

Abstract
SNARE [soluble NSF (N-ethylmaleimide-sensitive fusion protein) attachment protein receptor] proteins are essential for membrane fusion and are conserved from yeast to humans. Sequence alignments of the most conserved regions were mapped onto the recently solved crystal structure of the heterotrimeric synaptic fusion complex. The association of the four α-helices in the synaptic fusion complex structure produces highly conserved layers of interacting amino acid side chains in the center of the four-helix bundle. Mutations in these layers reduce complex stability and cause defects in membrane traffic even in distantly related SNAREs. When syntaxin-4 is modeled into the synaptic fusion complex as a replacement of syntaxin-1A, no major steric clashes arise and the most variable amino acids localize to the outer surface of the complex. We conclude that the main structural features of the neuronal complex are highly conserved during evolution. On the basis of these features we have reclassified SNARE proteins into Q-SNAREs and R-SNAREs, and we propose that fusion-competent SNARE complexes generally consist of four-helix bundles composed of three Q-SNAREs and one R-SNARE.
Keywords: membrane fusion, neurotransmission, clostridial neurotoxins
Intracellular membrane fusion involves conserved sets of membrane proteins that are commonly referred to as SNARE proteins [soluble NSF (N-ethylmaleimide-sensitive fusion protein) attachment protein receptor proteins] (1–4). SNARE proteins can be grouped into several small protein families with a growing number of members. The similarity between distant members of these protein families is rather limited, but it is thought that they all operate by means of a common mechanism. The variants functioning in neuronal exocytosis are among the best characterized; they include the synaptic vesicle protein synaptobrevin (also referred to as VAMP) and the synaptic plasma membrane proteins SNAP-25 and syntaxin-1A. These proteins readily assemble into a stable ternary complex whose core structure has been recently solved by x-ray crystallography (5). The SNARE complex can be reversibly disassembled by the ATPase NSF in conjunction with soluble cofactors termed SNAPs (soluble NSF attachment proteins) (6, 7). The formation of the assembled complex is now believed to be a critical step leading to membrane fusion. Assembly proceeds spontaneously from less structured monomers and results in a stoichiometric and elongated complex with all membrane anchor domains located at one side of the rod-shaped particle (2, 5, 8, 9). These findings led to a model that assembly juxtaposes membranes, thus overcoming the free energy barrier for fusion (2, 5, 9, 10). However, it remains to be established whether more distantly related SNARE proteins form similar complexes and which of the structural features of the neuronal complex are generally relevant for SNARE protein function.
METHODS
Sequence Analysis.
Sequences were aligned by using the ClustalW software available at http://www2.ebi.ac.uk/clustalw (11). A nearest-neighbor dendrogram of the SNAP-25, syntaxin, and synaptobrevin families was obtained and used to define subgroups consisting of syntaxin-1 through syntaxin-4, all known homologues of SNAP-25 excluding the distant yeast Sec9p and Sto20p homologues, and synaptobrevin-I, synaptobrevin-II and cellubrevin. The sequence variation was computed as follows: The amino acids were separated into five classes, hydrophobic (Ala, Val, Phe, Ile, Leu, Pro, Met), positively charged (Lys, Arg), negatively charged (Asp, Glu), hydrophilic (Ser, Thr, Tyr, Cys, Asn, Gln, His, Trp), and glycine. The sequence variation was defined as
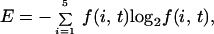 |
where f(i, t) is the frequency of the particular amino acid class at position i (12).
Modeling.
The accessible surface area of the synaptic fusion complex was computed by the method of Lee and Richards (13). The syntaxin-4 substitution was modeled, based on the synaptic fusion complex crystal structure (5), by substituting the residues that differ between syntaxin-4 and syntaxin-1, using the programs o and sod (14). The side-chain rotamer was transferred from the original residue to the substituted residue where possible; otherwise the most favorable side chain rotamer was picked. The model was regularized by using energy minimization as implemented in the Crystallography and NMR System (CNS) (15).
RESULTS AND DISCUSSION
Functional Significance and Evolutionary Conservation of the Central Layer Domain.
Primary sequence comparison with closely and distantly related homologues shows that the amino acids in the ionic layer (designated as “0” layer, ref. 5 and Fig. 1B) at the center of the synaptic fusion complex are the most highly conserved residues in all SNARE proteins (16) (Fig. 1A). This layer is composed of Arg-56 of synaptobrevin-II, Gln-226 of syntaxin-1A, and Gln-53 and Gln-174 of SNAP-25B (5). Amino acid substitutions in this layer severely disrupt membrane traffic. For example, Sec22p, a distantly related yeast synaptobrevin homologue, participates in the fusion of transport vesicles with the cis face of the Golgi apparatus (17). Replacing the conserved arginine with glycine results in defects that are comparable to a deletion of the gene (18, 19). Trafficking is also perturbed by a double mutation in the yeast SNARE Vti1p that affects both the 0 layer and the −5 layer, Gln-158 → Arg and Ala-141 → Ser, respectively (20) (Fig. 1A).
Figure 1.

(A) Sequence alignment (16) of the four-helix bundle region of the synaptic fusion complex (5) for a representative subset of the entire SNARE family. The sequence analysis was restricted to 16 layers (blue) of the four-helix bundle in the synaptic fusion complex (5), including 7 layers upstream (layers −1 to −7) and 8 layers downstream (layers +1 to +8) of the ionic layer (layer 0). Conserved residues are shaded in gray. The conserved glutamine and arginine residues forming the ionic 0 layer are indicated in red and green, respectively. The mutations discussed in the text are indicated by black shaded boxes. There are two alignment tables for the SNAP-25 family that correspond to the two SNAP-25 α-helices in the synaptic fusion complex (5). GenBank accession numbers for the syntaxin family are as follows: sx1a, RN, P32851; unc-64b, CE, AF047885; sx3, RN, Q08849; sx4, RN, Q08850; Sso1, SC, P32867; sed5, SC, Q01590; sx5, RN, Q08851; vam3, SC, Q12241. Accession numbers for the SNAP-25 family are SNAP-25B, HS, P13795; syndet, MM, U73143; SNAP-25, DM, U81153; Y22F5A.5, CE, AL021479; sec9, SC, L34336; and spo20, SC, Z49211. Accession numbers for the bos-group are bet1, SC, P22804; mbet1, MM, AF007552; vti1, HS, AF035824; vti1, SC, AF006074; membrin, RN, U91539; bos1, SC, P25385; and vam7, SC, P32912. Accession numbers for the synaptobrevin/vamp family (R-SNAREs) are sb2, RN, M24105; cb/cellubrevin, RN, S63830; sb1 CE AF003281; sb, HM, U85805; Snc1, SC, M91157; sb5/6, HS, AA222692; sb7, MM, X96737; sec22 (Sly2), SC, L8479; sec22b, MM, U91538; nyv1, SC, Z73265; and tomosyn, RN, U92072. The two-letter species abbreviations after the protein name are as follows: HS, Homo sapiens; MM, Mus musculus; RN, Rattus norvegicus; SC, Saccharomyces cerevisiae; DM, Drosophila melanogaster; TM, Torpedo marmorata; CE, Caenorhabditis elegans; and HM, Hirudo medicinalis. (B) Layers of the synaptic fusion complex crystal structure (5). Indicated are Cα traces (gray), local helical axes (blue, red, and green for synaptobrevin-II, syntaxin-1A, and SNAP-25b, respectively), and layers (black) by virtual bonds between corresponding Cα positions.
The layers flanking the ionic 0 layer are maintained primarily by hydrophobic interactions (5). The functional relevance of these layers is highlighted by phenotypes in different species that are caused by mutations resulting from single amino acid substitutions. Interactions in the +1 layer are disrupted by a loss-of-function mutation in the sec9–7 allele, Leu-627 → His (Ile-178 in SNAP-25B) (21, 22). Another example of a mutation in the +1 layer is provided by the bos1–1 allele of a yeast homologue of the SNARE family (Fig. 1A). In this case, a Leu-190 → Ser mutation results in lower stability of the SNARE complex in vitro and in a disruption of membrane traffic in vivo. In terms of the synaptic fusion complex, this hydrophilic substitution could destabilize the hydrophobic leucine zipper interactions in the +1 layer. There are also known single-site mutations that map to the outer layers of the fusion complex: C. elegans unc-64 Ala-241 → Val (Ala-240 in syntaxin-1A) and Ala-248 → Val (Ala-247 in syntaxin-1A), and C. elegans synaptobrevin Leu-62 → Phe (Leu-70 in synaptobrevin-II), and Ala-66 → Gly (Ala-74 in synaptobrevin-II). These single-site mutations in the +4, +5, and +6 layers affect synaptic transmission, resulting in lethargic animals with locomotory abnormalities. Interestingly, double mutants involving neighboring layers produced synergistic effects with severe defects in neurotransmission (23). These more severe phenotypes may be explained by disruption of packing interactions between the layers. Another example of a single amino acid substitution is the exchange of Thr-254 for Ile-254 in layer +7 of Drosophila syntaxin-1A (Thr-251 in rat syntaxin-1A). This mutation is characterized by a temperature-induced block of synaptic transmission, the lack of detectable SNARE complexes in neuronal extracts, and reduced binding to synaptobrevin in vitro (24).
The complementary nature of the ionic 0 layer reflects its ability to form strong hydrogen bonds between the three glutamine residues and the guanidino group of the arginine side chain. A different type of asymmetric “complementarity” is found in layers −3, −2, and +6 (5), where bulky side chains are packed together with smaller ones. These layers may enforce the correct register between the α-helical components of the fusion complex. Some of these layers could also enforce the correct relative orientation of the α-helices (25). An example of a complementary packing layer is the −3 layer of the synaptic fusion complex, in which a bulky phenylalanine residue, Phe-216, of syntaxin-1A occupies the hydrophobic core (Fig. 2). This packing arrangement is accommodated by Gly-43 of SNAP-25B. These residues are conserved in Sec9p and Sso1p, the yeast homologues of SNAP-25B and syntaxin-1A, respectively. The sec9–4 mutant involves the replacement of the glycine residue by aspartic acid, and it is characterized by defects in exocytosis and decreased complex formation (18, 21). A corresponding mutation in the leech homologue of SNAP-25B disrupted binding to syntaxin (8). An insertion of an arginine residue at this position in the synaptic fusion complex would result in significant steric and electrostatic penalties (Fig. 2). Another mutation that is associated with the −3 layer is the Met-46 → Ala substitution of neuronal synaptobrevin-II (26). The phenotype of the mutation is characterized by missorting and reduced endocytosis. It may be explained by a packing defect within the core of the four-helix bundle upon replacement of bulky methionine with a residue having a smaller side chain.
Figure 2.

Picture of the −3 layer in the synaptic fusion complex crystal structure (5). This layer consists of synaptobrevin-II Met-46, syntaxin-1A Phe-216, and SNAP-25B Gly-43 and Ala-164. The interactions within this layer explain the destabilizing effect of a mutation involving the corresponding glycine residue in the sec9–4 allele. Side-chain atoms in the layer are shown as “licorice bonds.” The polypeptide backbone is shown as a ribbon (blue, synaptobrevin-II; red, syntaxin-1A; green, SNAP-25B). This figure was prepared by Bobscript (39) and rendered using PovRay (http://www.povray.org).
To gain more insight into the biological significance of the central layers, sequence variation analysis was carried out for the entire family of SNARE proteins and representative subsets (Fig. 3A). Sequence variation is lowest for amino acids forming the core of the synaptic fusion complex. Two conclusions can be drawn from this analysis. First, the four-helix bundle structure is probably conserved among the entire SNARE family and is essential for the function of these proteins in membrane fusion. Second, the preservation of interacting amino acids explains why, at least in vitro, assembly can be promiscuous. For example, both syntaxin-1A and syntaxin-4 can form a complex with synaptobrevin-II and SNAP-25B in vitro (27). Modeling of this complex based on the crystal structure of the syntaxin-1A⋅SNAP-25B⋅synaptobrevin-II complex (5) required no major rearrangements because most substitutions occur on the surface of the complex (Fig. 3B). Furthermore, several of the yeast homologues involved in trafficking to and from the Golgi apparatus appear to function in complexes with different SNARE partners (28). Therefore, in contrast to one of the postulates of the “SNARE-hypothesis” (3), other factors may determine SNARE binding specificity. It should be mentioned, however, that there are nonconserved core residues in the carboxyl-terminal region of syntaxin and synaptobrevin (Fig. 3A). These residues may affect membrane fusion activity by reducing structural integrity in the carboxyl-terminal region when mismatched SNAREs interact.
Figure 3.

Conservation of SNARE complex structure. (A) Sequence variation (black line) for selected syntaxin (SX), synaptobrevin (SB), and SNAP-25 (residues 27–82, SN1; residues 148–203, SN2) families and surface accessibilities (gray bars) in the synaptic fusion complex crystal structure. The analysis was restricted to the 16 layers (indicated by arrowheads) of the synaptic fusion complex crystal structure (5) consisting of syntaxin-1A residues 201–255 (SX), SNAP-25B residues 28–82 (SN1) and residues 149–203 (SN2), and synaptobrevin residues 31–85 (SB). The layer numbers refer to Fig. 1B. The GenBank accession numbers used for the syntaxin-1 through -4 family are as follows: sx-1b, RN, P32853; sx1b, BS, P41414; sx-1a, RN, P32851; sx-1a, HS, L37792; sx-1a, MM, D45208; sx-A, HS, U12918; sx1a, BS, P32850; sx1, HM, U85807; sx, AC, U03123; sx1a, DM, L37732; unc-64b, CE, AF047885; sx, SP, AF014122; sx2, RN, P50279; sx3, RN, Q08849; sx3a, HS, AJ002076; sx4, RN, Q08850; sx4, HS, X85784; and sx4, MM, U76832. The accession numbers used for the SNAP-25 family are SNAP-25B, HS, P13795; SNAP-25, TM, P36976; syndet, MM, U73143; SNAP-25, SP, AF036902; SNAP-25, DM, U81153; SNAP-25, HM, U85806; SNAP-25, GG, L09253; SNAP-25A, CA, L22973; SNAP-25D, CA, L22976; and Y22F5A.5, CE, AL021479. The accession numbers used for the synaptobrevin family members in the second branch of the dendrogram (not shown) are Sb1, RN, M24104; Sb1B, RN, U74621; Sb1, MM, AF007167; Sb1, TM, P13701; Syb-B, DM, L14270; Syb-A, DM, L14270; N-Syb, DM, S66686; Sb, AC, P35589; cellubrevin (Sb3), MM U61751; cellubrevin (Sb3), RN S63830; Sb, SP, AF014119; Sb2, RN, M24105; Sb2, HS, AJ5044; Sb2, MM, AF007168; Sb, HM, U85805; Sb, FR, AF016494; Sb, LP, X74748; XSybI, XL, AF035016; Syb2, XL, U16801; Sb, BT, X76199; Snb1, CE, AF003281; and Sb, SM, U30182. The following two-letter species abbreviations are used: CA, Carrassius auratus; HS, Homo sapiens; MM, Mus musculus; RN, Rattus norvegicus; SC, Saccharomyces cerevisiae; DM, Drosophila melanogaster; TM, Torpedo marmorata; CE, Caenorhabditis elegans; SP, Strongylocentrotus purpuratus; HM, Hirudo medicinalis; GG, Gallus gallus; BT, Bos taurus; AC, Aplysia californica; SM, Schistosoma mansoni; FR, Fugu rubripes; XL, Xenopus laevis; and LP, Loligo pealei. (B) Model of a synaptic fusion complex consisting of syntaxin-4, SNAP-25B, and synaptobrevin-II, based on the crystal structure of the synaptic fusion complex (5), which contains syntaxin-1A. Syntaxin-4 residues are shown in black and red for matching and different residue types between syntaxin-1A and syntaxin-4, respectively. Most substituted residues occur on the surface of the complex.
Conservation of Surface Features and the Potential Role of Divalent Cation Binding Sites.
While most surface residues of the synaptic fusion complex are highly variable (Fig. 3A), some surface features are conserved among subgroups of SNARE homologues. For instance, the acidic residues forming a characteristic acidic patch in the middle of the synaptic fusion complex structure (5) are conserved among several homologues, but are not found in the yeast family members. Distinct acidic, basic, and hydrophobic patches on the four-helix bundle surface may determine the ability of the SNARE complex to interact with different effector proteins, such as synaptotagmin, complexin, α-SNAP, and NSF. The binding sites of these proteins remain to be clarified.
The SNARE proteins are the target for the clostridial neurotoxins, including botulinum (BoNT) and tetanus (TeNT) neurotoxins. As outlined before, the parallel orientation of both SNAP-25B α-helices in the crystal structure places all neurotoxin cleavage sites (10) into two distinct regions between the carboxyl-terminal membrane anchors and the ionic 0 layer (5). The crystal structure of synaptic fusion complex revealed a number of potential divalent cation binding sites on the surface (5). One such site is close to the cleavage sites of TeNT and BoNT/A (Fig. 4). It is possible that this potential cation binding site is involved in Ca2+-mediated triggering of exocytosis. Indeed, shortening of SNAP-25B by BoNT/A results in a partial block of exocytosis that can be overcome by increased Ca2+ levels, suggesting a possible link between Ca2+ binding and fusion complex function (29–32).
Figure 4.

A binding site for Sr2+ is close to the TeNT and BoNT/A toxin cleavage sites of the synaptic fusion complex (5). This binding site for Sr2+ is found in two of the three complexes in the asymmetric unit of the crystal structure of the synaptic fusion complex (5). The divalent cation is coordinated by the conserved (among the synaptobrevin family selected in Fig. 3A) synaptobrevin-II Ser-75, Glu-78, and partially conserved Thr-79 residues. The protein backbone is represented as a ribbon drawing. Blue, synaptobrevin-II; red, syntaxin-1A; green, SNAP-25B. The indicated toxin cleavage sites are located at the peptide bonds between Gln-76 and Phe-77 (TeNT and BoNT/B), and between Ala-81 and Ala-82 (BoNT/G) for synaptobrevin-II, between Lys-253 and Ala-254 (BoNT/C) for syntaxin-1A, and between Gln-197 and Arg-198 (BoNT/A) for SNAP-25B. The residues adjacent to the scissile bond are shown as licorice bonds. This figure prepared by Bobscript (36) and rendered by using PovRay.
Structure-Based Reclassification of SNARE Proteins into “Q-SNAREs” and “R-SNAREs.”
The conserved properties of SNARE complexes provide a structural basis for a regrouping of SNARE proteins. Previously, SNARE proteins were divided into “v-SNAREs” (including proteins homologous to synaptobrevin) and “t-SNAREs” (including proteins homologous to syntaxin-1 and SNAP-25), based on their preferred localization on either the trafficking vesicle (v) or the target membrane (t), respectively (3, 6). As outlined below, this classification scheme may not be accurate for all vesicular transport steps. Furthermore, it does not cover homotypic fusion events—i.e., fusion between vesicles that are functionally and structurally equivalent. Here we propose a reclassification of the SNARE proteins based on their contributions to the ionic 0 layer. R-SNAREs would provide an arginine (R) to this ionic layer and Q-SNAREs would provide complementary glutamines (Q) (Fig. 1A). Presently, there are only two exceptions to this convention. Assuming correct primary sequence alignment, yeast Bet1p would provide a serine to the layer. Leech synaptobrevin would provide a lysine to the layer instead of an arginine, resulting in a modified layer geometry. Although the R-SNAREs include most of the proteins previously classified as v-SNAREs, there are no structural reasons why the α-helices provided by the trafficking vesicles need to be derived only from R-SNAREs, as in the case of the synaptic fusion complex.
At present, only few SNARE complexes have been characterized in which all partners have been identified and localized, such as the SNARE complex involved in yeast exocytosis (21, 22). For other trafficking steps, relevant SNARE proteins are known, but it is unclear which of them interact in a particular fusion step. A well studied example is the vesicular traffic between the endoplasmic reticulum and the Golgi apparatus. Despite the assignment of several SNARE proteins it remains to be established which of them function in anterograde vs. retrograde traffic and whether intermediate fusion steps are involved (33). Anterogradely transported vesicles contain both an R-SNARE (Sec22p) and a Q-SNARE (Bos1p), which appear to form a complex with other SNAREs, probably involving Sed5p, a Q-SNARE. If the neuronal SNARE structure is conserved, the complex would consist of Sec22p (R-SNARE)/Bos1p (Q-SNARE)/Sed5p (Q SNARE) and a fourth Q-SNARE helix which may be contributed either by an additional SNARE or by a second copy of Bos1p or Sed5p. When applied to a SNARE complex that functions in “homotypic” vacuolar fusion, consisting of Nyv1p (R-SNARE), Vam3p (Q-SNARE), Vti1p (Q-SNARE), and Vam7p (Q-SNARE) (34), our convention predicts the formation of a tetrameric complex. Each Q-SNARE would contribute only a single α-helix to the four-helix bundle of this putative complex.
Although all characterized SNARE complexes involved in membrane fusion are of the 3 Q-SNARE/1 (R-SNARE) type, defined complexes have been reported that consist only of Q-SNAREs (9, 35, 36). It remains to be established whether these complexes are also four helix bundles and whether they play any role in membrane fusion events.
The recently discovered syntaxin-binding protein tomosyn (37, 38) exhibits similarity to R-SNAREs at its carboxyl terminus. In particular, tomosyn contains an arginine residue flanked by hydrophobic layers as found in all other R-SNAREs (Fig. 1A). Tomosyn binds the H3 domain of syntaxin-1 and immunoprecipitates with syntaxin-1A and syntaxin-1B, SNAP-25, and synaptotagmin. We predict that the R-SNARE domain in tomosyn forms a similar four-helix bundle with the Q-SNAREs syntaxin and SNAP-25. Tomosyn does not possess a transmembrane domain and is therefore unlikely to participate in membrane fusion. It is more likely that the R-SNARE domain enables tomosyn to bind to Q-SNAREs, thereby playing a regulatory role in SNARE complex assembly.
Our results support a model in which complex formation promotes membrane fusion (2, 5, 9, 36). Indeed, the α-helices are closely packed in the region directly adjacent to the transmembrane domains, and this region is characterized by a conserved group of basic and aromatic residues (5). Furthermore, this region is sensitive to neurotoxin cleavage. Such packing may be completed only after the fusion reaction when the membrane anchors are aligned in parallel in the same membrane. Consequently, partial assembly states may occur during membrane docking in which the membrane anchors point away from each other (figure 5 in ref. 5). It remains to be established whether the free energy released by these intermediate assembly states suffices to induce lipid mixing. Perhaps this process is assisted or regulated by accessory proteins, such as synaptotagmin, which could link Ca2+-dependent exocytosis and the synaptic fusion complex.
Acknowledgments
The authors thank Mark Gerstein and Paul D. Adams for expert advice; Wolfram Antonin for drawing our attention to the putative R-SNARE tomosyn; Klaus M. Fiebig, Christian Ostermeier, Gabi Fischer von Mollard, Rainer Ossig, and Martin Margittai for stimulating discussions; and Lothar Esser for assistance in figure preparation. Support by the National Institutes of Health to A.T.B. and by the Deutsche Forschungsgemeinschaft to R.J. (SFB 523) is gratefully acknowledged.
ABBREVIATIONS
- NSF
N-ethylmaleimide-sensitive fusion protein
- SNARE
soluble NSF attachment protein receptor
- SNAP
soluble NSF attachment protein
- BoNT
botulinum neurotoxin
- TeNT
tetanus neurotoxin
References
- 1.Ferro-Novick S, Jahn R. Nature (London) 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- 2.Hanson P I, Heuser J E, Jahn R. Curr Opin Neurobiol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 3.Rothman J E. Nature (London) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 4.Südhof T C. Nature (London) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 5.Sutton R B, Fasshauer D, Jahn R, Brunger A T. Nature (London) 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 6.Söllner T, Whiteheart S W, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman J E. Nature (London) 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof T, Niemann H. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fasshauer D, Bruns D, Shen B, Jahn R, Brunger A T. J Biol Chem. 1997;272:4582–4590. doi: 10.1074/jbc.272.7.4582. [DOI] [PubMed] [Google Scholar]
- 9.Fasshauer D, Otto H, Eliason W K, Jahn R, Brunger A T. J Biol Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 10.Jahn R, Niemann H. Ann NY Acad Sci. 1994;733:245–255. doi: 10.1111/j.1749-6632.1994.tb17274.x. [DOI] [PubMed] [Google Scholar]
- 11.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerstein M, Altman R B. J Mol Biol. 1995;251:161–175. doi: 10.1006/jmbi.1995.0423. [DOI] [PubMed] [Google Scholar]
- 13.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 14.Jones T A, Zou J Y, Cowan S W, Kjeldgaard Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 15.Brunger A T, Adams P D, Clore G M, Gros P, Grosse-Kunstleve R W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, Read R J, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 16.Weimbs T, Mostov K E, Low S H, Hofmann K. Trends Cell Biol. 1998;8:260–262. doi: 10.1016/s0962-8924(98)01285-9. [DOI] [PubMed] [Google Scholar]
- 17.Lian J P, Ferro-Novick S. Cell. 1993;73:735–745. doi: 10.1016/0092-8674(93)90253-m. [DOI] [PubMed] [Google Scholar]
- 18.Novick P, Field C, Schekman R. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- 19.Ossig R, Dascher C, Trepte H H, Schmitt H D, Gallwitz D. Mol Cell Biol. 1991;11:2980–2993. doi: 10.1128/mcb.11.6.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer von Mollard G, Stevens T H. J Biol Chem. 1998;273:2624–2630. doi: 10.1074/jbc.273.5.2624. [DOI] [PubMed] [Google Scholar]
- 21.Brennwald P, Kearns B, Champion K, Keranen S, Bankaitis V, Novick P. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 22.Rossi G, Salminen A, Rice L M, Brunger A T, Brennwald P. J Biol Chem. 1997;272:16610–16617. doi: 10.1074/jbc.272.26.16610. [DOI] [PubMed] [Google Scholar]
- 23.Saifee O, Wei L, Nonet M L. Mol Biol Cell. 1998;9:1235–1239. doi: 10.1091/mbc.9.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Littleton J T, Chapman E R, Kreber R, Garment M B, Carlson S D, Ganetzky B. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 25.Oakley M G, Kim P S. Biochemistry. 1998;37:12603–12610. doi: 10.1021/bi981269m. [DOI] [PubMed] [Google Scholar]
- 26.Hao J C, Salem N, Peng X R, Kelly R B, Bennett M K. J Neurosci. 1997;17:1596–1603. doi: 10.1523/JNEUROSCI.17-05-01596.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pevsner J, Hsu S C, Braun J E, Calakos N, Ting A E, Bennett M K, Scheller R H. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 28.Götte M, Fischer von Mollard G. Trends Cell Biol. 1998;8:215–218. doi: 10.1016/s0962-8924(98)01272-0. [DOI] [PubMed] [Google Scholar]
- 29.Xu T, Binz T, Niemann H, Neher E. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- 30.Capogna M, McKinney R A, O’Connor V, Gahwiler B H, Thompson S M. J Neurosci. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee A, Barry V A, DasGupta B R, Martin T F J. J Biol Chem. 1996;271:20223–20226. doi: 10.1074/jbc.271.34.20223. [DOI] [PubMed] [Google Scholar]
- 32.Trudeau L E, Fang Y, Haydon P G. Proc Natl Acad Sci USA. 1998;95:7163–7168. doi: 10.1073/pnas.95.12.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nichols B J, Pelham H R B. Biochim Biophys Acta. 1998;1404:9–31. doi: 10.1016/s0167-4889(98)00044-5. [DOI] [PubMed] [Google Scholar]
- 34.Ungermann C, Wickner W. EMBO J. 1998;17:3269–3276. doi: 10.1093/emboj/17.12.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fasshauer D, Eliason W K, Brunger A T, Jahn R. Biochemistry. 1998;37:10354–10362. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- 36.Fiebig, K., Rice, L. M., Pollock, E. & Brunger, A. T. Nature Structural Biology, in press. [DOI] [PubMed]
- 37.Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama S, Nishioka H, Matsuura Y, Mizoguchi A, Scheller R H, Takai Y. Neuron. 1998;20:905–915. doi: 10.1016/s0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- 38.Masuda E S, Huang B C B, Fisher J M, Luo Y, Scheller R H. Neuron. 1998;21:479–480. doi: 10.1016/s0896-6273(00)80559-0. [DOI] [PubMed] [Google Scholar]
- 39.Esnouf R M. J Mol Graph Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. , 112–113. [DOI] [PubMed] [Google Scholar]