

Abstract
The components of complex peptide mixtures can be separated by liquid chromatography, fragmented by tandem mass spectrometry, and identified by the SEQUEST algorithm. Inferring a mixture's source proteins requires that the identified peptides be reassociated. This process becomes more challenging as the number of peptides increases. DTASelect, a new software package, assembles SEQUEST identifications and highlights the most significant matches. The accompanying Contrast tool compares DTASelect results from multiple experiments. The two programs improve the speed and precision of proteomic data analysis.
Keywords: proteomics, protein identification, SEQUEST, data analysis, software, differential analysis, subtractive analysis, MudPIT, assembly
1. Introduction
Protein identification through mass spectrometry has become a major application of genetic sequence information. Two forms of protein identification have emerged. “Mass mapping” enzymatically digests an unknown protein and then identifies it by the masses of the peptides produced. Tandem mass spectrometry of peptides, on the other hand, fragments peptides to reveal their sequences.1,2 Both methods are effective for the identification of proteins isolated by polyacrylamide gels.3,4 Tandem mass spectrometry, however, is applicable to protein mixtures before isolation as well. “Shotgun proteomics” describes the process of digesting and separating a complex protein mixture, acquiring spectra from its peptides, and identifying the proteins originally found in the sample.5–9
Liquid chromatography is commonly used to separate mixtures of peptides for shotgun proteomics. Single-dimension liquid chromatography coupled to tandem mass spectrometry (LC/MS/MS) has been used to identify the components of protein complexes and subcellular compartments.5–12 This technique is capable of identifying hundreds of peptides in an hour.13 Two dimensions of liquid chromatography (LC/LC) enable many thousands of peptides to be identified through improved resolution. Link et al. used this approach, called multidimensional protein identification technology or “MudPIT”, to identify the components of large protein complexes such as yeast and human ribosomes, and Washburn et al. performed an analysis of whole yeast cells.8,9
Amino acid sequences of peptides can be inferred from tandem mass spectra.14 The SEQUEST algorithm automates this process.7 The software begins by enumerating candidate database peptides that match the observed peptide's mass. The sequences are quickly checked against the spectrum by a preliminary scoring algorithm, and obvious nonmatches are removed. A more extensive scoring algorithm cross-correlates sequence-derived theoretical spectra against the observed spectrum, and sequences are ranked on the basis of these scores. SEQUEST's automation of this process has helped the use of tandem mass spectrometry to expand from analytical chemistry to biology.
Because SEQUEST functions at the level of individual spectra, additional software is necessary to assemble information at the protein level. SEQUEST SUMMARY15 is broadly used, but it can only coordinate the results from a single dimension of chromatography. AUTOQUEST,15 a more extensive algorithm, can handle multidimensional results, but it has several limitations. As the number of peptides increases, the output grows in size very rapidly. It categorizes identifications by score, but the range of scores in each category is too broad for adequate discrimination. The program cannot organize results from DNA database searches. In addition, AUTOQUEST will not install alongside the commercial version of SEQUEST. Neither program allows much latitude for users to set standards on which identifications should be reported and which should not.
The DTASelect package was developed to assemble and evaluate shotgun proteomics data. The program applies multiple layers of filtering to SEQUEST search results and supplies several analytical reports in text and HTML files. The Contrast program is an accompanying tool for comparing multiple experiments and/or criteria sets. It is particularly useful for comparing results from different experimental conditions. This paper describes these two programs as demonstrated by an analysis of the purified yeast 26S proteosome using LC/MS/MS12 and MudPIT.16
2. Experimental Section
2.1. DTASelect Algorithm
DTASelect functions in three phases: summarization, evaluation, and reporting. The summarization phase reads individual spectrum identifications from the SEQUEST output (.out) files and assembles information for each locus (the database identity of the protein or gene from which the peptide derives). The evaluation phase applies the criteria indicated by the user for spectra and proteins, removing those that do not qualify. The reporting phase creates files to be read in a web browser or spreadsheet. At each step, the user is notified of DTASelect's progress.
Summarization collects the most important information from SEQUEST identification. Each identification's output file is read to acquire the cross-correlation score (XCorr), normalized difference between first and second match scores (DeltCN), rank by preliminary score (Sp rank), identified sequence, precursor m/z, protein or genetic locus from which this peptide derives, intensity, and percentage of fragment ions matched between predicited and observed spectra. The peptides are sorted by locus, and peptides for each locus are sorted by sequence. Next, the same database used by SEQUEST is consulted for the full protein sequence, descriptive name, and peptide positions. The information from the output files and database lookup is stored to the DTASelect.txt file so that this step can by bypassed in subsequent analyses of this sample.
Evaluation applies user-selected criteria to the matches. An identification is included only when it passes all specified spectrum criteria (see Table 1). Selection is next conducted at a higher level, retaining proteins that have a sufficient number of different peptides or that have at least one peptide showing up several times (see Table 2). Two filters reduce the redundancy of results. DTASelect can choose the best example of multiple spectra matching the same sequence by score or by signal intensity. In addition, many proteins may be indicated by exactly the same sets of peptides; DTASelect groups together proteins that are identical in sequence coverage. These selections highlight peptides and proteins that represent the entire sample.
Table 1.
DTASelect Spectrum Filtersa
| option | default | individual spectrum filters |
|---|---|---|
| -1 # | 1.8 | set lowest +1 XCorr |
| -2 # | 2.5 | set lowest +2 XCorr |
| -3 # | 3.5 | set lowest +3 XCorr |
| -d # | 0.08 | set lowest DeltCN |
| -c # | 1 | set lowest charge state |
| -C # | 3 | set highest charge state |
| -i # | 0.0 | set lowest proportion of fragment ions observed |
| -s # | 1000 | set maximum Sp ranking |
| -m 0 | require peptides to be modified | |
| -m 1 | X | include peptides regardless of modification |
| -m 2 | exclude modified peptides | |
| -y 0 | X | include peptides regardless of tryptic status |
| -y 1 | include only half- or fully tryptic peptides | |
| -y 2 | include only fully tryptic peptides | |
| option | extended filters (off by default) | |
| -Sic $ | sequences must contain all of these characters (excludes C terminal residue) | |
| -Sip $ | sequences must contain this pattern | |
| -Sec $ | sequences must not contain any of these characters (excludes C terminal residue) | |
| -Stn $ | preceding residue must be one of these | |
| -Stc $ | C terminal residue must be one of these | |
| -X1 # | set highest +1 XCorr | |
| -X2 # | set highest +2 XCorr | |
| -X3 # | set highest +3 XCorr | |
# symbols indicate that a numerical parameter follows while $ symbols indicate a character or string parameter follows. These filters are employed in the first pass of the filtering step. Each spectrum must pass all the standard filters to be included in the DTASelect report. Changes can be made at the command line or in a DTASelect configuration file.
Table 2.
DTASelect Locus Filters and Utilitiesa
| option | default | locus filters |
|---|---|---|
| -t 0 | do not purge duplicate spectra for each sequence | |
| -t 1 | purge duplicate spectra on basis of total intensity | |
| -t 2 | X | purge duplicate spectra on basis of XCorr |
| -u | false | include only loci with uniquely matching peptides |
| -o | false | remove proteins that are subsets of others |
| -e $ | remove proteins with names matching this string | |
| -E $ | include only proteins with names matching this string | |
| -l $ | remove proteins with descriptions including this word | |
| -L $ | include only proteins with descriptions including this word | |
| -M # | 0 | set minimum modified peptides per locus criterion |
| -r # | 10 | show all loci with peptides that appear this many times |
| -p # | 2 | set minimum peptides per locus criterion |
| option | utilities | |
| --nofilter, -n | do not apply any criteria | |
| --GUI | report through GUI instead of output files | |
| --compress | create .IDX and .SPM files from spectra | |
| --copy | print commands to copy selected IDs | |
| --XML | save XML report of filtered results | |
| --DB | save in format for database import | |
| --chroma | save chromatography report | |
| --similar | save protein similarity table | |
| --mods | save modification report | |
| --align | save sequence alignment report | |
| --help, -h | print this list of options | |
| --here, -. | include only IDs in current directory | |
# symbols indicate that a numerical parameter follows, while $ symbols indicate a character or string parameter follows. Filtering is conducted at the locus level after it has been performed for individual spectra. A protein must pass either the -p or the -r filter to be retained in the DTASelect reports.
The reporting phase generates several different files. The primary report is the DTASelect.html file, which enumerates the proteins and the spectra in evidence for each (see Figures 1 and 2). Hyperlinks from this report lead to sequence coverage for individual proteins, a view of each spectrum, and the output file for each spectrum. This information is also recorded in DTASelect-filter.txt, a tab-delimited text file suitable for review in a spreadsheet. For better data portability, DTASelect includes a graphical user interface, which can be used independently of SEQUEST's support programs (see Figure 3). This interface provides superior support for identifying sequence ions from spectra, especially those that come from triply charged precursors. In another mode, it displays the selected protein's sequence coverage. These files and the graphical user interface present the primary information produced by DTASelect.
Figure 1.
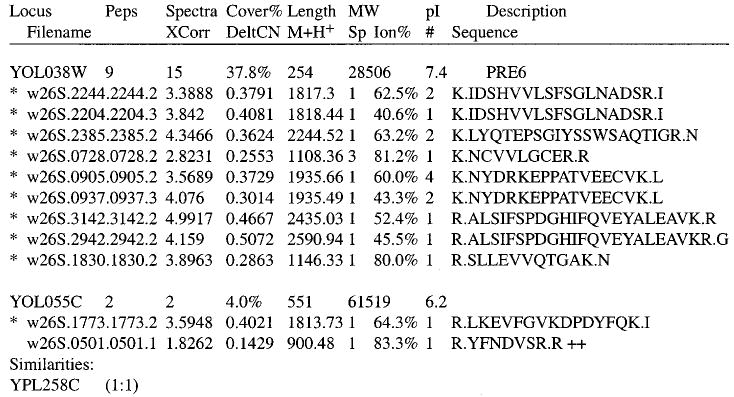
Sample DTASelect.html fragment. Each protein identity is printed beside the count of peptide sequences associated with it. The number of spectra representing those sequences is also shown, along with the protein's sequence coverage, length in residues, molecular weight, calculated pI, and description from the specified database. If multiple proteins in the database correspond to the same set of peptide sequences, the proteins are grouped together. The peptides found for each collection of loci are listed beneath it. Spectra matching the same sequences but possessing different charge states (discernible by the “.2” vs “.3” suffixes on filenames) are not considered duplicates. Peptides that are uniquely found at a particular locus are indicated with asterisks. The fields enumerated for each peptide include file name, XCorr, DeltCN, precursor ion mass, Sp rank, percentage of fragment ions found, copy count, and sequence. Addition symbols (as seen with w26S.0501.0501.1) link to other proteins in the report that also contain the indicated peptide. The similarity for protein YOL055C to YPL258C is reported, showing that one peptide present for YOL055C matches to the other protein and one peptide does not.
Figure 2.
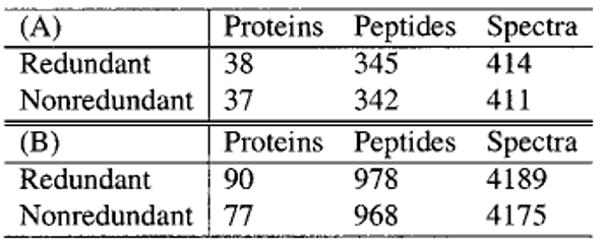
Summary tables from DTASelect output for LC/MS/MS and MudPIT analysis of purified 26S protesomes: (A) DTASelect summary output for LC/MS/MS analysis on 4 μg of purified 26S proteosome. Shown are total counts for proteins, peptides, and spectra. The difference between the nonredundant and redundant protein counts reflects that some proteins have been grouped together because of identical sequence coverage. When used with databases that contain a large number of related proteins (such as the human database), DTASelect's grouping functionality is a timesaver. (B) As in (A) except that results are for a MudPIT analysis of 40 μg of purified 26S proteosome.
Figure 3.
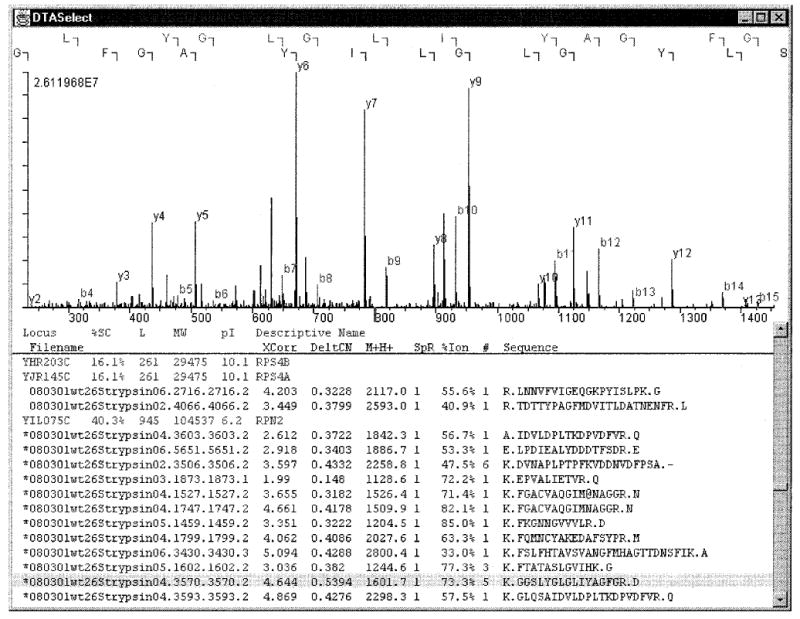
DTASelect graphical user interface. Identified peaks are color-coded blue for y ions or red b ions. The letters along the top of the window show the correspondence between fragment ions and sequence. Clicking on a peptide will cause its spectrum to be shown. Selecting a protein will show sequence coverage.
Supplementary information is presented in two auxiliary files. The first contains information about protein similarity. Multiple proteins may feature a particular peptide, but this is not always an indicator of substantial similarity among the proteins. Similarity is calculated to be the number of peptides shared between two proteins minus the number of nonmatching peptides; a score of zero indicates that the number of peptides that match equals the number that do not match. DTASelect includes all similarity scores greater than or equal to zero in its primary report and in a separate similarity table. Another DTASelect report evaluates chromatography. After filters have been applied, the number of singly, doubly, and triply charged peptides remaining from each round of chromatography is given, along with a profile of where in each cycle the peptides eluted. This information can help users optimize their separations for both completeness and efficiency.
2.2. Contrast Algorithm
Contrast extends DTASelect across multiple samples or multiple sets of criteria. Its three phases are reading, comparison, and reporting. The program is configured by changes in the Contrast.params file, and the specified DTASelect results are read from the disk. The presence of each protein is compared between the described sets, and the similarities and differences are reported in HTML and text formats.
The reading phase begins with the Contrast.params file, which allows the user to specify the DTASelect results of interest and to enumerate criteria sets to be applied to the results. Contrast requires at least one sample and set of criteria to be specified, but more can be included as long as the number of samples multiplied by the number of criteria sets does not exceed 63. Each DTASelect result is processed under each set of criteria, so four samples and two criteria sets will result in eight “data sets,” corresponding to eight columns in the output. Once the Contrast.params file sets up the run, Contrast will read the DTASelect.txt file for each sample and apply the specified filters as in DTASelect.
To compare the data sets, Contrast develops a master list of proteins including each locus appearing in any of the data sets. Each data set is assigned a power of two. Each locus' pattern of presence or absence is stored by summing the powers of two for the data sets in which the protein is found. Finally, the master list of loci is sorted by these power sums and then by protein name. The effect is that loci are grouped by patterns of appearance across data sets.
Contrast produces extensive output. The major report is Contrast.html, which lists the proteins in each group and includes links to the corresponding DTASelect.html files for each data set (see Figures 4 and 5). To permit Contrast results to be analyzed by spreadsheet, they are exported in a tab-delimited text file. For users who want to see how individual peptides for each protein differ among data sets, Contrast offers a “verbose” mode, which provides this level of detail (see Figure 6). Although a complex comparison can yield a lengthy report, Contrast's output organization ensures that details can be found in predictable locations.
Figure 4.
Sample Contrast.html fragment. This represents a group of proteins that appear in the new MudPIT sample but not the previous experiment when the same criteria are used against each. Each row in the table represents one protein, and the numbers in the columns are the sequence coverage percentages found in each data set (or, in the Total column, the cumulative sequence coverage across multiple columns). The percentages link to each protein's location in a corresponding DTASelect.html file. If multiple proteins have identical sequence coverage, they are grouped together (for example, NRL_1IKFH and NRL_1INDH). Several such sections appear in each Contrast output file, one for each combination of presence and absence.
Figure 5.
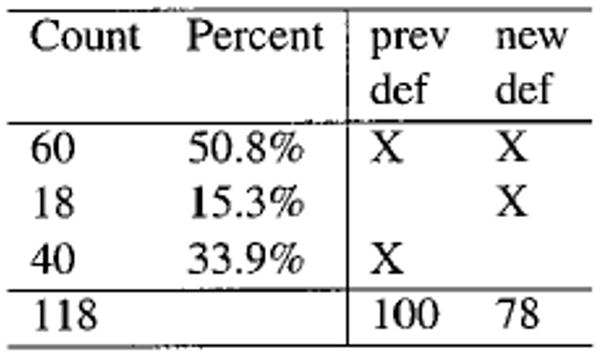
Sample Contrast.html summary. Each row in this table represents a particular combination of presence and absence in each of the data sets, with the “X” marks indicating this pattern. Each row's count links back to the appearance of the group above it in the Contrast.html file. Of the 118 proteins appearing, 60 were present in both samples, 18 were present only in the “new” analysis, and 40 were found only in the “prev” experiment.
Figure 6.
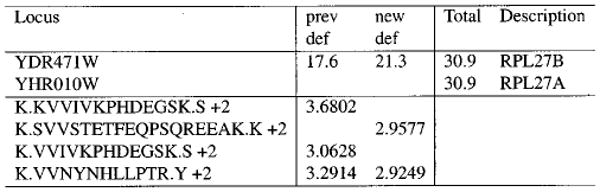
Sample Verbose Contrast.html fragment. Proteins YDR471W and YHR010W were found in both samples under this criteria set, though with different sequence coverages (17.6% and 21.3%, respectively). One peptide was found in both samples, but the other peptides were found in only one. The highest XCorr for each peptide in each sample is shown beside its sequence. Cumulatively, these peptides add up to 30.9% sequence coverage. The sequence coverage percentages for each sample lead to the relevant sections in the respective DTASelect output files. The cumulative sequence coverage links to a view of the protein's sequence overlaid with the peptide sequences.
3. Results and Discussion
Analyses of purified yeast 26S proteosomes by both single dimension LC/MS/MS12 and by MudPIT16 have been reported previously. All experiments were performed on a Thermo Finnigan LCQ or LCQ Deca using standard procedures.4,8,9 The resulting peptide identifications were processed by DTASelect and Contrast.
3.1. DTASelect Application: Single-Dimension LC/MS/MS
Digested peptides from 4 μg of purified 26S proteosome were resolved and analyzed during a 2 h LC/MS/MS experiment.12 After a SEQUEST search against a database of predicted yeast open reading frames and common contaminants, the resulting 6587 output files were filtered and summarized using the default criteria of DTASelect (see Tables 1 and 2). The filtering yielded 411 spectra corresponding to 342 peptides and 37 distinct proteins (see Figure 2A).
All but two of these 37 proteins had been observed before: 32 had been identified as part of the 26S proteosome, one was known to associate with the complex,12 and two were exogenous contaminants.8 DTASelect prepared its report in minutes (see Figure 1). The DTASelect output compiled the important metrics for each spectrum into a single file, enabling further study through hypertext links.
3.2. DTASelect Application: MudPIT
Digested peptides from 36 μg of the same preparation of 26S proteosome as for the single-dimension experiment were analyzed by MudPIT9 and searched against the database. After filtering and summarization, the 33 841 output files were pared down to 4157 spectra representing 968 distinct peptides and 77 proteins (see Figure 2B). Upon manual evaluation of the results, only four of these 77 proteins (5.2%) passed through the filters as false positives. Such percentages are typical of the results for these experiments. The percentage of false postives, however, is a function of the filters employed by the user; DTASelect allows considerable latitude in the stringency applied to the analysis.
3.3. Contrast Application: MudPIT versus MudPIT
The most common application for Contrast is for the comparison of multiple LC/MS/MS or MudPIT experiments. The recent MudPIT analysis was compared to a previous MudPIT analysis of 60 μg of purified proteosome.16 The summary table generated by Contrast allows the researcher to evaluate how many proteins were found in both samples (see Figure 5). Manual evaluation confirmed that none of the 60 shared proteins were passed through the filters as false positives. For this group of proteins, all were either components of the proteosome,32 associated with the proteosome,8 highly abundant yeast proteins that are found in numerous purifications,18 or exogenous contaminants such as immunoglobulin and trypsin.2
Contrast produces an analysis within a few minutes that would take a few hours to produce manually. As in DTASelect output, important information is directly available, including locus name, percent sequence coverage, and locus description/protein name (see Figure 4). The proteins are sorted by the experiments in which they are found, and hypertext links provide access to more detailed information for evaluative purposes (see section 2.2). When multiple positive and negative controls are part of the experiment, Contrast's automation becomes even more valuable.
3.4. DTASelect and Contrast Applications: Miscellaneous
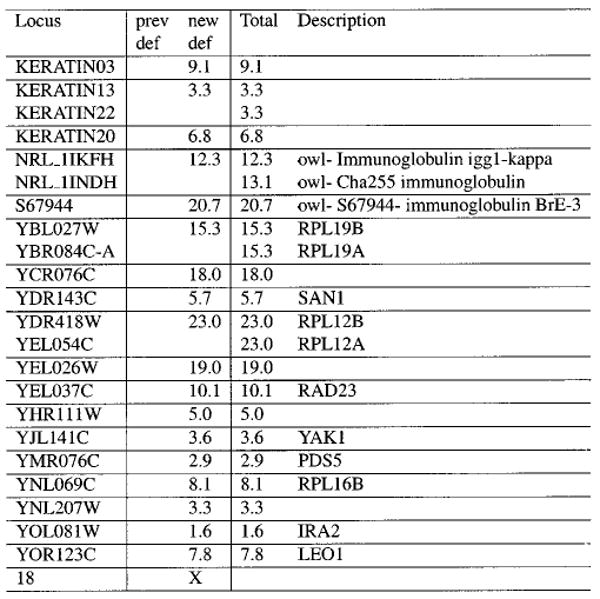
Since a large variety of customizable filters are available in DTASelect and thus Contrast, individual researchers can adapt the programs to their own needs. Contrast can be used for a “step analysis” in which progressively more stringent criteria are applied to data from a single experiment. Those proteins that meet the highest stringency requirements are sorted to the top of the output file, and those passing only the lowest criteria are sorted to the end. This approach is well suited for stratifying the protein identifications in a complex sample. For projects focusing on a particular protein, the verbose mode of Contrast can show how individual peptides vary among samples. If modifications like phosphorylation or methylation are included in SEQUEST search parameters, DTASelect can be used to list only those peptides found to be modified, saving time for researchers interested in this subset of the data. It can also be used for chemical labeling tags such as the ICAT reagent.17 The open-ended design of the programs accommodates many purposes.
4. Conclusions
DTASelect assembles protein-level information from peptide data more quickly, flexibly, and uniformly than existing tools. DTASelect focuses attention on peptides of interest by sweeping away less likely identifications. By streamlining data analysis for proteomics, DTASelect makes more complex experiments feasible. Contrast provides extensive sample comparison capabilities. Whether stepping through multiple levels of criteria to triage the proteins identified from a single sample or differentiating the protein content of multiple, complex samples, Contrast is a potent tool for the molecular biologist. Most users of SEQUEST can benefit from DTASelect and Contrast. Just as SEQUEST automated the time-intensive process of spectrum identification, DTASelect and Contrast automate the process of SEQUEST result interpretation.
Acknowledgments
We thank Rati Verma and Raymond Deshaies of the Division of Biology and Howard Hughes Medical Institute at Caltech for graciously providing proteosome samples used to demonstrate the software. D.L.T. was supported by a National Science Foundation Graduate Research Fellowship. W.H.M. was supported by the Merck Genome Research Institute. Partial support was derived from NIH Grant Nos. RR11823 and R33CA81665. Suggestions from the members of the Yates Lab contributed many features to the development of these programs.
References
- 1.Yates JR, III, McCormack AL, Eng JK. Anal Chem. 1996;68:534A–540A. doi: 10.1021/ac962050l. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR., III Electrophoresis. 1998;19:893–900. doi: 10.1002/elps.1150190604. [DOI] [PubMed] [Google Scholar]
- 3.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gatlin CL, Kleeman GR, Hays LG, Link AJ, Yates JR., III Anal Biochem. 1998;263:93–101. doi: 10.1006/abio.1998.2809. [DOI] [PubMed] [Google Scholar]
- 5.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, Yates JR., III Anal Chem. 1997;69:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 6.Link AJ, Carmack E, Yates JR., III Intl J Mass Spectrom. 1997;160:303–316. [Google Scholar]
- 7.Eng JK, McCormack AL, Yates JR., III J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 8.Link AJ, Eng JK, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 9.Washburn MP, Wolters D, Yates JR., III Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 10.Meeusen S, Tieu Q, Wong E, Weiss E, Schieltz DM, Yates JR, III, Nunnari J. J Cell Biol. 1999;145:291–304. doi: 10.1083/jcb.145.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goode BL, Wong JJ, Butty AC, Peter M, McCormack AL, Yates JR, III, Drubin DG, Barnes G. J Cell Biol. 1999;144:83–98. doi: 10.1083/jcb.144.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma R, Chen S, Feldman R, Schieltz D, Yates JR, III, Dohmen J, Deshaies RJ. Mol Biol Cell. 2000;11:3425–3439. doi: 10.1091/mbc.11.10.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates JR., III J Mass Spectrom. 1998;33:1–19. doi: 10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CR. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabb DL, Eng JK, Yates JR., III . Protein Identification by SEQUEST. In: James P, editor. Proteome Research: Mass Spectrometry. Vol. 1. Springer; New York: 2001. pp. 125–142. [Google Scholar]
- 16.Verma R, McDonald WH, Yates JR, III, Deshaies RJ. Mol Cell. 2001;8:439–48. doi: 10.1016/s1097-2765(01)00308-2. [DOI] [PubMed] [Google Scholar]
- 17.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH. Nat Biotechnol. 2000;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]