.8) | 8.5 (7.0) | 3.2 (2.6) | 9.9 (9.0) | 2.7 (2.9) |
| Number of audiograms | 3.4 (1.2) | 4.6 (3.1) | 9.4 (4.5) | 8.0 (6.2) | 6.2 (3.3) | 3.5 (1.6) |
| Age (y) at baseline | 11.2 (12.2) | 11.4 (14.4) | 5.2 (5.0) | 3.8 (1.6) | 5.6 (1.6) | 6.4 (4.0) |
| Baseline PTA* (dB HL) | 67.1 (32.2) | 66.6 (31.6) | 59.4 (26.4) | 56.6 (19.2) | 78.1 (27.0) | 62.2 (36.4) |
| Final PTA* (dB HL) | 61.6 (30.7) | 76.5 (30.6) | 78.1 (24.9) | 55.3 (20.2) | 64.8 (21.8) | 52.3 (33.9) |
0.5/1/2/4-kHz pure-tone average.
Number of mutant SLC26A4 alleles.
We applied a linear mixed-effects model to longitudinal audiological data for ears with normal, mild, or moderate hearing loss at baseline. This revealed that duration of follow-up had a small (0.96 dB/y) but significant (p = .046) relationship with hearing loss progression. There were no significant effects of number of mutant alleles of SLC26A4 or the presence/absence of cochlear anomalies on longitudinal hearing.
Discussion
We observed that ears with EVA in individuals with two mutant alleles of SLC26A4 have significantly greater hearing loss than those with one or no mutant alleles of SLC26A4 (Figure 1a). This is consistent with reports 14, 15 of two different Caucasian-majority cohorts, but differs from those of some other studies 11, 16, 17, 18, 20. For example, there was no detectable relationship of hearing level with number of mutant (p.H723R) alleles of SLC26A4 in a small Japanese cohort with probable ceiling effects on hearing levels 16, 17. Wu et al 18 detected no similar relationship in a Taiwanese cohort, but their metric for hearing was not defined. Azaiez et al 11 found no relationship between number of SLC26A4 mutations and a binaural mean pure-tone threshold average in a Caucasian-majority cohort, but this discrepancy may reflect the misclassification of SLC26A4 polymorphisms as mutations 20 or the use of a binaural measure of hearing in a cohort with a high prevalence of unilateral hearing loss. We cannot rule out potential associations of number of mutant SLC26A4 alleles with sensory hearing because we had an insufficient number of audiograms with bone conduction data. This reflects, in part, a bias introduced by ceiling effects in individuals with severe and profound hearing loss in whom there is no response to bone-conduction stimuli at the limits of the test equipment.
We also observed that the presence of a cochlear anomaly in addition to EVA is not associated with more severe hearing loss when the contribution of other covariates (e.g., genotype) is taken into account. We are unable to directly compare our findings to those of a similar analysis by Wu et al 19 because they classified and grouped their ears with cochlear anomalies differently. Okumura et al 26 reported better hearing in 23 ears with additional cochlear anomalies among Japanese EVA patients, but they defined EVA as a midpoint diameter > 4mm and did not report their metric for hearing loss.
Although we detected a suggestive relationship between number of SLC26A4 mutant alleles and cochlear radiological status, the relationship was not statistically significant. This is consistent with two 14, 18 previous independent reports finding no correlation of number of mutant SLC26A4 alleles with pattern of inner ear malformations. In contrast, Azaiez et al 11 reported that individuals with biallelic SLC26A4 mutations have a more severe radiological phenotype, but ambiguities in their genotypic and phenotypic classification preclude a direct conclusion or comparison 20. We did not observe a significant relationship between size of the EVA and number of mutant SLC26A4 alleles, in contrast to the study of Madden et al 15 that more broadly defined EVA as a midpoint diameter >0.9 mm or an operculum diameter >1.8 mm. The strong inverse correlation of two mutant alleles of SLC26A4 with a vestibular aqueduct midpoint diameter <1.5 mm 12 may account for their observed relationship and the apparent discrepancy with our conclusion.
Our data suggest that neither the number of mutant alleles of SLC26A4 nor cochlear radiologic phenotype significantly affect longitudinal changes in hearing. Albert et al 14 reported a similar result. Although Madden et al 15 found that ears with monoallelic or biallelic SLC26A4 mutations are more stable than those with no mutation, the difference was not significant among ears with >36 months of follow-up. Our correlation of number of mutant alleles with severity of hearing loss but not longitudinal hearing changes may reflect an inadequate duration of follow-up in our study. We indeed observed a small but significant effect of duration of follow-up on the amount of deterioration in hearing in ears with less than severe hearing loss at baseline. It may also reflect the uncontrolled contribution of environmental factors (e.g., trauma) associated with changes in hearing in ears with EVA3.
We did not detect a significant relationship between the size of the air-bone gap (ABG) and either the number of SLC26A4 mutations, the presence of a cochlear anomaly, or the size of the vestibular aqueduct. The frequent association of EVA with ABGs and normal tympanometry is thought to reflect the existence of a third mobile window in the labyrinth 27. This theory is supported by the observation of abnormally low thresholds for the vestibular-evoked myogenic potential (VEMP) in a subpopulation of children with EVA 28. The lack of correlation of VA diameter with ABG size does not invalidate this hypothesis because the midpoint diameter of the VA may not be mechanistically relevant to this model. We did not measure VEMP thresholds on a sufficient number of our subjects to test this hypothesis 28.
Conclusion
Our study indicates that the number of mutant alleles of SLC26A4 has a significant association with severity of hearing loss in ears with EVA. Furthermore, the presence of structural anomalies of the cochlea is not associated with more severe hearing loss when other covariates (e.g., genotype) are taken into account. This information will be useful for prognostic counseling of patients and families with EVA.
Acknowledgments
This work was supported by NIH intramural research funds Z01-DC000039, Z01-DC000060 and Z01-DC000064, and the Intramural Research Program of the National Human Genome Research Institute. There are no financial disclosures to report.
This work was supported by intramural funds from the NIDCD. We thank the study families for their participation, our NIDCD colleagues for critical review of the manuscript, Ning Hu for data entry, and Tom Friedman and Barb Biesecker for support. Andrew Griffith had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Preliminary data presented during a poster session at the American Academy of Audiology Convention, Denver, CO, April 20, 2007
Contributor Information
Kelly A. King, Email: kingke@nidcd.nih.gov.
Byung Yoon Choi, Email: choiby@nidcd.nih.gov.
Christopher Zalewski, Email: zalewski@nidcd.nih.gov.
Anne C. Madeo, Email: madeoa@mail.nih.gov.
Ani Manichaikul, Email: amanicha@virginia.edu.
Shannon P. Pryor, Email: pryors@gmail.com.
Anne Ferruggiaro, Email: aferruggiaro@hesp.umd.edu.
David Eisenman, Email: deisenman@smail.umaryland.edu.
H. Jeffrey Kim, Email: hk7@georgetown.edu.
John Niparko, Email: jnipark1@jhmi.edu.
James Thomsen, Email: jthomsen@childrensent.com.
John A. Butman, Email: jbutmana@cc.nih.gov.
Andrew J. Griffith, Email: griffita@nidcd.nih.gov.
Carmen C. Brewer, Email: brewerc@nidcd.nih.gov.
References
- 1.Arcand P, Desrosiers M, Dube J, Abela A. The large vestibular aqueduct syndrome and sensorineural hearing loss in the pediatric population. J Otolaryngol. 1991;20(4):247–250. [PubMed] [Google Scholar]
- 2.Okumura T, Takahashi H, Honjo I, Takagi A, Azato R. Magnetic resonance imaging of patients with large vestibular aqueducts. Eur Arch Otorhinolaryngol. 1996;253(7):425–428. doi: 10.1007/BF00168496. [DOI] [PubMed] [Google Scholar]
- 3.Colvin IB, Beale T, Harrop-Griffiths K. Long-term follow-up of hearing loss in children and young adults with enlarged vestibular aqueducts: relationship to radiologic findings and Pendred syndrome diagnosis. Laryngoscope. 2006;116(11):2027–2036. doi: 10.1097/01.mlg.0000240908.88759.fe. [DOI] [PubMed] [Google Scholar]
- 4.Madden C, Halsted M, Benton C, Greinwald J, Choo D. Enlarged vestibular aqueduct syndrome in the pediatric population. Otol Neurotol. 2003;24(4):625–632. doi: 10.1097/00129492-200307000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Naganawa S, Koshikawa T, Fukatsu H, et al. Enlarged endolymphatic duct and sac syndrome: relationship between MR findings and genotype of mutation in Pendred syndrome gene. Magn Reson Imaging. 2004;22:25–30. doi: 10.1016/j.mri.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Reardon W, CF OM, Trembath R, Jan H, Phelps PD. Enlarged vestibular aqueduct: a radiological marker of pendred syndrome, and mutation of the PDS gene. Qjm. 2000;93(2):99–104. doi: 10.1093/qjmed/93.2.99. [DOI] [PubMed] [Google Scholar]
- 7.Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 8.Madeo AC, Pryor SP, Brewer C, et al. Pendred syndrome. Seminars in Hearing. 2006;27:160–170. [Google Scholar]
- 9.Li XC, Everett LA, Lalwani AK, et al. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18:215–217. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- 10.Usami S, Abe S, Weston MD, et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–192. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- 11.Azaiez H, Yang T, Prasad S, et al. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122(5):451–457. doi: 10.1007/s00439-007-0415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pryor SP, Madeo AC, Reynolds JC, et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42:159–165. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai CC, Shiao AS. Chronological changes of hearing in pediatric patients with large vestibular aqueduct syndrome. Laryngoscope. 2004;114(5):832–838. doi: 10.1097/00005537-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Albert S, Blons H, Jonard L, et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14:773–9. doi: 10.1038/sj.ejhg.5201611. [DOI] [PubMed] [Google Scholar]
- 15.Madden C, Halsted M, Meinzen-Derr J, et al. The influence of mutations in the SLC26A4 gene on the temporal bone in a population with enlarged vestibular aqueduct. Arch Otolaryngol Head Neck Surg. 2007;133:162–8. doi: 10.1001/archotol.133.2.162. [DOI] [PubMed] [Google Scholar]
- 16.Sato E, Nakashima T, Miura Y, et al. Phenotypes associated with replacement of His by Arg in the Pendred syndrome gene. Eur J Endocrinol. 2001;145:697–703. doi: 10.1530/eje.0.1450697. [DOI] [PubMed] [Google Scholar]
- 17.Sugiura M, Sato E, Nakashima T, et al. Long-term follow-up in patients with Pendred syndrome: vestibular, auditory and other phenotypes. Eur Arch Otorhinolaryngol. 2005;262:737–43. doi: 10.1007/s00405-004-0884-z. [DOI] [PubMed] [Google Scholar]
- 18.Wu CC, Chen YS, Chen PJ, et al. Common clinical features of children with enlarged vestibular aqueduct and Mondini dysplasia. Laryngoscope. 2005;115:132–7. doi: 10.1097/01.mlg.0000150691.85387.3f. [DOI] [PubMed] [Google Scholar]
- 19.Wu CC, Yeh TH, Chen PJ, et al. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: a unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope. 2005;115:1060–4. doi: 10.1097/01.MLG.0000163339.61909.D0. [DOI] [PubMed] [Google Scholar]
- 20.Choi BY, Stewart AK, Madeo AC, et al. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat. 2009;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madeo AC, Manichaikul A, Reynolds J, et al. Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Archives of Otolaryngology. 2009;135:670–6. doi: 10.1001/archoto.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemmerling MM, Mancuso AA, Antonelli PJ, et al. Normal modiolus: CT appearance in patients with large vestibular aqueduct. Radiology. 1997;204:213–9. doi: 10.1148/radiology.204.1.9205250. [DOI] [PubMed] [Google Scholar]
- 23.ANSI. S3.1-1999 American National Standard Maximum Permissible Ambient Noise Levels for Audiometric Test Rooms (Standard S3.1) New York: American National Standards Institute; 2003. [Google Scholar]
- 24.ANSI. S3.6 – 1996 American National Standard Specification for Audiometers (Standard S3.6) New York: American National Standards Institute; 2004. [Google Scholar]
- 25.King KA, Makisima T, Zalewski CK, et al. Analysis of auditory phenotype and karyotype in 200 females with Turner syndrome. Ear and Hearing. 2007;28(6):831–841. doi: 10.1097/AUD.0b013e318157677f. [DOI] [PubMed] [Google Scholar]
- 26.Okumura T, Takahashi H, Honjo I, et al. Sensorineural hearing loss in patients with large vestibular aqueduct. Laryngoscope. 1995;105:289–93. doi: 10.1288/00005537-199503000-00012. discussion 93–4. [DOI] [PubMed] [Google Scholar]
- 27.Merchant SN, Nakajima HH, Halpin C, et al. Clinical investigation and mechanism of air-bone gaps in large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol. 2007;116(7):532–541. doi: 10.1177/000348940711600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou G, Gopen Q, Kenna MA. Delineating the hearing loss in children with enlarged vestibular aqueduct. The Laryngoscope. 2008;118:2062–2066. doi: 10.1097/MLG.0b013e31818208ad. [DOI] [PubMed] [Google Scholar]