

Abstract
Accumulation of the synaptic protein α-synuclein (α-syn) is a hallmark of Parkinson's disease (PD) and Lewy body disease (LBD), a heterogeneous group of disorders with dementia and parkinsonism, where Alzheimer's disease and PD interact. Accumulation of α-syn in these patients might be associated with alterations in the autophagy pathway. Therefore, we postulate that delivery of beclin 1, a regulator of the autophagy pathway, might constitute a strategy toward developing a therapy for LBD/PD. Overexpression of α-syn from lentivirus transduction in a neuronal cell line resulted in lysosomal accumulation and alterations in autophagy. Coexpression of beclin 1 activated autophagy, reduced accumulation of α-syn, and ameliorated associated neuritic alterations. The effects of beclin 1 overexpression on LC3 and α-syn accumulation were partially blocked by 3-MA and completely blocked by bafilomycin A1. In contrast, rapamycin enhanced the effects of beclin 1. To evaluate the potential effects of activating autophagy in vivo, a lentivirus expressing beclin 1 was delivered to the brain of a α-syn transgenic mouse. Neuropathological analysis demonstrated that beclin 1 injections ameliorated the synaptic and dendritic pathology in the tg mice and reduced the accumulation of α-syn in the limbic system without any significant deleterious effects. This was accompanied by enhanced lysosomal activation and reduced alterations in the autophagy pathway. Thus, beclin 1 plays an important role in the intracellular degradation of α-syn either directly or indirectly through the autophagy pathway and may present a novel therapeutic target for LBD/PD.
Introduction
Alzheimer's (AD) and Parkinson's (PD) diseases are the leading causes of dementia and movement disorders in the aging population. The spectrum of disorders where AD and PD overlap is called Lewy body disease (LBD) (Kosaka et al., 1984; Hansen and Galasko, 1992; McKeith, 2000). The major component of the Lewy bodies, the hallmark of LBD, is α-synuclein (α-syn) (Wakabayashi et al., 1997; Takeda et al., 1998; Trojanowski and Lee, 1998), a major synaptic protein (Iwai et al., 1995) involved in vesicular release (Murphy et al., 2000).
Progressive accumulation of aggregated α-syn has been implicated in neurological disorders with parkinsonism, including PD (Polymeropoulos et al., 1997), LBD (Spillantini et al., 1997), and multiple systems atrophy (Wakabayashi et al., 1998). α-syn is a natively unfolded protein with little tertiary structure (Weinreb et al., 1996). In its pathologic state it is folded to allow the formation of dimer and higher-order toxic oligomer species (Conway et al., 2000; Tsigelny et al., 2007). It is these states that proceed to accumulation within neurons and lead to dysfunction and neuronal degeneration (Rochet et al., 2000).
Overexpression, misfolding, or decreased degradation are all pathways that may lead to the accumulation of α-syn in neurons. One pathway of protein degradation in cells is by autophagic targeting to the lysosome (Mariño and López-Otín, 2004). This pathway is divided into chaperone-mediated autophagy (CMA), macroautophagy, and microautophagy (Cuervo, 2004). The macroautophagy pathway (hereafter referred to as autophagy) is involved in nutrient starvation as well as functioning in the degradation of long-lived proteins and organelles. Autophagy of protein and organelles is initiated by one of three pathways: (1) direct involvement of mTor (mammalian Target of Rapamycin), (2) indirect involvement of mTor with Atg1, or (3) VPS34-beclin 1 complex (for review, see Mariño and López-Otín, 2004).
Recent evidence suggests that dysfunction in the autophagy pathway is common in numerous neurodegenerative diseases (Ravikumar et al., 2004; Nixon et al., 2005; Sarkar et al., 2007; Pan et al., 2008), including AD, PD/LBD, and Huntington's disease (HD). In PD/LBD, α-syn accumulation has been linked to alterations in chaperone-mediated autophagy and lysosomal functioning (Cuervo et al., 2004; Martinez-Vicente et al., 2008; Xilouri et al., 2009). Clearly the autophagy pathway is disrupted in patients and animal models that involve accumulation of α-syn, and induction of this pathway can be used to reduce levels of α-syn in neurons. For example, in a previous study we showed that activation of autophagy via immunotherapy might contribute to α-syn clearance in neurons (Masliah et al., 2005).
In patients with AD, beclin 1 has been linked with alterations in Aβ clearance (Pickford et al., 2008). Beclin 1 is a coiled-coil, 60 kDa protein that is analogous to the yeast autophagy protein Apg6 (Liang et al., 1998, 1999). Overexpression of beclin 1 reduces apoptosis and increases autophagy (Erlich et al., 2006; Hamacher-Brady et al., 2006). Recently, we found that lentivirus vector (LV) delivery of beclin 1 into a mouse model of AD (Pickford et al., 2008) ameliorated the neuropathology. To determine whether “jump-starting” the autophagy pathway could reduce α-syn accumulation and the associated pathology, we generated an LV expressing the beclin 1 cDNA. The LV-Beclin 1 activated autophagy, reduced the accumulation of α-syn, and ameliorated the associated neuritic alterations, thus providing a directed gene therapy approach for the reduction of accumulated α-syn without any off-target pathology that may be associated with generalized increase in autophagy.
Materials and Methods
Construction of lentivirus vectors.
The mouse beclin 1 cDNA (Open Biosystems) was PCR amplified and cloned into the third-generation self-inactivating lentivirus vector (Naldini et al., 1996) with the cytomegalovirus (CMV) promoter driving expression producing the vector LV-Beclin 1. The lentivirus vector expressing the human wild-type α-syn has been previously described (Bar-On et al., 2008). Lentiviruses expressing beclin 1, α-synuclein (α-syn), β-synuclein (β-syn), green fluorescent protein (GFP), or empty vector (as controls) were prepared by transient transfection in 293T cells (Naldini et al., 1996; Tiscornia et al., 2006).
Establishment of a neuronal cell line expressing α-syn and Beclin 1.
For these experiments we used the rat neuroblastoma cell line B103. This model was selected because overexpression of α-syn in these cells results in mitochondrial alterations, reduced cell viability, defective neurite outgrowth, and abnormal accumulation of oligomeric α-syn (Takenouchi et al., 2001; Hashimoto and Masliah, 2003). For all experiments, cells were infected with LVs expressing wt α-syn at a multiplicity of infection (MOI) of 40. Cells were coinfected with LV-Beclin 1, GFP, or empty vector (as controls). Additional control experiments were performed in cells infected with LV-β-syn. After infection, cells were incubated in a humidified, 5% CO2 atmosphere at 37°C. All experiments were conducted in triplicate to ensure reproducibility.
Immunoblot analysis.
Cells were infected with lentivirus vectors for 72 h and then lysed in TNE buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA; all from Sigma-Aldrich) containing 1% Nonidet P-40 (Calbiochem) with protease and phosphatase inhibitor cocktails (Roche). Total cell extracts were centrifuged at 6000 × g for 15 min, and the protein concentration of supernatants was assayed with a BCA protein assay kit (Pierce Biotechnology). For Western blot analysis, 20 μg of lysate per lane was loaded into 4–12% Bis-Tris SDS-PAGE gels and blotted onto polyvinylidene fluoride (PVDF) membranes. Blots were incubated with antibodies against α-syn (Millipore Bioscience Research Reagents), beclin 1 (Novus), cathepsin D (DAKO), LAMP2 (Santa Cruz Biotechnology, C-20), Atg5 (Abcam), Atg7 (Abcam), LC3 (Abcam), GFP (Millipore Bioscience Research Reagents), and actin (Millipore Bioscience Research Reagents) followed by secondary antibodies tagged with horseradish peroxidase (Santa Cruz Biotechnology), visualized by enhanced chemiluminescence and analyzed with a Versadoc XL imaging apparatus (Bio-Rad). Analysis of actin levels was used as a loading control.
Analysis of autophagy.
The B103 cells were grown as described above and were then plated onto poly-l-lysine-coated glass coverslips at a density of 5 × 104 cells per coverslip. Five hours after plating, cells were infected with the LV-α-syn and/or LV-Beclin 1 at an MOI of 40 and incubated for 48 h. All coverslips were also coinfected with a lentiviral vector expressing LC3-GFP. Most control experiments were performed with cells infected with LV-control and in some cases with LV-β-syn. To examine the effects on the autophagy pathway, rapamycin (200 nm) or bafilomycin A1 (200 nm) was added to cultures for 6 h or 3 methyladenine (3-MA) (10 mm) (Sigma-Aldrich) was added for 24 h before fixation. Other cultures were washed 2× with serum-free DMEM and then fed either complete media or serum-free media for 12 h before fixation with 4% PFA. Briefly as previously described (Pickford et al., 2008), coverslips were treated with Prolong Gold antifading reagent with DAPI (Invitrogen) and imaged with the laser-scanning confocal microscope (LSCM, MRC1024, Bio-Rad) to determine the number of GFP-positive granular structures consistent with autophagosomes. In each case, an average of 50 cells were analyzed using the ImageQuant software. Analysis was performed in duplicate with samples blind coded. Further analysis included estimates of the average numbers of LC3-GFP-positive structures below or over 1 μm in diameter.
Additional studies were performed in the same neuronal cells infected with LV-α-syn and/or LV-Beclin 1 and analyzed by Western blot for LC3 protein. The membranes were blocked with PBS with 0.2% Tween 20 (PBST) containing 3% skim milk or bovine serum albumin (BSA), followed by incubation with antibody against LC3 (Abcam). After washing with PBS, the membranes were incubated with secondary antibodies and analyzed with enhanced chemiluminescence (ECL, PerkinElmer) and analyzed with the VersaDoc gel imaging system (Bio-Rad) to determine the ratio of the LCII to actin. Loading values were normalized in the same blots reacted with an antibody against actin.
LDH, TUNEL, caspase 3, and neurite length assays.
Cell death was evaluated by the LDH assay as previously described (Langford et al., 2004). Cells were plated on 96-well plates in complete media. After treatments, assays were then performed following the manufacturer's instructions (Promega). Results were expressed as percentage cell death. We have previously reported that impaired plasticity in this neuronal cell line promoted by accumulation of α-syn is associated with reduced neurite outgrowth and adhesion (Takenouchi et al., 2001). For this purpose, an average of 100 neurons per condition were imaged with phase-contrast microscopy and neurite length estimated with the ImageQuant software in a blinded manner.
Additional neuronal cells infected with LV-α-syn with or without LV-Beclin 1 were plated onto coated coverslips, and after 3 d they were fixed with 4% paraformaldehyde in PBS buffer and assayed with the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (ApopTag In Situ Apoptosis Detection Kit, Millipore Bioscience Research Reagents) or immunostained with an antibody against activated caspase-3 (Cell Signaling Technology) detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences) followed by imaging with the confocal laser-scanning microscope. Coverslips were covered with the Prolong Gold antifading reagent with DAPI (Invitrogen).
Real-time analysis of RNA expression.
Neuronal cells were infected with LV-α-syn with or without LV-Beclin 1 at an MOI of 40. Seventy-two hours after infection, total RNA was isolated with the RNeasy Total RNA kit (Qiagen). cDNA was generated with the qScript cDNA synthesis kit (Quanta Biosciences) according to the manufacturer's directions. cDNA was then quantified with the 2× SYBR Green (Quanta Biosciences) with primers specific for α-syn (TGT TGG AGG AGC AGT GGT GA). A standard curve was generated from an α-syn plasmid.
Immunocytochemical analysis and confocal microscopy.
To verify expression levels of α-syn and beclin 1 in cells infected with the different LV vectors, neurons were seeded onto poly-l-lysine-coated glass coverslips, grown to 60% confluence, and fixed in 4% PFA for 20 min. Coverslips were pretreated with 0.1% Triton X-100 in TBS for 20 min and then incubated overnight at 4°C with antibodies against human α-syn (Millipore Bioscience Research Reagents) and beclin 1 (Novus). The following day, the beclin 1 signal was detected with the FITC-conjugated secondary antibody (Vector Laboratories), and the α-syn signal was detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences). Control samples included empty vector- (referred hereafter as LV-control) or GFP-infected cells, and immunolabeling in the absence of primary antibodies. Coverslips were mounted with Prolong Gold antifading reagent with DAPI (Invitrogen). Cells were analyzed with a digital epifluorescence microscope (Olympus BX51) to estimate the percentage of total cells (DAPI stained) that displayed GFP, α-syn, or beclin 1 immunoreactivity.
To verify the coexpression in neuronal cells coinfected with the different LV vectors, coverslips were double labeled with antibodies against α-syn (or β-syn) (Millipore Bioscience Research Reagents) and beclin 1 (Novus) as previously described (Crews et al., 2008). Coverslips were air dried, mounted on slides with anti-fading media (Vectashield, Vector Laboratories), and imaged with a confocal microscope. An average of 50 cells were imaged per condition, and the individual channel images were merged and analyzed with the ImageJ program to estimate the extent of colocalization between α-syn and beclin 1.
Transgenic mouse lines and intracerebral injections of lentiviral vectors.
For this study, mice overexpressing α-synuclein from the platelet-derived growth factor β (PDGF-β) promoter (line D) were used (Masliah et al., 2000; Rockenstein et al., 2002). This model was selected because mice from this line develop intraneuronal α-synuclein aggregates distributed throughout the neocortex and hippocampus similar to what has been described in LBD. A total of 48 hα-synuclein tg mice from line D (9 months old) were injected with 3 μl of the lentiviral preparations (2.5 × 107 TU) into the temporal cortex and hippocampus (using a 5 μl Hamilton syringe). Briefly, as previously described (Marr et al., 2003), mice were placed under anesthesia on a Koft stereotaxic apparatus and coordinates (hippocampus: AP −2.0 mm, lateral 1.5 mm, depth 1.3 mm; and cortex: AP −.5 mm, lateral 1.5 mm, depth 1.0 mm) were determined as per the Franklin and Paxinos (1997) atlas. The lentiviral vectors were delivered using a Hamilton syringe connected to a hydraulic system to inject the solution at a rate of 1 μl every 2 min. To allow diffusion of the solution into the brain tissue, the needle was left for an additional 5 min after the completion of the injection. Mice received unilateral injections (right side) to allow comparisons against the contralateral side, with either LV-Beclin 1 (n = 12), or LV-control (n = 6). Additional controls were performed by injecting nontransgenic littermates with either LV-Beclin 1 (n = 6), or LV-control (n = 6). Mice survived for 3 months after the lentiviral injection. As an additional control for LV injection, age matched littermates were injected with LV-luciferase. Since no differences were observed between the LV-control and the LV-luciferase all data presented in this paper are shown with the LV-control vector. Following NIH guidelines for the humane treatment of animals, mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline.
Brains and peripheral tissues were removed and divided sagittally. The right hemibrain was postfixed in phosphate-buffered 4% PFA, pH 7.4, at 4°C for 48 h for neuropathological analysis, while the left hemibrain was snap-frozen and stored at −70°C for subsequent RNA and protein analysis.
Immunocytochemical and neuropathological analyses.
Analysis of α-synuclein accumulation was performed in serially sectioned, free-floating, blind-coded vibratome sections from tg and non-tg mice treated with LV-Beclin 1 and control vectors (Masliah et al., 2000). Sections were incubated overnight at 4°C with an anti-α-synuclein antibody (affinity-purified rabbit polyclonal, Millipore Bioscience Research Reagents) (Masliah et al., 2000), followed by biotinylated goat anti-rabbit IgG (Vector Laboratories), Avidin D-HRP (1:200, ABC Elite, Vector), and detection with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences) to determine the number of hα-synuclein-immunoreactive inclusions. For each case, three sections were analyzed by the dissector method using the Stereo-Investigator System (MBF Bioscience), and the results were averaged and expressed as numbers per cubic millimeter.
To determine efficiency of transduction, double-labeling experiments with beclin 1 and the neuronal marker NeuN (Millipore Bioscience Research Reagents) were performed, as previously described (Hashimoto et al., 2001). To determine the colocalization between α-synuclein immunolabeled neurons and beclin 1, 40-μm-thick vibratome sections were immunolabeled with the rabbit polyclonal antibodies against α-synuclein (affinity-purified rabbit polyclonal, Millipore Bioscience Research Reagents) (Masliah et al., 2000) and beclin 1 (Novus). The α-syn-immunoreactive structures were detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences), while beclin 1 was detected with the goat anti-rabbit fluorescein isothiocyanate (FITC) antibody (Vector Laboratories). This system allows the simultaneous detection of signals from antibodies from the same species.
To determine whether beclin 1 gene transfer ameliorated the neurodegenerative alterations associated with the expression of α-syn, briefly as previously described (Rockenstein et al., 2005a,b), blind-coded, 40-μm-thick vibratome sections from mouse brains fixed in 4% paraformaldehyde were immunolabeled with the mouse monoclonal antibodies against synaptophysin (synaptic marker, Millipore Bioscience Research Reagents), microtubule-associated protein-2 (MAP2, dendritic marker, Millipore Bioscience Research Reagents), or glial fibrillary acidic protein (GFAP, astroglial marker, Millipore Bioscience Research Reagents) (Mucke et al., 1995). After overnight incubation with the primary antibodies, sections were incubated with FITC-conjugated horse anti-mouse IgG secondary antibody (Vector Laboratories), transferred to SuperFrost slides (Fisher Scientific), and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. The immunolabeled blind-coded sections were serially imaged with the LSCM and analyzed with the Image 1.43 program (NIH), as previously described (Toggas et al., 1994; Mucke et al., 1995). For each mouse, a total of three sections were analyzed, and for each section, four fields in the frontal cortex and hippocampus were examined. For synaptophysin and MAP2, results were expressed as percentage area of the neuropil occupied by immunoreactive terminals and dendrites.
All sections were processed simultaneously under the same conditions and experiments were performed twice to assess the reproducibility of results. Sections were imaged with a Zeiss 63× (numerical aperture 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 LSCM system (Bio-Rad) (Masliah et al., 2000).
Electron microscopy.
Briefly, B103 neuronal cells were plated in 35 mm dishes with a coverslip in the bottom and infected with LVs. After 48 h, cells were fixed in 2% paraformaldehyde and 1% glutaraldehyde, then fixed in osmium tetroxide and embedded in epon Araldite. Blocks with the cells were detached from the coverslips and mounted for sectioning with an ultramicrotome (Leica). Similarly, sections from the α-syn tg mice treated with the LV-Beclin 1 or LV-control were fixed, embedded, and sectioned with the ultramicrotome. Grids were analyzed with a Zeiss OM 10 electron microscope as previously described (Rockenstein et al., 2001). To analyze the relative changes in average numbers of autophagic vesicles (AVs) following LV-α-syn and/or LV-Beclin 1 infections, a total of 25 cells were analyzed per condition. Cells were randomly acquired from three grids, and electron micrographs were acquired at a magnification of 25,000×.
Statistical analysis.
All experiments were done blind coded and in triplicate. Values in the figures are expressed as means ± SEM. To determine the statistical significance, values were compared by using the one-way ANOVA with post hoc Dunnett's test when comparing the LV-α-syn and Beclin 1 to LV-control. Additional comparisons were done using Tukey–Kramer or Fisher post hoc tests. The differences were considered to be significant if p values were <0.05.
Results
Overexpression of α-syn in a neuronal cell line recapitulates α-syn accumulation and autophagy alterations
To investigate the effects of beclin 1 delivery in α-syn related pathology, we first established an in vitro model system in which α-syn was overexpressed in the B103 neuronal cells line by infection with a lentivirus expressing the wild-type α-syn (LV-α-syn) (Fig. 1). The B103 neuronal cell line was derived from a rat neuroblastoma and shares many typical neuronal properties with other commonly used neuronal cell lines, including outgrowth of neurites upon differentiation, synthesis of neurotransmitters, possession of neurotransmitter receptors, and electrical excitability of surface membranes (Schubert et al., 1974). Compared with LV-control (Fig. 1A), overexpression of α-syn resulted in extensive protein accumulation that was observed primarily in cytoplasmic granular structures as well as in punctate structures located along the neuronal processes (Fig. 1B). In contrast, B103 cells infected with LV-GFP (Fig. 1C) virus or LV-β-syn (Fig. 1D) exhibited a diffuse cytoplasmic labeling. Moreover, this experiment confirmed that with the LV system, >95% of the cells were infected. The LV-control used in this and all subsequent experiments is the identical lentivirus backbone including the CMV promoter (Fig. 1A). The LV-control vector was used instead of the LV-GFP vector because GFP overexpression is known to increase reactive oxygen species in the cell and lead to abnormal DNA methylation (Liu et al., 1999; Hong et al., 2001). The LV-β-syn was also used as a control for protein overexpression. Consistent with the immunocytochemical analysis, Western blot studies with total cell lysates showed overexpression of wild-type α-syn or β-syn. In these lysates, phosphorylated α-syn, beclin 1, LAMP2, and cathepsin D were also detectable (Fig. 1E).
Figure 1.

Lentivirus delivery of α-syn to B103 neuronal cells. A–D, B103 neuronal cells infected with empty vector (LV-control) (A), wild-type α-syn (LV-α-syn) (B), GFP (LV-GFP) (C), or β-synuclein (LV-β-syn) (D). Cells were immunostained with antibodies against α-syn (A, B) or were directly visualized for GFP expression (C) or for β-syn (D). Scale bar, 25 μm. E, Total protein from uninfected B103 cells or cells infected with the α- or β-syn vectors was examined by Western blot for expression levels of α- or β-syn and lysosomal proteins. F, Schematic representation of the third-generation lentivirus vector that was used to overexpress the human wild-type α-syn or β-syn gene. LTR, Long terminal repeat; ppt, central poly-purine tract; CMV, human cytomegalovirus immediate early promoter/enhancer; WPRE, woodchuck hepatitis post-transcriptional regulatory element.
To further investigate whether the α-syn aggregates (Fig. 2A) observed in neuronal cells infected with LV-α-syn might be associated with lysosomes and represent alterations in the autophagy pathway, double-immunolabeling and electron microscopic studies were performed. Ultrastructural analysis showed that the alterations in neuronal cells infected with the LV-α-syn corresponded to electrodense aggregates surrounded by single and double membranes (Fig. 2B,C). This material was localized to enlarged vesicles that appeared to be atypical autophagic vesicles. Quantitative analysis of the electron micrographs showed significantly more double-membrane, enlarged vesicles in those neurons that overexpressed the wild-type α-syn compared with LV-control and LV-β-syn (Fig. 2D,E). Consistent with the ultrastructural analysis, double-labeling studies showed that the α-syn aggregates colocalized with lysosomal/autophagy markers such as cathepsin D and LC3 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), indicating localization to the lysosome. To confirm whether the neuronal cells were collecting aggregated α-syn, total cell lysates were examined by Western blot and probed with a α-syn antibody that recognizes oligomeric species of α-syn. Increased accumulation of oligomeric α-syn was observed in the 60–80 kDa range in cells infected with the LV-α-syn wt (Fig. 2F). Quantification of these bands indicated a 10-fold increase in oligomeric α-syn compared with cells infected with the LV-control virus.
Figure 2.

Accumulation of α-syn in autophagic structures in neuronal cells infected with LV-α-syn. A, B103 cells were infected with the LV-α-syn and examined by immunofluorescence for α-syn (red, arrows) and tubulin (green). Scale bar, 5 μm. B–D, Neuronal cells infected with LV-α-syn (B, C) or LV-β-syn (D) were examined by electron microscopy showing electrodense autophagic vesicles (arrows). n, Nucleus; m, mitochondria. Scale bars, 1 μm (B–D). E, The total number of electrodense AVs was counted per cell and compared with control-infected B103 cells. F, Total protein from LV-infected B103 cells was extracted and examined for total α-syn accumulation by Western blot.
Autophagy is a major mechanism of clearance of accumulated proteins in the CNS (Williams et al., 2006). We examined whether autophagy was disrupted in the neuronal cell cultures that overexpressed α-syn. B103 cells that had been infected with either the LV-α-syn or the LV-control vector were coinfected with a lentivirus expressing the LC3-GFP fusion protein. LC3 activation is a marker of autophagy, and in the activated state, will form punctate structures within the cytoplasm that correspond to autophagic vesicles. With this method, we detected two populations of these vesicles, punctate of normal characteristics usually ∼1 μm or less in diameter and enlarged structures >1 μm in diameter. Under baseline conditions, levels of LC3-GFP punctate formation (<1 μm in diameter) were low in the LV-control (Fig. 3A,G), with a slight upregulation in the neuronal cells infected with LV-α-syn (Fig. 3D,G). However, upon serum starvation (conditions that normally activate the autophagy pathway) neuronal cells infected with the LV-control (Fig. 3B) and LV-α-syn (Fig. 3E) showed a significant increase in the numbers of LC3-GFP punctate. The increase in the LC3-GFP punctate (<1 μm in diameter) was more prominent in the LV-control than in the LV-α-syn cells (Fig. 3G), suggesting a disruption in the autophagy response when excess α-syn was present. In support of this possibility, compared with LV-control, in the serum starved LV-α-syn neuronal cells, there were abundant, enlarged LC3-GFP structures >1 μm in diameter (Fig. 3H) similar to the abnormal autophagic vesicles observed under ultrastructural microscopy. Treatment with rapamycin, an autophagy inducer, resulted in a fivefold increase in LC3-GFP punctate (<1 μm in diameter) in LV-control (Fig. 3C) and LV-α-syn cells (Fig. 3F). The upregulation of LC3-GFP structures >1 μm in diameter was less prominent when the LV-α-syn neuronal cells were treated with rapamycin (Fig. 3H). This suggests that accumulation of the α-syn protein in the neuronal cells alters the autophagic degradation pathway and that activation of autophagy with rapamycin might ameliorate this problem.
Figure 3.

Confocal analysis of the alterations in autophagy in neuronal cells infected with LV-α-syn and LC3-GFP. The B103 cells were infected with lentivirus expressing the LC3-GFP fusion protein and kept under baseline (serum fed) (A, D) or starvation (serum starved) (B, E) for 12 h or treated with rapamycin (C, F) for 6 h and then visualized for GFP by confocal microscopy. A–F, Neuronal cells were infected with LV-LC3-GFP and LV-control (A–C) or LV-α-syn (D–F). G, H, Image analysis of serial confocal frames to determine the average number of LC3-GFP grains below or above 1 μm in diameter. Serum fed (baseline) LC3-GFP granules (arrows) were quantified and compared with serum starved (starvation) or rapamycin treated. Scale bar, 10 μm. *Statistical significance compared with baseline. #Statistical significance compared with control-treated cells (p < 0.05, 1-way ANOVA, post hoc Fisher test).
Lentivirus-mediated delivery of beclin 1 reduces α-syn accumulation and related autophagy deficits
Since α-syn accumulation is associated with alterations in the autophagy pathway in our cell model, and beclin 1 overexpression activates autophagy (Pickford et al., 2008), then overexpression of beclin 1 might ameliorate these defects and reduce α-syn accumulation in neuronal cells. For this purpose, B103 cells were infected with either LV-α-syn, LV-Beclin 1, or both (Fig. 4). Immunocytochemical analysis demonstrated that compared with controls (supplemental Fig. 2A–C, available at www.jneurosci.org as supplemental material) levels of beclin 1 were higher in LV-Beclin 1-infected cells (supplemental Fig. 2D–F, available at www.jneurosci.org as supplemental material). Double labeling studies confirmed that beclin 1 and α-syn immunoreactivity colocalized in >90% of the neurons infected with both LVs (supplemental Fig. 2J–L, available at www.jneurosci.org as supplemental material). Consistent with the immunocytochemical analysis, Western blot confirmed the expression of beclin 1 and α-syn in the LV-infected neuronal cell line (Fig. 4A,B). Wild-type α-syn was reduced ∼50% when beclin 1 was coexpressed (Fig. 4A,B). In addition, a reduction in the phosphorylated α-syn was observed in those neuronal cells coinfected with LV-Beclin 1 (Fig. 4A). The reduction in α-syn protein was not due to a reduction in mRNA as real-time analysis confirmed no difference in expression levels of α-syn in the presence or absence of LV-Beclin 1 infection (Fig. 4C). Immunocytochemical analysis confirmed that compared with controls (Fig. 4D–F,H), in neuronal cells coinfected with LV-Beclin 1 and LV-α-syn, there was a significant reduction in α-syn accumulation (Fig. 4G,H). Similar experiments with LV-β-synuclein and LV-Beclin 1 showed no reduction in levels of β-synuclein, indicating that beclin 1 expression specifically reduced the accumulated levels of α-syn (data not shown).
Figure 4.

Overexpression of beclin 1 from the lentivirus reduces levels of α-syn and restores neurite outgrowth. B103 neuronal cells were infected with LV-α-syn alone or in combination with LV-Beclin 1 and analyzed by immunoblot, PCR, and confocal analysis. A, Total protein was extracted and examined by Western blot for α-syn, phosphorylated (Ser129) α-syn, and beclin 1. B, Analysis of levels of α-syn immunoreactivity. C, Total RNA was extracted and examined by real-time PCR for expression of α-syn mRNA. D–G, Neuronal cells were infected with LV-α-syn alone or in combination with LV-Beclin 1 and double labeled with antibodies against α-syn (red) and tubulin III (green). H, I, Analysis of the levels of α-syn and tubulin III immunoreactivity from digital images acquired by confocal microscopy. J, Computer-aided image analysis of neurite length. Scale bar, 10 μm. *Statistical significance (p < 0.05, 1-way ANOVA, post hoc Fisher test).
We have previously shown that in stably transfected B103 neuronal cells, α-syn overexpression results in decreased adhesion, neurite outgrowth (Takenouchi et al., 2001), and alterations in the tubulin cytoskeleton (Kawahara et al., 2008). To determine whether coexpression of beclin 1 could reverse these deficits, the levels of tubulin III immunoreactivity and neurite outgrowth were determined. While in neuronal cells infected with LV-α-syn there was a 50% reduction in tubulin III levels (Fig. 4I) and neurite length (Fig. 4J) compared with LV-control, in cells coinfected with LV-Beclin 1, levels were nearly identical to control neuronal B103 cells (Fig. 4I,J), indicating that reducing the accumulation of α-syn with LV-Beclin 1 had functional beneficial effects.
To determine whether overexpression of beclin 1 would affect cell survival or growth, we analyzed the cells with the LDH, TUNEL, and cleaved caspase-3 assays. We did not observe a significant difference in cell survival following infection with the LV-Beclin 1 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), indicating that beclin 1 overexpression does not appear to have any detrimental effects. Moreover, in our system, overexpression of α-syn was not accompanied by an increase in markers of cell death (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) but rather by alterations in neurite plasticity (Fig. 4).
Beclin 1 is essential for the initial steps of autophagy (Furuya et al., 2005; Takacs-Vellai et al., 2005; Erlich et al., 2006; Hamacher-Brady et al., 2006), so to determine whether the reductions in α-syn accumulation were associated with activation of the autophagy pathway in cells infected with LV-Beclin 1, we coinfected cells with a lentivirus expressing LC3 fused to GFP. Expression of α-syn and LC3-GFP in the neuronal cells resulted in increased punctate staining of the α-syn protein as we had previously observed and LC3-GFP in the cytoplasm (Fig. 5A,E). Addition of the LV-Beclin 1 increased the punctate of LC3 and resulted in considerable colocalization of LC3 and α-syn (Fig. 5A,E). Image analysis of the LC3-GFP showed a fourfold increase in LC3 granules or punctate structures only when beclin 1 was coexpressed, indicating that delivery of the LV-Beclin 1 promoted autophagy even in cells coexpressing α-syn (Fig. 5E). This increase in autophagy promoted by LV-Beclin 1 was accompanied by a reduction in the accumulation of α-syn (Fig. 5E). Consistent with the confocal imaging, immunoblot analysis for LC3 showed a moderate increase in LC3-II or activated LC3 protein in those neuronal cultures that received LV-α-syn alone and a prominent increase in those cells expressing both LV-α-syn and LV-Beclin 1 (Fig. 6A,D). This was accompanied by decreased levels of α-syn by confocal microscopy (Fig. 5A,E) and by immunoblot (Fig. 6A,G).
Figure 5.

Laser-scanning confocal imaging of the effects LV-Beclin 1 expression in neuronal cells expressing LC3-GFP and α-synuclein and treated with modulators of the autophagy pathway. B103 neuronal cells were infected with LV-α-syn alone or with LV-Beclin 1 and then further infected with LV-LC3-GFP and treated with blockers or activators of the autophagy pathway. A–D, Confocal images of vehicle control group (A); effects of 3-MA (10 mm) (B); effects of bafilomycin A1 (200 nm) (C); and effects of rapamycin (200 nm) (D). Average numbers of LC3-GFP grains per cell and levels of α-syn immunofluorescence were analyzed using confocal images and the ImageQuant system. E–H, Vehicle control group (E); effects of 3-MA (10 mm) (F); effects of bafilomycin A1 (200 nm) (G); and effects of rapamycin (200 nm) (H).*Statistical significance compared with uninfected control cultures; #statistical significance compared with untreated LV-α-syn-infected cultures (p < 0.05, 1-way ANOVA, post hoc Fisher test).
Figure 6.

Immunoblot analysis of the effects of LV-Beclin 1 expression on LC3 II formation and α-syn accumulation in cells treated with modulators of autophagy. B103 neuronal cells were infected with LV-α-syn alone or with LV-Beclin 1 and then were treated with blockers or activators of the autophagy pathway. A–C, Representative Western blots with antibodies against LC3, α-syn, and actin. A–I, Effects of 3-MA (A, D, G), bafilomycin-A1 (B, E, H), or rapamycin (C, F, I). D–F, Levels of LC3 protein were estimated as a ratio of LC3-II to actin levels. G–I, Levels of α-syn immunoreactivity were estimated as a ratio to levels of actin. *Statistical significance compared with uninfected control cultures; #statistical significance compared with untreated LV-α-syn-infected cultures (p < 0.05, 1-way ANOVA, post hoc Fisher test).
Although overexpression of beclin 1 appeared to induce autophagy, to show that beclin 1 was directly responsible for the increased autophagy, we used three different autophagy modulators in the context of beclin 1 overexpression. The autophagy inhibitor, 3-MA, which acts to block PI3-kinase, significantly reduced the numbers of LC3-GFP puncta (Fig. 5B,F) and the formation of LC3-II by immunoblot (Fig. 6A,D) and thus resulted in increased accumulation of α-syn (Figs. 5B,F, 6A,G). Moreover, addition of 3-MA partially blocked the effects of LV-Beclin 1 at inducing the formation of LC3-GFP puncta by confocal microscopy (Fig. 5B,F) and LC3 II by immunoblot in control and cells expressing α-syn and beclin 1 (Fig. 6A,D). Consistent with these findings, 3-MA interfered with the effects of LV-Beclin 1 at reducing the accumulation of α-syn by confocal microscopy (Fig. 5B,F) and by immunoblot (Fig. 6A,G).
Next we tested the effects of the autophagy inhibitor bafilomycin A1, which acts by blocking the fusion of the autophagosome and the lysosome. As expected, this compound increased the formation of LC3-GFP grains by confocal microscopy (Fig. 5C,G) and LC3 II by immunoblot in control cells and in cells expressing α-syn and beclin 1 (Fig. 6B,E). Treatment with bafilomycin A1 interfered with the effects of LV-Beclin 1, resulting in greater accumulation of α-syn by confocal microscopy (Fig. 5C,G) and immunoblot (Fig. 6B,H).
In contrast, the addition of rapamycin, an activator of autophagy, increased the formation of LC3-GFP grains by confocal microscopy (Fig. 5D,H) and LC3 II by immunoblot in controls and cells expressing α-syn and beclin 1 (Fig. 6C,F). Treatment with rapamycin resulted in considerable clearance of α-syn-immunoreactive aggregates by confocal microscopy (Fig. 5D,H) and immunoblot (Fig. 6C,I). The clearance of α-syn was further enhanced in neuronal cells infected with LV-Beclin 1 (Figs. 5D,H, 6C,I).
Double-immunolabeling studies confirmed that neuronal cells expressing beclin 1 and α-syn showed increased colocalization with the lysosomal membrane marker LAMP2 (supplemental Fig. 4A–C, available at www.jneurosci.org as supplemental material). Examination of other autophagy/lysosomal markers such as Atg5 (supplemental Fig. 4D–F, available at www.jneurosci.org as supplemental material), Atg7 (supplemental Fig. 4G–I, available at www.jneurosci.org as supplemental material), and cathepsin D (supplemental Fig. 4J–L, available at www.jneurosci.org as supplemental material) showed similar colocalization of α-syn when beclin 1 was expressed.
Lentivirus delivery of beclin 1 rescues α-syn accumulation and neuronal deficits in tg mice
Overexpression of beclin 1 from the lentivirus reduced the accumulation of α-syn and related deficits in neurite outgrowth in B103 neuronal cells by inducing the autophagy pathway. To determine whether this might prove to be a viable possibility in vivo, we delivered the beclin 1 lentivirus via stereotaxic injection to the temporal cortex and hippocampus of transgenic mice overexpressing the α-syn under the regulatory control of the PDGF-β promoter (line D) (Masliah et al., 2000). This line of mice was selected because they develop neuronal accumulation of α-syn in the limbic system associated with behavioral deficits and have lysosomal defects (Rockenstein et al., 2005b).
Compared with the LV-control (Fig. 7A,B), delivery of LV-Beclin 1 resulted in increased expression in beclin 1 in pyramidal neurons in the hippocampus and in areas adjacent to the injection track in the neocortex (Fig. 7C,D). Beclin 1 immunostaining was localized primarily to the neuronal perykaria with some extension to the apical dendrites (Fig. 7D). Consistent with the immunocytochemistry, immunoblot analysis with tissues micro-dissected from the site of the injection confirmed that beclin 1 levels were increased by 75% in mice that received the LV-Beclin 1 injections compared with LV-control (Fig. 7E). Mice injected with the LV-control virus contained abundant intracellular aggregates of α-syn (Fig. 7F–H), in contrast, following LV-Beclin 1 injection, there was a considerable reduction in the intraneuronal α-syn accumulation; which was now localized to smaller, punctate structures, suggestive of lysosome or autophagic vesicles (Fig. 7I–K). Double-immunocytochemical analysis confirmed that in α-syn tg mice that received the LV-Beclin 1 injections, α-syn and beclin 1 were colocalized in discrete perinuclear, granular structures (Fig. 7L–N).
Figure 7.
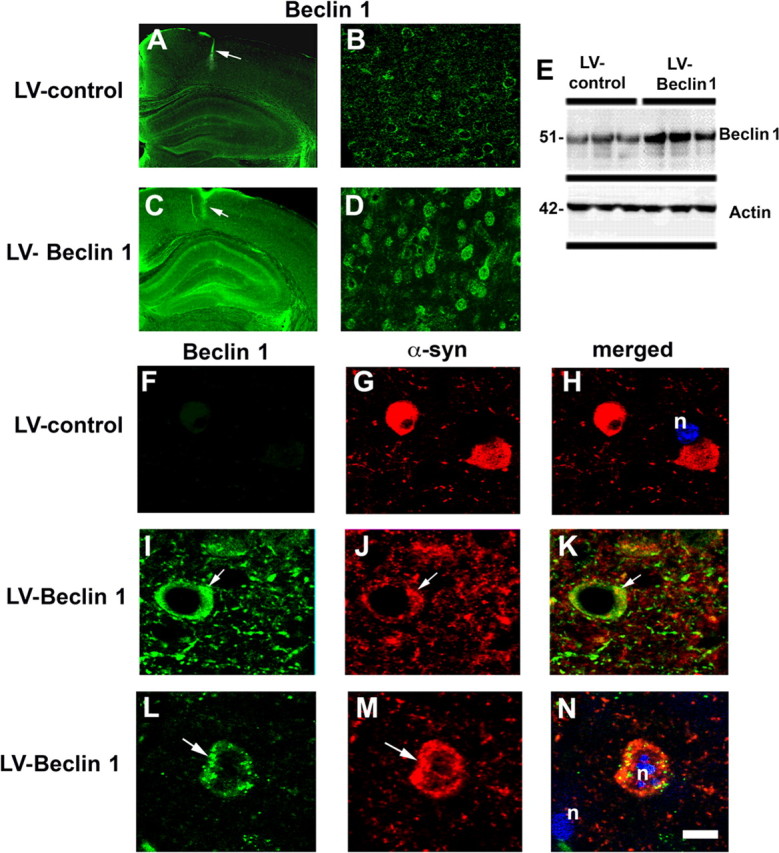
Lentivirus-mediated delivery of beclin 1 to α-syn tg mice reduces α-syn accumulation. Immunocytochemical and confocal analysis 3 months after injection. A, B, Levels of beclin 1 immunoreactivity in α-syn tg mice (line D) that were injected with LV-control. C, D, Beclin 1 immunoreactivity in α-syn tg mice treated with LV-Beclin 1. Arrows (A, C) indicate injection tract in the neocortex. Low-power images of the brain (A, C) were taken with a 5× objective, and higher-magnification images (B, D) were taken with a 40× objective. E, Immunoblot analysis of brain tissue from the injection site to confirm expression of beclin 1. F–N, Confocal analysis of sections double immunolabeled with antibodies against α-syn (red) and beclin 1 (green). F–H, α-syn tg mice that were injected with LV-control. I–N, Colocalization of α-syn and beclin 1 in discrete granular structures in tg mice injected with LV-Beclin 1. Samples were further stained with DAPI to visualize nuclei (n) (blue). Arrows indicate areas of beclin 1 and α-syn colocalization. Scale bar, 5 μm.
Analysis of the levels of α-syn immunoreactivity by confocal microscopy showed a 60% reduction in the α-syn in tg mice that had received the LV-Beclin 1 compared with the LV-control (Fig. 8A–D). In association with the reduction in α-syn accumulation, immunolabeling for the presynaptic marker protein synaptophysin showed an increase in the area covered by immunoreactive presynaptic terminals of α-syn tg mice that received the LV-Beclin 1 injections compared with the LV-control (Fig. 8E–H). Similarly, analysis of the postsynaptic marker MAP2 showed an increase in the percentage area of the neuropil covered by dendrites in mice that received the LV-Beclin 1 virus (Fig. 8I–L) supporting the possibility that the reduction of α-syn accumulation in the LV-Beclin 1-treated animals ameliorated the structural damage to neurons of α-syn tg mice. Importantly, no significant deleterious effects on neuronal or synaptodendritic content were observed in non-tg mice that received injections with LV-Beclin 1 (Fig. 8H,L). To investigate the effects of LV-Beclin 1 in activating autophagy, sections were analyzed by immunocytochemistry with an antibody against LC3 (Fig. 8M). This analysis showed that compared with mice injected with the LV-control, mice that received LV-Beclin 1 displayed increased levels of LC3 immunoreactivity (Fig. 8N–P). In the α-syn tg mice injected with LV-control there was a moderate increase in LC3 immunoreactivity compared with non-tg controls.
Figure 8.
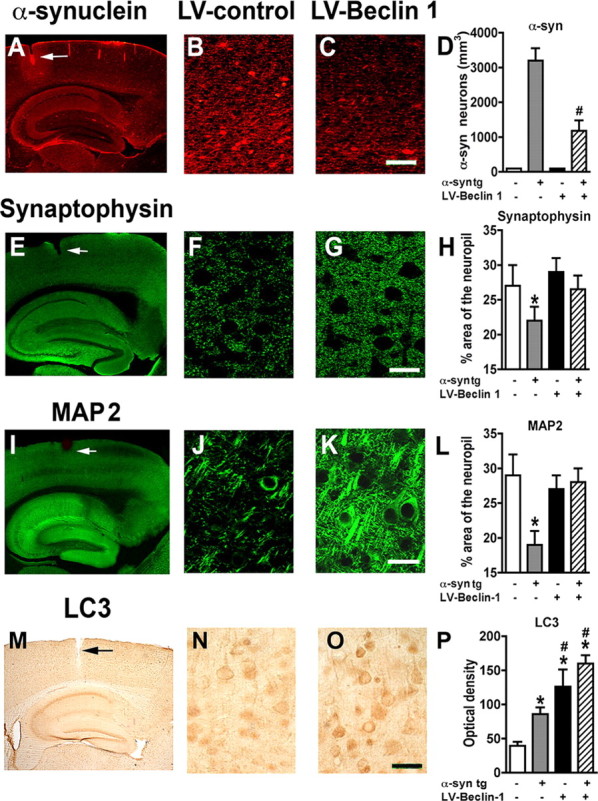
Delivery of LV-Beclin 1 into transgenic mice ameliorates α-synuclein accumulation and related neurodegenerative pathology. Non-tg and α-syn tg mice were injected with LV-control or LV-Beclin 1 and after 3 months of the injection, mice were immunostained and analyzed with confocal microscopy or bright-field microscopy. A–C, Effects of LV-Beclin 1 on patterns of α-syn immunoreactivity. D, Stereological analysis using the disector method. E–G, Immunostaining with the presynaptic protein synaptophysin. H, Percentage area of neuropil occupied by synaptophysin immunolabeled terminals. I–K, Immunostaining with the postsynaptic marker MAP2. L, Percentage area of neuropil occupied by MAP2 immunolabeled dendrites. M–O, Immunocytochemical analysis with an antibody against the autophagy marker LC3. P, Semiquantitative analysis of levels of LC3 immunostaining expressed as optical density. Arrows indicate injection tract. Scale bar, 10 μm for high-power images. #Statistical significance (p < 0.05, 1-way ANOVA, post hoc Fisher test) compared with α-syn tg mice. *Statistical significance (p < 0.05, 1-way ANOVA, post hoc Dunnett's test) compared with non-tg mice.
To further confirm these effects, ultrastructural analysis was performed. Compared with non-tg controls (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material), in the α-syn tg mice there was extensive cytoplasmic accumulation of electrodense deposits that was associated with single- and double-membrane vesicles (supplemental Fig. 5C,D, available at www.jneurosci.org as supplemental material), consistent with the alterations in autophagy. In contrast, in the α-syn tg mice that received the LV-Beclin 1 injections, there was a considerable reduction in the intraneuronal accumulation of electrodense material (supplemental Fig. 5E,F, available at www.jneurosci.org as supplemental material). In addition, normal-looking autophagosomes and multivesicular bodies were identified. Consistent with these effects, confocal microscopy analysis of α-syn tg mice that received LV-Beclin 1 showed extensive α-syn colocalization with LC3 granular structures (supplemental Fig. 5G–I, available at www.jneurosci.org as supplemental material). Together, these data suggest that delivery of LV-Beclin 1 to the α-syn tg mice reduced the accumulation of α-syn and related neuronal pathology by inducing a physiological autophagic response.
Discussion
The present study showed that in a neuronal cell line and in a transgenic mouse model of LBD, lentivirus-mediated overexpression of beclin 1 reduced the abnormal accumulation of α-syn and related neuronal pathology by inducing autophagy. In neuronal cultures, α-syn aggregates colocalized with lysosomal markers and were associated with alterations in the autophagy pathway that lead to reduced neuronal plasticity. The effect of beclin 1 overexpression was related to the autophagy pathway because blocking with 3-MA or bafilomycin A1 resulted in greater accumulation of α-syn, while rapamycin had the opposite effect. Previous studies (Berger et al., 2006; Sarkar et al., 2007) have emphasized the upregulation of autophagy by pharmacological approaches (e.g.: rapamycin), but this is the first study to show a specific gene therapy-directed method for upregulation of autophagy as a therapeutic tool for the treatment of accumulated α-syn. This approach can be specifically targeted to regions of the brain most affected by accumulation of α-syn aggregates.
In agreement with previous studies (Xilouri et al., 2009), we found that neuronal cells with accumulated α-syn showed some increase of LC3 II levels but with impaired lysosomal function. More detailed analysis by confocal microscopy showed that the increased numbers of LC3-GFP autophagic structures were enlarged and irregular. Ultrastructural analysis revealed these electrodense structures to be extremely abnormal. In particular, there were numerous structures with single membranes, and most structures were much larger than normal autophagic vesicles. This coupled with the alterations in LC3-GFP punctate staining suggests that these may be immature autophagic vesicles. This suggests interference with the normal maturation pathway of the autophagic vesicles.
The mechanisms through which accumulation of wild-type α-syn might impair macroautophagy in our neuronal cells are not completely clear. However, a previous study using differentiated SH-SY5Y and primary neurons showed that the alterations in macroautophagy in wild-type α-syn-expressing cells were attributed to the blockage of CMA (Xilouri et al., 2009). Moreover, studies have shown that mutant α-syn inhibits CMA and triggers increased macroautophagy and lysosomal dysfunction independent of CMA (Cuervo et al., 2004; Xilouri et al., 2009).
A therapeutic approach with rapamycin or beclin 1 might act to reestablish the physiological autophagy pathway. This view is supported by the studies with LC3-GFP, immunoblot with LC3 and ultrastructural studies in the α-syn tg mice treated with LV-Beclin 1 where we detected the presence of autophagic vesicles of normal appearance in contrast to α-syn tg mice treated with LV-control where abnormal and enlarged autophagic vesicles were observed.
This is consistent with recent studies showing that α-syn aggregates are cleared via autophagy and that alterations in the lysosomal pathway might participate in the mechanisms of α-syn-mediated neurodegeneration (Stefanis et al., 2001; Meredith et al., 2002; Webb et al., 2003; Cuervo et al., 2004; Rideout et al., 2004; Nakajima et al., 2005; Rockenstein et al., 2005b). Mutant forms of α-syn found in familial PD patients have been shown to block autophagy and α-syn contains a consensus sequence for CMA targeting (Cuervo et al., 2004). Further supporting a role for autophagy dysfunction in these disorders, recent studies have shown that in lysosomal storage diseases such as Gaucher's disease (Tayebi et al., 2001; Várkonyi et al., 2003) and Niemann-Pick disease (Saito et al., 2004), there is increased susceptibility to develop parkinsonism and α-syn accumulation.
Therefore, activation of the autophagy pathway by pharmacological means or gene transfer might reduce the pathology associated with the neuronal accumulation of α-syn aggregates. We have previously shown that beclin 1 is capable of inducing autophagy and reducing the neuropathological alterations in APP tg models of AD (Pickford et al., 2008). In the present study, we showed that beclin 1 gene transfer is also capable of reducing the accumulation of α-syn aggregates and associated neuronal deficits. Delivery of the LV-Beclin 1 virus in the limbic system of α-syn tg mice led to improvements in expression of the presynaptic marker synaptophysin and the postsynaptic marker MAP2. The decrease in accumulated α-syn was observed in areas of autophagy induction as determined by increases in expression of LC3. Ultrastructural analysis of neurons in the region of LV injection revealed decreased accumulated α-syn and increased markers of autophagy. Thus, the delivery of the beclin 1 by the lentivirus appears to reverse the accumulation of α-syn probably by increasing activation of the autophagy pathway.
These findings are in agreement with recent studies showing that rapamycin or rapamycin analogs are capable of reducing the accumulation of α-syn oligomers via the autophagy pathway (Sarkar et al., 2007). Treatment of cells with rapamycin, which blocks the mTOR protein activity, results in upregulation of the autophagy pathway (Meijer and Codogno, 2004). Rapamycin acts by binding to FK506 binding protein 12, which then inhibits phosphorylation of mTor. Moreover, rapamycin treatment has been shown to be protective in animal models of other neurodegenerative disorders such as Huntington's disease (Ravikumar et al., 2004).
Similar to overexpression of beclin 1, rapamycin ultimately induces autophagy and reduces overall levels of misfolded proteins. However, in addition to inducing autophagy, rapamycin (via mTor) downregulates overall protein synthesis and cell proliferation (Wang and Proud, 2006; King et al., 2008; Wyttenbach et al., 2008). These undesirable side effects of rapamycin make its use in neurodegenerative diseases involving misfolded proteins problematic. This, coupled with the fact that rapamycin poorly crosses the blood–brain barrier to exert its effect on neurons (Pong and Zaleska, 2003), makes the identification of alternative autophagy inducers desirable. Gene delivery of beclin 1 might alleviate some of these problems. The lentivirus, or other viral vector, containing the gene for beclin 1 can be delivered to the exact site of α-syn accumulation, for instance, the caudate–putamen or substantia nigra in PD or, as in this study, the limbic system in case of LBD.
Although the role of beclin 1 in autophagy and cell maintenance is clear, ample evidence suggests that beclin 1 is required not only for cell viability but also for neuronal function in the face of aggregated proteins. Beclin 1 is a coiled-coil, 60 kDa protein that is analogous to the yeast autophagy protein Apg6 (Liang et al., 1998, 1999). Beclin 1 binds Bcl2 to block apoptosis (Liang et al., 1998, 2006; Takacs-Vellai et al., 2005) and functions through its interactions with Vps34 and PI3 kinase to induce autophagy in an mTor-independent pathway (Furuya et al., 2005; Takacs-Vellai et al., 2005). Recently identified interacting proteins, UVRAG and Ambra1, also act as regulators of autophagy (Liang et al., 2006; Fimia et al., 2007). Beclin 1 has been localized in some instances to the Golgi apparatus of the ER and may play a role in sorting proteins for degradation via the autophagy pathway (Furuya et al., 2005; Takacs-Vellai et al., 2005). Thus, beclin 1 may be playing a role in neurons in PD and LBD in targeting excess α-syn to the autophagy pathway thereby reducing the potential for accumulation.
The specific molecular defects in the autophagy pathway leading to PD and LBD are not completely understood yet; however, recent studies have shown that reduced Atg5 or 7 expression in knock out mice results in loss of neurons and accumulation of protein in the remaining neurons (Hara et al., 2006; Komatsu et al., 2006).
Together, these studies support the view that alterations in the autophagy pathway play an important role in the pathogenesis of disorders with parkinsonism, dementia, and α-syn accumulation and that activation of the autophagy pathway might ameliorate the associated deficits. Thus, beclin 1 plays an important role in the intracellular degradation of α-syn either directly or indirectly through the autophagy pathway and may present a novel therapeutic target for LBD/PD.
Footnotes
This work was supported by National Institutes of Health Grants AG10435, AG022074, AG18440, and AG5131.
References
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson's disease. J Neurochem. 2008;105:1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C, Winner B, Winkler J, Masliah E. Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci. 2008;28:4250–4260. doi: 10.1523/JNEUROSCI.0066-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Erlich S, Shohami E, Pinkas-Kramarski R. Neurodegeneration induces upregulation of Beclin 1. Autophagy. 2006;2:49–51. doi: 10.4161/auto.2156. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Galasko D. Lewy body disease. Curr Opin Neurol Neurosurg. 1992;5:889–894. [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Masliah E. Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer's and dementia with Lewy bodies. Neurochem Res. 2003;28:1743–1756. doi: 10.1023/a:1026073324672. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. β-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- Hong K, Sherley J, Lauffenburger DA. Methylation of episomal plasmids as a barrier to transient gene expression via a synthetic delivery vector. Biomol Eng. 2001;18:185–192. doi: 10.1016/s1389-0344(01)00100-9. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HAR, Kittel A, Saitoh T. The precursor protein of non-Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, Rockenstein E, Imam SZ, Masliah E. alpha-Synuclein aggregates interfere with Parkin solubility and distribution: role in the pathogenesis of Parkinson disease. J Biol Chem. 2008;283:6979–6987. doi: 10.1074/jbc.M710418200. [DOI] [PubMed] [Google Scholar]
- King MA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052–1063. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree—a new disease? Clin Neuropathol. 1984;3:185–192. [PubMed] [Google Scholar]
- Langford D, Grigorian A, Hurford R, Adame A, Crews L, Masliah E. The role of mitochondrial alterations in the combined toxic effects of human immunodeficiency virus Tat protein and methamphetamine on calbindin positive-neurons. J Neurovirol. 2004;10:327–337. doi: 10.1080/13550280490520961. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem Biophys Res Commun. 1999;260:712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- Mariño G, López-Otín C. Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci. 2004;61:1439–1454. doi: 10.1007/s00018-004-4012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- McKeith IG. Spectrum of Parkinson's disease, Parkinson's dementia, and Lewy body dementia. Neurol Clin. 2000;18:865–902. doi: 10.1016/s0733-8619(05)70230-9. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC, Jr, Lau YS. Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res. 2002;956:156–165. doi: 10.1016/s0006-8993(02)03514-x. [DOI] [PubMed] [Google Scholar]
- Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, Alford M, Masliah E. Protection against HIV-1 gp120-induced brain damage by neuronal overexpression of human amyloid precursor protein (hAPP) J Exp Med. 1995;181:1551–1556. doi: 10.1084/jem.181.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM-Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Takauchi S, Ohara K, Kokai M, Nishii R, Maeda S, Takanaga A, Tanaka T, Takeda M, Seki M, Morita Y. Alpha-synuclein-positive structures induced in leupeptin-infused rats. Brain Res. 2005;1040:73–80. doi: 10.1016/j.brainres.2005.01.099. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Pong K, Zaleska MM. Therapeutic implications for immunophilin ligands in the treatment of neurodegenerative diseases. Curr Drug Targets CNS Neurol Disord. 2003;2:349–356. doi: 10.2174/1568007033482652. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-β proteins deposits in a mutant APP transgenic model depends on levels of Aβ1–42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, Masliah E. High beta-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-beta levels: implications for the treatment of Alzheimer disease. J Biol Chem. 2005a;280:32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005b;80:247–259. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- Saito Y, Suzuki K, Hulette CM, Murayama S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J Neuropathol Exp Neurol. 2004;63:323–328. doi: 10.1093/jnen/63.4.323. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Müller F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513–1517. doi: 10.1016/j.cub.2005.07.035. [DOI] [PubMed] [Google Scholar]
- Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Sisk A, Masliah E. Abnormal distribution of the non-Aβ component of Alzheimer's disease amyloid precursor/α-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. 1998;78:1169–1177. [PubMed] [Google Scholar]
- Takenouchi T, Hashimoto M, Hsu LJ, Mackowski B, Rockenstein E, Mallory M, Masliah E. Reduced neuritic outgrowth and cell adhesion in neuronal cells transfected with human alpha-synuclein. Mol Cell Neurosci. 2001;17:141–150. doi: 10.1006/mcne.2000.0923. [DOI] [PubMed] [Google Scholar]
- Tayebi N, Callahan M, Madike V, Stubblefield BK, Orvisky E, Krasnewich D, Fillano JJ, Sidransky E. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab. 2001;73:313–321. doi: 10.1006/mgme.2001.3201. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JX, Masliah E. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007;274:1862–1877. doi: 10.1111/j.1742-4658.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- Várkonyi J, Rosenbaum H, Baumann N, MacKenzie JJ, Simon Z, Aharon-Peretz J, Walker JM, Tayebi N, Sidransky E. Gaucher disease associated with parkinsonism: four further case reports. Am J Med Genet A. 2003;116A:348–351. doi: 10.1002/ajmg.a.10028. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson's disease. Neurosci Lett. 1997;239:45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180–182. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Hands S, King MA, Lipkow K, Tolkovsky AM. Amelioration of protein misfolding disease by rapamycin: translation or autophagy? Autophagy. 2008;4:542–545. doi: 10.4161/auto.6059. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]