

Abstract
A biased-potential molecular dynamics simulation method, accelerated molecular dynamics (AMD), was combined with the chemical shift prediction algorithm SHIFTX to calculate 1HN, 15N, 13Cα, 13Cβ, and 13C′ chemical shifts of the ankyrin repeat protein IκBα (residues 67−206), the primary inhibitor of nuclear factor κ-B (NF-κB). Free-energy-weighted molecular ensembles were generated over a range of acceleration levels, affording systematic enhancement of the conformational space sampling of the protein. We have found that the predicted chemical shifts, particularly for the 15N, 13Cα, and 13Cβ nuclei, improve substantially with enhanced conformational space sampling up to an optimal acceleration level. Significant improvement in the predicted chemical shift data coincides with those regions of the protein that exhibit backbone dynamics on longer time scales. Interestingly, the optimal acceleration level for reproduction of the chemical shift data has previously been shown to best reproduce the experimental residual dipolar coupling (RDC) data for this system, as both chemical shift data and RDCs report on an ensemble and time average in the millisecond range.
Chemical shifts, the primary observables in NMR spectroscopy, have long been recognized as sensitive probes of protein structure and dynamics.(1) Despite their complex dependence on multiple geometric and electronic factors, chemical shifts have been employed with some considerable success in the determination of protein structures in solution.(2) The success of such structure determination protocols can be attributed to the remarkable advances made in the accurate prediction of chemical shifts given a representative static structure. Numerous algorithms, such as SHIFTS,(3) SHIFTX,(4) and SPARTA,(5) allow for the rapid and automated prediction of chemical shifts. These algorithms generally employ a combination of quantum-chemical, semiempirical, and homology/chemical shift database approaches.(6) The interpretation and prediction of chemical shift data is complicated by the fact that this experimental observable represents both an ensemble and time average. Proteins are intrinsically flexible systems that display a broad range of dynamics over a hierarchy of time scales. The experimentally measured chemical shift for a given nucleus therefore reports on a free-energy-weighted average over all conformational substates explored by the protein up to the millisecond time scale (the so-called chemical shift coalescence limit). In light of this, there is a fundamental limitation on how accurately one can interpret or predict chemical shift data using a “single-copy” or static-structure representation of the system. Although a homology/chemical shift database approach affords an approximate representation of the effect of short-time-scale vibrational motion, an accurate chemical shift analysis for nuclei found in highly flexible loops or regions of the protein that adopt multiple conformational substates can be problematic, as has been previously reported in the literature.(7)
Chemical shifts are not the only NMR observables that are sensitive to the effects of dynamic averaging: scalar J couplings and residual dipolar couplings (RDCs) also report on averages up to the millisecond range and therefore encode key information for understanding slower protein motions.(8) In recent years, considerable effort has been focused on improving the interpretation of such NMR observables, including the effect of dynamic averaging: focusing predominantly on RDC data, simultaneous structure−dynamics determination protocols have been developed using molecular-modeling approaches,(9) or in the framework of ensemble-averaged molecular dynamics (MD) simulation.(10) We have recently proposed an alternative approach for the study of RDCs using a biased-potential MD simulation method called accelerated molecular dynamics (AMD).(11) Using this approach, we have previously demonstrated that enhanced conformational space sampling provides a representation of both RDCs and scalar J couplings considerably better than that provided by standard MD trajectories.(12) In the present work, we show that enhanced conformational space sampling allows for an improvement in the prediction of chemical shift data, focusing on the ankyrin repeat protein IκBα (residues 67−206), the primary inhibitor of nuclear factor κ-B (NF-κB),(13) because of its heterogeneous distribution of dynamics.
A detailed account of the simulation protocol employed has been presented elsewhere(12a) and is included in the Supporting Information (SI). The atomic coordinates for IκBα(67−206) were obtained from the X-ray crystal structure (PDB entry 1NFI).(14) After a standard equilibration procedure, five 10 ns MD simulations were performed. These simulations acted as a control set and were used as a starting point for AMD simulations. Twenty “dual-boost” AMD simulations were performed for 10 million steps at increasing levels of torsional acceleration. An initial free-energy “pre-pruning” for each AMD trajectory was performed in which the high-energy structures (some 80% of the trajectory) were stripped out and the remaining 20% were used to perform the clustering analysis. To obtain accurate free-energy statistics, a reduced set of structures representing the conformational space sampled in the AMD trajectory were used to seed classical MD simulations, which were then subjected to a MM/PBSA analysis.(15) Multiple free-energy-weighted molecular ensembles were constructed at each acceleration level. All of the simulations were performed using a modified version of the AMBER10 simulation suite.(16) SHIFTX(4) was employed to obtain chemical shifts for all of the 1HN, 15N, 13Cα, 13Cβ, and 13C′ nuclei. Chemical shifts were calculated for each member of the free-energy-weighted molecular ensemble and then averaged. The resulting predicted chemical shift data were then averaged over all ensembles at a given acceleration level. In this way, we obtained an ensemble- and time-averaged representation of the chemical shift data. The accuracy of the predicted chemical shifts was assessed by calculating the root-mean-square-difference (RMSD) with respect to the experimental data.
IκBα exhibits a strongly heterogeneous distribution of dynamics over a broad hierarchy of time scales. Relative to experimental spin-relaxation data (which report on internal dynamics up to 6 ns), we observed a dramatic enhancement of conformational space sampling in the N-terminal tail and the flexible loops (residues 98−101 and 162−175) at weak acceleration levels. At more aggressive acceleration levels, enhanced conformational space sampling was observed in residues 86−89, 108−115, 136−145, and 180−185, which correlate well with those residues exhibiting exchange relaxation (μs-to-ms dynamics) as obtained using R1/R2/het-NOE analysis.(12a) Similarly, we observed a broad variation in the accuracy of the predicted chemical shifts across the different acceleration levels, as shown in the inset of Figure 1. The X-ray crystal structure provided a rather poor prediction of the experimental data. Chemical shift RMSDs for the X-ray crystal structure for HN, N, Cα, Cβ, and C′ nuclei were 0.62, 2.89, 1.45, 1.31, and 1.92 ppm, respectively, giving a cumulative RMSD of 8.19 ppm. The results obtained from the trajectory-averaged 10 ns MD simulations were slightly better (0.57, 2.60, 1.31, 1.18, and 1.78 ppm, respectively). The optimal reproduction of the chemical shift data was obtained from trajectory-averaged molecular ensembles generated using the acceleration parameters [Eb(dih) − V(dih) = 600 kcal/mol, α(dih) = 120 kcal/mol]. Interestingly, this coincides with the acceleration level that best reproduced the experimental RDC data for this system,(12a) as both chemical shift data and RDCs report on an ensemble and time average over the millisecond range. The optimal conformational space sampling gave corresponding chemical shift RMSDs of 0.44, 1.84, 1.01, 1.01, and 1.62 ppm, yielding a cumulative RMSD of 5.92 ppm, which represents a 20% improvement relative to the standard MD simulations and a 28% improvement relative to the static X-ray crystal structure.
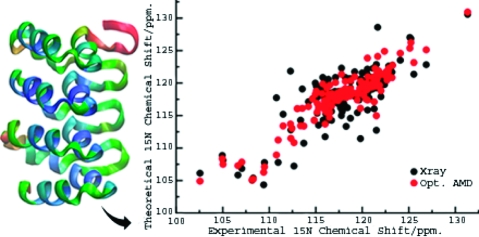
Figure 1.

Correlation between the experimental and predicted 15N chemical shifts for the X-ray crystal structure (black circles) and the trajectory-averaged optimal conformational space sampling ensembles (red circles). The inset shows the variation of the cumulative RMSD as a function of the acceleration level, Eb(dih) − V(dih), with α(dih) = 120 kcal/mol.
The most notable improvement in the chemical shift results was obtained for the 15N nuclei. This result is shown graphically in Figure 1, which depicts the correlation between the experimental and predicted 15N chemical shifts for the X-ray crystal structure and the trajectory-averaged optimal conformational space sampling ensembles. The extensive averaging procedure is very important: no individual molecular ensemble obtained at the optimal acceleration level gave a chemical shift prediction as good as the trajectory-averaged result.
The improvement in the predicted chemical shift data is directly correlated to those regions of the protein that exhibit substantially enhanced conformational space sampling. This result is presented graphically in Figure 2. The top panel compares NH order parameters obtained from experimental spin-relaxation data with those calculated from the molecular ensembles obtained at the optimal acceleration level for reproduction of the chemical shift data. The bottom panel compares 15N chemical shift RMSDs for the X-ray crystal structure and the trajectory-averaged optimal molecular ensembles. Significant improvement in the predicted 15N chemical shift data clearly coincides with those regions of the protein that exhibit backbone dynamics on longer time scales. Graphical results for HN, Cα, Cβ, and C′ are presented in the SI. The strong correlation between the improvement in the predicted chemical shifts and the regions of enhanced conformational space sampling is also observed for the 13Cα and 13Cβ nuclei. The improvement in the 1HN chemical shifts is smaller and not as strongly correlated with the backbone dynamics. An identical analysis was performed on ubiquitin, and a similar improvement in the calculated chemical shifts commensurate with its less dynamical behavior was observed (see the SI). Thus, for proteins with a heterogeneous distribution of dynamics over a hierarchy of time scales, chemical shift prediction from free-energy-weighted AMD ensembles provides substantial improvement.
Figure 2.

(top) NH order parameters from experimental spin relaxation data (black) and calculated from the molecular ensembles that best reproduced the chemical shift data (red). (bottom) 15N chemical shift RMSDs for the X-ray crystal structure (black) and the optimal conformational space sampling molecular ensembles (red).
Acknowledgments
We gratefully acknowledge Prof. H. Jane Dyson, Dr. G. Kroon, and the Scripps Research Institute NMR facility.
Supporting Information Available
Complete refs (2c) and (16); MD simulation protocol; HN, Cα, Cβ, and C′ results for IκBα; and tabulated results for ubiquitin. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- a Wishart D. S.; Case D. A. Methods Enzymol. 2001, 338, 3–34. [DOI] [PubMed] [Google Scholar]; b Hunter C. A.; Packer M. J.; Zonta C. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 47, 27–39. [Google Scholar]; c Berjanskii M. V.; Wishart D. S. J. Am. Chem. Soc. 2005, 127, 14970–14971. [DOI] [PubMed] [Google Scholar]
- a Osapay K.; Theriault Y.; Wright P. E.; Case D. A. J. Mol. Biol. 1994, 244, 183–197. [DOI] [PubMed] [Google Scholar]; b Cavalli A.; Salvatella X.; Dobson C. M.; Vendruscolo M. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 9615–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shen Y.; et al. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.-P.; Case D. A. J. Biomol. NMR 2001, 21, 321–333. [DOI] [PubMed] [Google Scholar]
- Neal S.; Nip A. M.; Zhang H.; Wishart D. S. J. Biomol. NMR 2003, 26, 215–240. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Bax A. J. Biomol. NMR 2007, 38, 289–302. [DOI] [PubMed] [Google Scholar]
- Case D. A. Curr. Opin. Struct. Biol. 1998, 8, 624–630. [DOI] [PubMed] [Google Scholar]; Case D. A. Curr. Opin. Struct. Biol. 2000, 10, 197–203. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. J. Biomol. NMR 2009, 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]; Page 213: “Large discrepancies between predictions and crystal structures are primarily limited to loop regions, and for the few cases where multiple X-ray structures are available such residues are often found in different states in the different structures.”
- a Tjandra N.; Bax A. Science 1997, 278, 1111–1114. [DOI] [PubMed] [Google Scholar]; b Prestegard J. H.; al-Hashimi H. M.; Tolman J. R. Q. Rev. Biophys. 2000, 33, 371–424. [DOI] [PubMed] [Google Scholar]
- a Tolman J. R. J. Am. Chem. Soc. 2002, 124, 12020–12031. [DOI] [PubMed] [Google Scholar]; b Bouvignies G.; Bernado P.; Meier S.; Cho K.; Grzesiek S.; Bruschweiler R.; Blackledge M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13885–13890. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bouvignies G.; Markwick P. R. L.; Bruschweiler R.; Blackledge M. J. Am. Chem. Soc. 2006, 128, 15100–15101. [DOI] [PubMed] [Google Scholar]; d Meiler J.; Peti W.; Prompers J.; Griesinger C.; Bruschweiler R. J. Am. Chem. Soc. 2001, 123, 6098–6107. [DOI] [PubMed] [Google Scholar]
- a Schwieters C. D.; Clore G. M. Biochemistry 2004, 43, 10678–10691. [DOI] [PubMed] [Google Scholar]; b Lange O. F.; Lakomek N. A.; Walter K. F.; Fares C.; Schroeder G. F.; Walter K. F.; Becker S.; Meiler J.; Grubmuller H.; Griesinger C.; de Groot B. L. Science 2008, 320, 1471–1475. [DOI] [PubMed] [Google Scholar]
- a Hamelberg D.; Mongan J.; McCammon J. A. J. Chem. Phys. 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]; b Hamelberg D.; McCammon J. A. J. Am. Chem. Soc. 2005, 127, 13778–13779. [DOI] [PubMed] [Google Scholar]; c Markwick P. R. L.; Bouvignies G.; Blackledge M. J. Am. Chem. Soc. 2007, 129, 4724–4730. [DOI] [PubMed] [Google Scholar]
- a Cervantes C. F.; Markwick P. R. L.; Sue S.-C.; McCammon J. A.; Dyson H. J.; Komives E. A. Biochemistry 2009, 48, 8023–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Markwick P. R. L.; Showalter S.; Bouvignies G.; Bruschweiler R.; Blackledge M. J. Biomol. NMR 2009, 45, 17–21. [DOI] [PubMed] [Google Scholar]; c Markwick P. R. L.; Bouvignies G.; Salmon L.; McCammon J. A.; Nilges M.; Blackledge M. J. Am. Chem. Soc. 2009, 131, 16968–16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle P. A.; Baltimore D. Science 1998, 242, 540–546. [DOI] [PubMed] [Google Scholar]
- Jacobs M. D.; Harrison S. C. Cell 1998, 95, 749–758. [DOI] [PubMed] [Google Scholar]
- Massova I.; Kollmann P. A. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar]
- Case D. A.; et al. AMBER10; University of California: San Francisco, 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.