

Abstract
The mechanism by which mutations in the superoxide dismutase (SOD1) gene cause motor neuron degeneration in familial amyotrophic lateral sclerosis (ALS) is unknown. Recent reports that neuronal death in SOD1-familial ALS is apoptotic have not documented activation of cell death genes. We present evidence that the enzyme caspase-1 is activated in neurons expressing mutant SOD1 protein. Proteolytic processing characteristic of caspase-1 activation is seen both in spinal cords of transgenic ALS mice and neurally differentiated neuroblastoma (line N2a) cells with SOD1 mutations. This activation of caspase-1 is enhanced by oxidative challenge (xanthine/xanthine oxidase), which triggers cleavage and secretion of the interleukin 1β converting enzyme substrate, pro-interleukin 1β, and induces apoptosis. This N2a culture system should be an instructive in vitro model for further investigation of the proapoptotic properties of mutant SOD1.
Amyotrophic lateral sclerosis (ALS) is a paralytic disorder caused by degeneration of motor neurons. Although the majority of the cases are sporadic, about 25–30% are associated with mutations in the gene encoding superoxide dismutase (SOD1) (1). More than 50 different mutations in SOD1 have been found exclusively in familial ALS (FALS) patients. Because SOD1 is an antioxidant protein, the observation that SOD1 mutations cause motor neuron disease suggests the hypothesis that SOD1-associated FALS is a consequence of disturbed free radical homeostasis and resulting oxidative stress (2).
Several arguments support the hypothesis that toxicity of the mutant SOD1 protein arises through a novel adverse function, as suggested by the dominant inheritance pattern. Although many FALS-associated mutations reduce total cellular activity of SOD1 (3), some retain normal catalytic activity (4). Mice devoid of SOD1 activity survive into adulthood with normal motor function (5). Transgenic mice with high levels of mutant SOD1 protein and activity develop a disease similar to ALS (6, 7). Moreover, the acquired property does not involve a dominant negative mechanism (8).
Two categories of hypotheses have been proposed for the gain of function of the mutant SOD1 protein, predicated on the observation that the mutant molecule is unstable and subject to conformational modifications (9, 10). One invokes novel enzymatic activities, causing increased intracellular concentrations of hydroxyl radicals or nitronium ions (11–13). A second set proposes that conformational alterations induce the mutant molecule to form toxic aggregates (14–16) or to bind proteins aberrantly (17).
Evidence is accumulating that the neuronal death process is partially apoptotic. In contrast with native SOD1, which is antiapoptotic in neurons (18–20) and a determinant of life span in Drosophila (21), mutant SOD1 proteins promote apoptosis (15, 22, 23). These findings in neuronal cultures are supported by analyses of motor neuron death in FALS mice. Survival is prolonged by overexpression of the antiapoptotic protein bcl-2 and inhibition of caspase-1 (interleukin 1β converting enzyme or ICE) (24, 25). No direct evidence for activation of apoptotic cell death genes (e.g., caspases) has yet been described in FALS nor has cleavage of a caspase substrate been reported.
We investigated apoptosis caused by mutant SOD1 protein, by using transgenic ALS mice and mouse neuroblastoma N2a cell lines stably transfected with wild-type (WT) and mutant (G37R, G41D, and G85R) SOD1 cDNAs. We report here that mutant SOD1 promotes apoptosis in differentiated, oxidatively stressed N2a cells by activating caspase-1/ICE; this process entails secretion of mature interleukin 1β (IL-1β). Analogous proteolytic cleavage and activation of ICE occur in two lines of transgenic mice expressing hSOD1 G37R and G85R mutations, but not WT SOD1.
MATERIALS AND METHODS
Materials.
Catalase, human SOD1, and xanthine were purchased from Sigma, and xanthine oxidase was from Boheringer Mannheim. Manganese (III) 4,4′,4′′,4′′′ (21H,2H-porphirine-5,10,15,20-tetrayl) tetrakis (benzoic acid) (MnTBAP) was purchased from Calbiochem. Benzyloxycarbonil-Val-Ala-Asp (O-methyl)-fluoromethylketone (ZVAD-FMK), acetyl-Tyr-Val-Ala-Asp-chloromethylketone (Ac-YVAD-CMK), and acetyl-Tyr-Val-Ala-Asp-aldeide (Ac-YVAD-CHO) were from Bachem. Antibodies to human SOD1, GAP-43, and mouse ICE-p20 were from Calbiochem, Sigma, and Santa Cruz Biotechnology.
N2a Cell Culture.
N2a monolayer cell cultures were grown in 50% DMEM and 50% OPTI-MEM with 5% fetal bovine serum (FBS), 1% antibiotic antimycotic, and 400 μg/ml of the neomycin analog G418. Differentiation was induced within 3 days in serum-free medium. Differentiated cells were incubated without FBS for 6 days before treatments. For experiments with caspase inhibitors, cells were incubated for 30 min with the inhibitor before adding xanthine/xanthine-oxidase (X/XO).
Human SOD1 DNA Mutagenesis and Expression of hSOD1 in Cultured N2a Cells.
The SOD1 mutations (G37R, G41D, and G85R) were introduced into a plasmid clone of human genomic SOD1 (pHGSOD-SVneo). Stably transfected lines of N2a cells were selected with the drug G418, after transfection with SOD1 pHGSOD-SVneo plasmids (8).
Western Blot Analysis.
N2a cells and spinal cords from transgenic mice were lysed in buffer (150 mM NaCl/1% Nonidet P-40/12 mM sodium deoxycolate/0.1% SDS/50 mM Tris⋅HCl, pH 7.2) with protease inhibitors. Proteins were electrophoresed and blotted to poly(vinylidene difluoride) membrane. Blots were probed with anti-human SOD1, GAP-43, or mouse ICE-p20 antibodies and visualized with peroxidase-linked secondary antibodies by using chemiluminescence detection (Amersham).
Cell Viability and Nuclear Staining.
Cell viability was determined by using a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and trypan blue exclusion (26). The mode of cell death was assessed with the Hoechst 33342 (Molecular Probes) nuclear stain (27). Stained nuclei were examined immediately after treatment with the dye by fluorescence microscopy, sometimes simultaneously with propidium iodide staining to assess membrane integrity.
Assay for IL-1β Secretion.
IL-1β secretion was quantified by ELISA by using the Duoset ELISA Kit (Genzyme) using recombinant IL-1β as a standard.
SOD-1 Enzyme Assay.
Total SOD1 activity in N2a lysates was determined by the ability of SOD1 to inhibit superoxide-mediated reduction of tetrazolium to formazan (28). Because it interferes with SOD1 activity, SDS was omitted from the buffer. Activity from 50 μl of total lysates was determined in triplicate for two different cell preparations; mutant SOD activities were normalized to naive cells.
RESULTS
N2a Cells Stably Express Mutant and WT Human SOD1.
To study neuronal cell death induced by mutations in SOD1, we have used a mouse neuroblastoma cell line (N2a) stably transfected to express mutant (G37R, G41D, and G85R) and WT SOD1. These cells are maintained as nondifferentiated, dividing cells (Fig. 1a) but can be induced to differentiate to form neural cells (Fig. 1b) by serum deprivation. This property allows comparison of effects of mutations in SOD1 in cells with neuronal and non-neuronal phenotypes. We first verified patterns of expression of human SOD1 (hSOD1) by using Western immunoblotting with an anti-human SOD1 polyclonal antibody. All transfected SOD1 cDNAs were expressed in the N2a cells (Fig. 1c). While hWT, hG37R, and hG41D (Fig. 1c, lanes 4–9) comigrate with the commercial hSOD1 (lane 1) at around 21 kDa on a 15% SDS/PAGE gel, the hG85R variant (lanes 10–11) has a faster electrophoretic mobility corresponding to that of the endogenous mouse SOD1. The levels of hSOD1 expressed were similar in WT-, G37R-, and G41D-expressing cells whereas lower levels were seen in the G85R cultures (Fig. 1c, lanes 10 and 11; the G85R lanes have twice the amount of total proteins loaded in other lanes). Moreover, comparable levels of hSOD1 protein were detected in undifferentiated and differentiated cells. Immunoblotting with a polyclonal antibody generated against a peptide identical in human and mouse allowed comparison of levels of hSOD1 to that of endogenous mouse SOD1 (mSOD1). hSOD1 (WT, G37R, and G41D) and mSOD1 protein levels were similar (data not shown). We examined total SOD1 activity in the undifferentiated N2a cell lines. When compared with that of the naive N2a cells, total SOD1 activity of the WT hSOD1-expressing cells was increased about 5-fold. The G37R-, G41D-, and G85R-positive cells showed 4.5-, 2.8-, and 1.4-fold increases, respectively. In each hSOD1-positive cell line, there was a correlation between enzyme concentration and activity.
Figure 1.

Expression of WT and mutant hSOD1 in undifferentiated and differentiated N2a cells. (a) Undifferentiated, WT hSOD1-expressing cells grown in medium containing 5% fetal bovine serum. (b) Differentiated WT hSOD1 cells 4 days after serum deprivation. (c) Lysates from undifferentiated and differentiated cells were electrophoresed on 15% SDS/PAGE gels and immunoblotted by using a sheep polyclonal antibody that recognizes human SOD1. Comparable levels of hSOD1 are detected before and after differentiation. The anti-human SOD1 antibody shows a slight cross-reactivity with endogenous mouse SOD1; thus there is a weak band corresponding to the electrophoretic mobility of mSOD1. (d) Immunoblots stained with a mouse mAb raised against GAP-43, a neuron-specific, growth-associated protein used as marker of neuronal differentiation. Lysates were electrophoresed on 10% SDS/PAGE gel. A positive band migrating at an apparent Mr of 46 kDa, corresponding to the electrophoretic mobility of GAP-43 on 10% gels, is detected only in differentiated cells.
We examined the degree of differentiation after serum withdrawal as indicated by expression of the neuron-specific protein GAP-43. Immunoblots demonstrated GAP-43 only in differentiated cells (Fig. 1d); GAP-43 protein levels were similar in all cell lines.
Mutant SOD1 Does Not Impair N2a Cell Viability.
Because mutant SOD1 protein is proapoptotic in neuronal cells in culture and causes motor neuron cell death in vivo, we examined the effects of mutant and WT hSOD1 on the viability of the N2a cells by using the MTT cytotoxicity assay (26). In both undifferentiated (Fig. 2a) and differentiated cells (Fig. 2b), mutant SOD1 does not affect cell viability. A slight decline in survival after expression of mutant SOD1 did not achieve statistical significance. Similar results were seen by using trypan blue exclusion.
Figure 2.

Expression of SOD1 mutations does not affect N2a cell viability. Effect of mutant and WT hSOD1 expression on cell viability of undifferentiated (a) and differentiated (b) N2a cells. MTT assay was performed as described in Materials and Methods, and results were expressed as percentage of naive cells. Viability estimates by trypan exclusion corresponded to those determined by MTT assay. Data represent the mean ± SEM of six observations of three independent experiments.
X/XO Impairs Viability of Differentiated N2a Cells with Mutant, But Not WT, SOD1.
While baseline viability of our N2a cells was not significantly affected by mutations in SOD1, we considered the possibility that oxidative stress might reduce N2a cell survival in the presence of mutant SOD1. We therefore re-examined cell viability after exposing the cells to X/XO to generate superoxide anion (O2−⋅) and hydrogen peroxide (H2O2) (29). Exposure of undifferentiated N2a cells to X/XO for 4 hr did not produce cell death, regardless of whether the cells possessed only the endogenous SOD1 or expressed WT or mutant hSOD1 (Fig. 3a). By contrast, in differentiated cell cultures expressing mutant SOD1, the same treatment increased cell death by 40–60% (Fig. 3b). Neither X nor XO alone affected cell viability. The effect of X/XO on viability in differentiated N2a cells expressing mutant SOD1 was concentration dependent. At the highest concentration tested (100 μM X, 10 milliunits/ml XO), a 40% reduction in cell viability was seen for G37R- and G41D-positive N2a cells, whereas a more robust decrease (60%) was evident in X/XO-treated G85R-positive cells.
Figure 3.

X/XO induces a concentration-dependent loss of viability in differentiated N2a cells expressing mutant SOD1. Cell viability of either undifferentiated (a) or differentiated (b) cells was determined by using the MTT assay after exposing the cells to different concentrations of X + 10 milliunits/ml XO for 4 hr at 37°C. Viability was expressed as a percentage of treated to untreated cells for each cell line. Five experiments performed in triplicate showed that in undifferentiated cells, X/XO did not induce loss of cell viability at any concentration tested (P > 0.5, Student’s t test). A concentration-dependent significant loss of viability was observed in differentiated cells expressing the three mutant SODs (P < 0.05 or <0.01, Student’s t test).
X/X0 Induces Apoptotic Morphology in Differentiated, Mutant SOD1-Positive N2a Cells.
We next examined the morphological properties of mutant SOD1-positive N2a cells dying after exposure to X/XO. As monitored with Hoechst 33342 nuclear stain, X/XO-induced death was characterized by apoptotic features: nuclear chromatin condensation, nuclear shrinkage, and cell dissolution without swelling. Apoptotic cell bodies remained adherent to the culture dish while their processes were easily detached. As shown in Fig. 4, after exposure to X/XO (100 μM, 10 milliunits/ml) for 4 hr, chromatin condensation and formation of small apoptotic nuclei were readily evident in the G85R cultures (Fig. 4f), whereas nuclear morphology was unchanged in naive and WT-expressing cells (Fig. 4 d and e). Results were similar for cells with the other two mutant SODs. After 2 hr of X/XO exposure, several G85R cells showed blue positive chromatin condensation but excluded propidium iodide, an indication that the cell membrane remained intact. However, after treatment with X/XO for 4 hr, nuclei of the G85R cells showed propidium iodide staining, consistent with loss of integrity of both the cell and nuclear membranes. At that time, the G85R cells also demonstrated apoptotic condensation of nuclear chromatin (data not shown).
Figure 4.
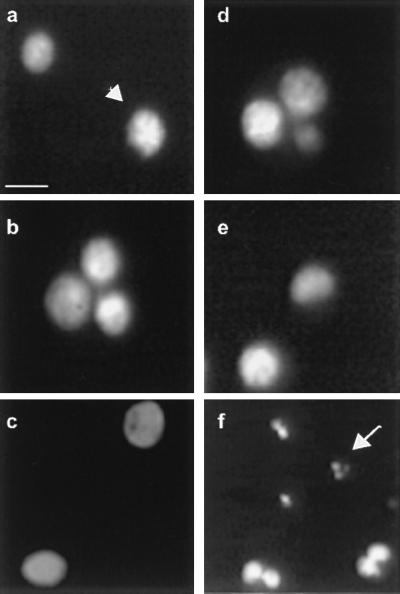
X/XO-induced cell death in G85R-expressing cells is accompanied by morphological changes typical of apoptosis. (a–c) Fluorescent images of Hoechst 33342 stained nuclei from differentiated cells under normal conditions (without X/XO). Nuclei from naive (a), WT hSOD1- (b), or G85R- (c) expressing cells were all round and uniformly stained with the blue dye. (d–f) Fluorescent images of Hoechst 33342 stained nuclei of differentiated cells exposed to X/XO (100 μM X, 10 milliunits/ml XO) for 4 hr. Naive (d) and WT-expressing cells (e) maintain the same nuclear morphology as in a and b, without signs of apoptosis. Small apoptotic nuclei with chromatin condensation are evident in the G85R-positive cells (f). The arrow and arrowhead, respectively, indicate apoptotic and nonapoptotic nuclei. (Bar: 20 μm).
X/XO Enhances Mature IL-1β Secretion.
Inhibition of ICE slows progression of the motor neuron disease in G93A transgenic ALS mice (25), suggesting that this caspase is involved in the death of neurons with mutant SOD1. To determine whether X/XO-induced apoptosis in differentiated N2a cells expressing SOD1 correlates with caspase-1/ICE activation, we measured levels of mature IL-1β in the medium of N2a cells exposed to different concentrations of X/XO. ICE is the only caspase identified so far to process pro-IL-1β into mature IL-1β in vitro and in vivo (30). Thus, secretion of mature IL-1β is a specific indication of ICE activity. In lines expressing all three mutant SODs, X/XO induced a concentration-dependent increase in mature IL-1β levels (Fig. 5a). Similar trends were seen after exposing the cultures to X/X0 for only 2 hr. Naive and WT-expressing cells treated with the same concentrations of X/XO showed no increase in IL-1β secretion as compared with untreated cells. The X/XO induction of IL-1β secretion was evident only when N2a cells were in the differentiated state (data not shown). Because processing of pro-IL-1β is a specific indication of ICE activation, these data argue that in the presence of mutant SODs X/XO induces ICE activation.
Figure 5.

X/XO induces a concentration-dependent increase in mature IL-1β secretion and enhances ICE cleavage in differentiated mutant SOD1-positive cells. (a) After exposure of the cells to three different concentrations of X/XO for 4 hr at 37°C, secreted IL-1β was determined by ELISA and expressed as pg/ml per cell number. Each column of the histogram represents the mean ± SEM value of released IL-1β obtained from 9–12 single determinations. As shown by ∗, a statistical difference between untreated and X/XO-treated groups is observed only in mutant SOD1-expressing cells (P < 0.05). (b) Differentiated cells expressing either the WT hSOD1 or mutant hSOD1 were exposed to X/XO (100 μM X, 10 milliunits/ml XO). After 4 hr, the processing of ICE was detected by Western blotting using a polyclonal antibody against the p20 subunit. In WT-expressing cells, the antibody recognized only the p45 proactive form of ICE. The cleavage product (p20) is present in G37R-, G41D-, and G85R-positive cells after treatment with X/XO. In G41D- and G85R-expressing cells, the p20 product is also present without X/XO treatment and slightly increases after X/XO.
Caspase-1/ICE Is Activated in Differentiated N2a Cells with Mutant, But Not WT, SOD1 But Does Not Necessarily Trigger Cell Death.
ICE is synthesized as a 45-kDa proform that is cleaved during activation to generate p10 and p20 subunits (30). In vitro, activation of p45 ICE occurs through a sequential series of proteolytic steps to generate p35, p22/p12, and p20/p10 fragments of progressively increasing catalytic activity (31). To test the hypothesis that ICE must be activated in our N2a cells concomitantly with IL-1β secretion, we assayed for ICE- cleavage products by using Western immunoblots with a polyclonal antibody against residues 276–294 (p20 subunit) of ICE. After exposure to X/XO (100 μM, 10 milliunits/ml) for 4 hr, cells expressing WT hSOD1 did not show evidence of the p20 fragment. By contrast, under similar conditions, all three mutant hSOD1 produced the p20 subunit. Indeed, in G41D- and G85R-expressing cells the p20 product was present even without X/XO treatment, even though these cells showed no loss in cell viability or increase in basal IL-1β secretion. In those lines, treatment with X/XO enhanced p20 production. In G37R-positive cells, the p20 fragment was detected only after treatment with X/XO (Fig. 5b). Positive immunostaining for ICE p35 was detected in all lysates from mutant SOD-expressing cells but not in WT-expressing cells. Immunostaining of positive bands was markedly diminished by preadsorption with the synthetic peptide used as immunogen to produce the antibody (data not shown).
Caspase-1/ICE Is Activated in G37R and G85R Transgenic Mice.
These data are uniformly consistent with the hypothesis that ICE is activated selectively in neurally differentiated N2a cells expressing mutant, but not WT, hSOD1. Moreover, in the face of exogenous oxidative challenge (X/XO), this activation leads to both IL-1β secretion and loss of viability. To determine whether this is relevant to the action of mutant SOD1 protein in motor neurons in vivo, we studied ICE activation in mice bearing the G37R (line 42) (9) and G85R (line 164) (32) transgenes. Three animals from each group were analyzed. At the time of sacrifice, the G37R were near the end stage of motor neuron disease (≈6 months), whereas all three G85Rs were analyzed at 4.5 months of age and were unaffected. The expected onset of disease for these G85R mice is around 10 months of age. Immunoblots (Fig. 6) revealed ICE-like cleavage products in extracts of lumbo-sacral spinal cord of both G37R and G85R mice. Similar fragments were evident in brain extracts of these two lines of mice (data not shown). No such fragments were detected in transgenic mice carrying WT hSOD1. While cleavage of the p45 precursor in G37R mice generated a p22 subunit, in the G85R mice the p20 fragment was preferentially formed.
Figure 6.

ICE is activated in spinal cords of G37R and G85R transgenic mice. Immunoblots of lumbo-sacral spinal cord extracts of (from left to right) WT, G37R, and G85R hSOD1 transgenic mice probed with the same polyclonal antibody anti-ICE p20 subunit as in the legend of Fig. 5.
Caspase Inhibitors Prevent X/XO-Induced Cell Death, Caspase-1/ICE Cleavage, and IL-1β Secretion.
These experiments are consistent with activation of ICE-mediated cell death pathways in the presence of mutant SOD1. Because the same activation process was evident in ALS mice in vivo and in neural N2a cells in vitro, we used the N2a line to determine whether oligopeptide inhibitors of caspases prevent cell death, ICE cleavage, and IL-1β secretion. We first examined the effect of ZVAD-FMK and Ac-YVAD-CHO on X/XO-induced cell death in the G85R-expressing cell cultures. ZVAD-FMK is an irreversible tripeptide inhibitor that has a broad specificity for ICE and CPP32-like proteases. Ac-YVAD-CHO is a reversible tetrapeptide inhibitor that specifically inhibits ICE in vitro (33); its sequence closely resembles the pro-IL-1β cleavage site (YVHD). While ZVAD-FMK completely protected the cells from X/XO-induced loss of viability, the effect of Ac-YVAD-CHO was more modest (≈50% protection) (Fig. 7a). Both inhibitors had no significant effect on the viability of untreated cells and when tested on naive and WT-hSOD1-positive cultures did not affect cell viability. It was not clear whether the different effects of these two caspases inhibitors reflected differing specificities of the compounds toward different caspases or the fact that ZVAD-FMK is an irreversible inhibitor while Ac-YVAD-CHO is not. In two subsequent experiments we therefore tested a third compound, acetyl-Tyr-Val-Ala-Asp-chloromethylketone (Ac-YVAD-CMK), an irreversible tetrapeptide inhibitor that is more selective for ICE-like than for CPP32- or Ich-1-like caspases (33). This compound protected about 60% of the G85R-positive N2a cells from death after X/XO-treatment, an effect comparable to that of Ac-YVAD-CHO. Thus, the differential effect of ZVAD-FMK and Ac-YVAD-CHO is not the result of their different mode of action. The data suggest that activation of ICE alone is not sufficient to induce cell death in cells expressing mutant SODs. It is therefore possible that oxidative stress induces activation of other caspase(s) or more than one apoptotic death pathway in these cells.
Figure 7.

Caspase inhibitors protect G85R-expressing cells against X/XO-induced loss of cell viability, caspase-1/ICE cleavage, and increased IL-1β secretion. (a) Differentiated G85R-expressing cells were preincubated for 30 min at 37°C with the inhibitor before addition of X/XO (100 μM X, 10 milliunits/ml XO), in the presence of inhibitors. Cell viability then was determined by the MTT assay as described and expressed as a percentage of untreated cells. Data are the mean ± SEM of five independent experiments for ZVAD-FMK and four independent experiments for Ac-YVAD-CHO. (b) Levels of IL-1β were measured as described above in the medium of G85R cells treated with X/XO in the presence or absence of caspase inhibitors. Data represent the mean ± SEM of nine observations of three independent experiments. (c) Differentiated G85R-positive cells were treated as described in a. After lysis, processing of ICE was detected by Western immunoblot as described above.
We then tested the effect of Ac-YVAD-CHO and ZVAD-FMK on levels of secreted IL-1β in differentiated N2a cells. Both compounds completely blocked the secretion of mature IL-1β in X/XO-treated G85R-expressing cells (Fig. 7b). Neither inhibitor had any effect on IL-1β levels in untreated G85R-positive cells or naive and WT-expressing cells. Concomitant with inhibition of mature IL-1β production, Ac-YVAD-CHO blocked ICE cleavage in the same cell line (Fig. 7c).
Catalase But Not Exogenous SOD1 Blocks X/XO-Induced Toxicity.
Extracellular application of X/XO leads to the formation of both O2−⋅ and H2O2 (29). A priori, it is not apparent which species mediates N2a cell death in the presence of mutant SOD1. If the primary toxicity arose from the generation of extracellular O2−⋅, then the addition of extracellular SOD1 might be palliative by diminishing O2−⋅ levels. Analogously, if the main toxic species is H2O2, addition of extracellular catalase might be protective. To distinguish these possibilities, we exposed the G85R-positive line to X/XO in the presence of either catalase or commercial hSOD1. Although exogenous hSOD1 did not protect the cells from X/XO toxicity, catalase completely restored cell viability (Fig. 8). In motor neurons deprived of brain-derived neurotrophic factor and ganglion cell cultures, hSOD1 has been shown to be protective only when delivered intracellularly (34). We therefore repeated the experiment by using manganese (III) 4,4′,4′′,4′′′ (21H,2H-porphirine-5,10,15,20-tetrayl) tetrakis (benzoic acid) (MnTBAP), a membrane-permeable scavenger of O2−⋅ (35). At 100 μM (a concentration shown to be protective against O2−⋅-mediated toxicity) (34), MnTBAP was not protective whereas catalase provided nearly complete protection against X/XO-induced cell death. These data argue that the mutant SOD1 molecule is cytotoxic through an interaction with H2O2. Consistent with this conclusion, we observed that direct addition of H2O2 induced a significant loss of cell viability only in the differentiated G85R-expressing cells but not in naive and WT-hSOD1-positive N2a cells (data not shown).
Figure 8.

Catalase protects G85R-positive cells against X/XO-induced loss of cell viability. The G85R-positive line was treated with X/XO in the presence of either catalase (100 units/ml) or hSOD1 (90 units/ml). The viability of each group, as determined by the MTT assay, is expressed as percentage of untreated cells. Each column represents the mean ± SEM of four independent experiments performed in triplicate.
DISCUSSION
Although mutations in the gene encoding SOD1 were described in patients with FALS more than 5 years ago (1), it remains unclear how the mutant protein leads to motor neuron cell death. The present model recapitulates a neural-specific death process triggered selectively by mutant, but not WT, SOD1 and thus provides a useful tool for dissecting the mechanism of mutant SOD1 cytotoxicity. Using this in vitro model and transgenic ALS mice, we report that neural cells and tissues expressing mutant SOD1 show cleavage of caspase-1/ICE. Activation of a specific caspase, and resulting substrate cleavage, have not previously been described in SOD1-mediated ALS.
Cleavage of ICE in human monocytic tumor cells (THP.1) begins with the p45-kDa pro-ICE precursor and proceeds via fragments of 35 and 33 kDa to the active forms of either 22 or 20 and 10 kDa (31); the active ICE is a tetramer with two monomers of the 20- (or 22) and 10-kDa forms (36). Our findings are consistent with this sequence. Thus, although there is a partial caspase-1 cleavage from the p45 to the p35 fragments in control and transgenic WT hSOD1 mice, the fully active p22 and p20 fragments are present only in transgenic mice and in neurally differentiated N2a cells with ALS-related mutations. This cleavage pattern is particularly striking in association with conditions favoring apoptotic neural cell death. In differentiated N2a cells, the relative levels of the p22 and p20 subunits increased after X/XO treatments that trigger apoptotic death. Analogously, in both the G37R and G85R transgenic mice, a prominent p22/p20 fragment is seen in the lumbo-sacral spinal cord, but it is not seen in the mice overexpressing WT hSOD1 protein.
In many paradigms, apoptotic cell death reflects activation of cascades of caspases initiated by diverse stimuli (37). Our findings highlight the complexity of these multistep pathways. In this system, mutant SOD1 is required for caspase-1 cleavage but is not sufficient to fully activate the death process. The latter requires an additional stimulus: the oxidative challenge imposed by extracellular X/XO. X/XO fully activates caspase-1 cleavage, causing IL-1β secretion and apoptosis. Moreover, it is likely that caspases other than just ICE are implicated in these events, as ICE-specific inhibitors that block ICE cleavage and fully suppress IL-1β secretion [Ac-YVAD-CHO and acetyl-Tyr-Val-Ala-Asp-chloromethylketone (Ac-YVAD-CMK)] do not completely block cell death. By contrast, inhibitors not selective for ICE (e.g., ZVAD-FMK) completely rescue the N2a cells after X/XO. In THP.1 and Jurkat cells, ZVAD-FMK blocks apoptosis induced by different stimuli by inhibiting activation of pro-CPP32 to its active form (38). Because we did not use a specific inhibitor of caspase-3/CPP32, we cannot rule out the possibility that CPP32, or caspase(s) other than ICE, are involved in X/XO-induced cell death.
The enhanced sensitivity of cells with mutant SOD1 to X/XO is not a consequence of diminished total cellular SOD1 activity. Although they possessed the lowest dismutase activity among the five cell lines analyzed, naive N2a cells did not die after treatment with X/XO. X/XO killed all three mutant SOD1-positive cells whose SOD1 activity exceeded that of the naive cells. Rather, the experiments here favor the view that the N2a cells died because of an adverse interaction mediated by the mutant SOD1 protein. The rescue of these cells by catalase but not by either exogenous SOD1 or a membrane-permeable SOD1 mimic suggests that H2O2 rather than O2−• is a primary factor in the death process.
Our observations are broadly consistent with several recent studies documenting that mutant, but not WT, SOD1 is proapoptotic in neural cell lines or primary neurons (15, 22, 23). A subtle difference in our differentiated N2a cells is that we see no loss of viability in the absence of an oxidative stimulus provoked by adding X/XO. We speculate that this finding reflects the fact that we used stably transfected cell lines, possibly selecting for cells inherently resistant to the toxicity of the mutant SOD1 protein. Indeed, a careful comparison of these stably transfected cells with cells dying after transient SOD1 transfection may provide insight into mechanisms that ameliorate the toxicity of the abnormal protein. Similarly, a comparative analysis of the neural and undifferentiated cells may reveal why the neural cells are more susceptible to apoptosis after X/XO. It is possible, for example, that one or more antiapoptotic proteins, as well as endogenous antioxidants, may be down-regulated after neural differentiation. Candidate proteins might include the antiapoptotic gene bcl-2, antioxidants like catalase or glutathione peroxidase, or other SOD proteins, such as manganese SOD. All are well documented to modify patterns of cell death in cultured neurons.
Our results from both differentiated N2a cells and transgenic ALS mice suggest a two-stage model of cell death in which an inherited molecular defect (SOD1 mutation) renders a neuron susceptible to an exogenous stimulus (X/XO). In turn, this model suggests the hypothesis that the expression of the ALS phenotype in patients with SOD1 gene mutations may require both the genetic defect and an additional event, with the added corollary that susceptibility to the additional, possibly exogenous, trigger is enhanced by aging. This N2a cell model should be an ideal resource to explore the proximal mechanisms whereby the mutant protein activates one or more cascades of caspases and the determinants of the final levels of programmed neuronal death.
Acknowledgments
We thank Drs. J. Yuan and L. Hayward for helpful discussions. This study was supported by the Amyotrophic Lateral Sclerosis Association, Muscular Dystrophy Association, National Institutes of Health Grants 1PO1NS31248–02 and 5F32HS10064, Telethon Italia, the Pierre L. de Bourgknecht ALS Research Foundation, and the Myrtle May MacLellan Fund.
ABBREVIATIONS
- SOD1
superoxide dismutase
- ALS
amyotrophic lateral sclerosis
- FALS
familial ALS
- ICE
interleukin 1β converting enzyme
- WT
wild type
- IL-1β
interleukin 1β
- X/XO
xanthine/xanthine-oxidase
- Ac-YVAD-CHO
acetyl-Tyr-Val-Ala-Asp-aldeide
- ZVAD-FMK
benzyloxycarbonil-Val-Ala-Asp (O-methyl)-fluoromethylketone
- MTT
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J O, Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Brown R H., Jr Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 3.Bowling A C, Schulz J B, Brown R H, Jr, Beal M F. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt D R, Lee M K, Slunt H S, Guarnieri M, Xu Z-S, Wong P C, Brown R H, Price D R, Sisodia S S, Cleveland D W. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reaume A G, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Lood D G, Beal M F, Brown R H, et al. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 6.Gurney M E, Pu H, Chiu A Y, Dal Canto M C, Polchow C Y, Alexander D D, Caliendo J, Hentati A, Kwon Y W, Deng H X, et al. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 7.Ripps M E, Huntley G W, Hof P R, Morrison J H, Gordon J W. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borchelt D R, Guarnieri M, Wong P C, Lee M K, Slunt H S, Xu Z-S, Sisodia S S, Price D L, Cleveland D W. J Biol Chem. 1995;270:3234–3238. doi: 10.1074/jbc.270.7.3234. [DOI] [PubMed] [Google Scholar]
- 9.Wong P C, Pardo C A, Borchelt D R, Lee M K, Copeland N G, Sisodia S S, Cleveland D W, Price D L. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 10.Lyons T J, Liu H, Goto J J, Nerissian A, Roe J A, Graden J A, Café C, Ellerby L M, Bredesen D E, Gralla E B. Proc Natl Acad Sci USA. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 12.Yim M B, Kang J-H, Yim H-S, Kwak H-S, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 14.Chou S, Wang H, Komai K J. Comp Neuropathol. 1995;10:249–258. [Google Scholar]
- 15.Durham H D, Roy J, Dong L, Figlewicz D A. J Neuropathol Exp Neurol. 1997;5:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Bruijn L I, Houseweart M, Kato S, Anderson K L, Anderson S D, Ohama E, Reaume A G, Scott R W, Cleveland D W. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 17.Kunst C B, Mezey E, Brownstein M J, Patterson D. Nat Genet. 1997;15:91–94. doi: 10.1038/ng0197-91. [DOI] [PubMed] [Google Scholar]
- 18.Rothstein J D, Bristol L A, Hosler B, Brown R H, Jr, Kuncl R W. Proc Natl Acad Sci USA. 1994;91:4155–4159. doi: 10.1073/pnas.91.10.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Troy C M, Shelanski M L. Proc Natl Acad Sci USA. 1994;91:6384–6387. doi: 10.1073/pnas.91.14.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlund L J S, Deckwerth T L, Johnsosn E M. J Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 21.Parkes T L, Elia A J, Dickinson D, Hilliker A J, Philips J P, Boulianne G L. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 22.Rabizadeh S, Butler Gralla E, Borchelt D R, Gwinn R, Valentine J S, Sisodia S, Wong P, Lee M, Hahn H, Bredesen D E. Proc Natl Acad Sci USA. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghadge G D, Lee J P, Bindokas V P, Jordan J, Ma L, Miller R J, Roos R P. J Neurosci. 1997;17:8756–8766. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 25.Friedlander R M, Wang J, Gagliardini V, Brown R H, Jr, Yan Y. Nature (London) 1997;388:31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- 26.Behl C, Davis J B, Lesley R, Schubert D. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson M D, Burne F F, Raff M C. EMBO J. 1994;33:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beauchamp C, Fridovich I. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 29.Olanow C W. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 30.Thornberry N A, Bull H G, Calaycay J R, Chapman K T, Howard A D, Kostura M J, Miller D K, Molineaux S M, Weidner J R, Aunins J, et al. Nature (London) 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 31.Yamin T-T, Ayala J M, Miller D K. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 32.Bruijn L I, Becher M, W, Lee M K, Anderson K L, Jenkins N A, Copeland N G, Sisodia S S, Rothstein J D, Borchelt D R, Price D L, et al. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 33.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, et al. Nature (London) 1996;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 34.Estevez A G, Spear N, Manuel S M, Radi R, Henderson C E, Barbeito L, Beckman J S. J Neurosci. 1997;18:923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faulkner K M, Liochev S I, Fridovich I. J Biol Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- 36.Wilson K P, Black J-A F, Thomson J A, Kim E E, Griffith J P, Navia M A, Murcko M A, Chambers S P, Aldape R A, Raybuck S A, et al. Nature (London) 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 37.Pettmann B, Henderson C E. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 38.Slee E A, Zhu H, Chow S C, MacFarlane M, Nicholson D W, Cohen G M. Biochem J. 1996;315:21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]