

Abstract
Pharmacological modulation of cellular proteins as a means to block virus replication has been proposed as an alternative antiviral strategy that may be less susceptible than others to the development of viral drug resistance. Recent evidence indicates that the ubiquitin-proteasome pathway interacts with different aspects of the hepatitis B virus (HBV) life cycle in cell culture models of virus replication. We therefore examined the effect of proteasome inhibition on HBV replication in vivo using HBV transgenic mice. The proteasome inhibitor bortezomib (Velcade) inhibits proteasome activity in vivo and is used therapeutically for the clinical treatment of multiple myeloma. We found that a single intravenous dose of 1 mg of bortezomib/kg of body weight reduced virus replication for as long as 6 days. The inhibition of HBV by bortezomib was dose dependent and occurred at a step in replication subsequent to viral RNA and protein expression. The reduction in HBV replication did not result from nonspecific hepatocellular toxicity and was not mediated indirectly through the induction of an intrahepatic interferon response. Thus, pharmacological manipulation of the ubiquitin-proteasome pathway may represent an alternative therapeutic approach for the treatment of chronic HBV infection.
Despite the availability of an effective vaccine, more than 350 million people worldwide are chronically infected with hepatitis B virus (HBV), and many of them develop serious liver diseases, such as cirrhosis and hepatocellular carcinoma. Several nucleoside or nucleotide analogs are currently approved for the treatment of chronic HBV infection; these interact with the viral polymerase (Pol) and act as competitive substrate inhibitors to block reverse transcription of the pregenomic viral RNA to DNA (reviewed in reference 5). However, Pol inhibitors, such as lamivudine, can be rendered ineffective due to resistance mutations arising within Pol that are subsequently selected for during antiviral treatment (2). One approach to potentially circumvent antiviral resistance is combination therapy with agents that target viral proteins and the cellular proteins required for completion of the viral life cycle, as in the case of maraviroc (Selzentry), a CCR5 antagonist that can be used in multidrug regimens to block HIV entry (6, 10).
The ubiquitin-proteasome pathway of protein degradation is important for the regulation of a number of cellular processes. This pathway is the major mechanism by which damaged or misfolded proteins are removed from the cell (42, 43). A variety of cellular transcription factors, such as p53 and NF-κB, are regulated by proteasome-mediated degradation (24, 26), and the proteasome is also critical for the generation of peptides that are presented by class I major histocompatibility complex (MHC) molecules (32). Given the importance of this pathway, it is not surprising that many viruses produce proteins that interact with or otherwise modulate ubiquitin-proteasome activity. For example, retrovirus Gag proteins interact with ubiquitin to mediate virus budding (34), and the HIV-1 Vif protein promotes the ubiquitination and degradation of cellular antiviral APOBEC3 deaminases (17, 21). Kaposi's sarcoma-associated herpesvirus encodes proteins that utilize the ubiquitin-proteasome pathway to alter the activity of cellular proteins such as MHC class I and IRF7 (18, 45). The human papillomavirus E6 protein targets p53 for degradation through the cellular E6-associated protein ubiquitin ligase (33). Thus, many viral proteins interact with the ubiquitin-proteasome pathway to produce a cellular environment that is favorable for viral replication.
Like other viruses, HBV interacts with the ubiquitin-proteasome pathway in multiple ways during its replication cycle. The HBx protein binds to a number of proteasome subunits, including XAPC7, PSMA7, and PSMC1 (14, 15, 35, 47), and this interaction has functional consequences for proteasome function and HBV replication. HBx decreases the chymotryptic and tryptic-like activities of the proteasome, resulting in reduced rates of degradation of ubiquitinated proteins (14). By binding to the α4/MC6 proteasome subunit, HBx inhibits the activation of the proteasome by the interferon (IFN)-induced regulatory subunit PA28 (35). The interaction of HBx with the proteasome may be functionally important for virus replication, since the replication defect of an HBx-deficient HBV mutant is suppressed by proteasome inhibition in cell culture (46). We have also recently found that the IFN-regulated proteasome catalytic subunits can influence the specificity of the HBV-specific CD8 T-cell response (30) and that depletion of free cellular ubiquitin through proteasome inhibition blocks HBV release in cell culture (7).
To study the role of proteasome activity in HBV replication using a more physiological model, we treated 1.3-genome-length HBV transgenic mice with a proteasome inhibitor that has been well characterized previously with respect to its activity in vivo (1, 16, 20). Bortezomib (Velcade) is a potent proteasome inhibitor that is currently used as a therapy for patients with multiple myeloma (29). Surprisingly, we found that a low dose of bortezomib rapidly reduced HBV replication in the mice. Therefore, inhibition of proteasome activity may represent an alternative or complementary therapeutic approach for chronic HBV infection.
MATERIALS AND METHODS
Animals.
HBV transgenic mice (strain 1.3.32) have been described previously (8, 9). These animals encode a 1.3-overlength copy of the HBV genome (serotype ayw) and reproduce the virus replication cycle from gene expression through virion release. Groups of mice in all experiments were matched for age (8 to 12 weeks), sex (male), and serum HBeAg levels as determined by an enzyme-linked immunosorbent assay (ELISA) (International Immunodiagnostics, Foster City, CA; Abbott Laboratories, Abbott Park, IL). All animals were housed in specific-pathogen-free rooms under strict barrier conditions. Mice were injected intravenously (i.v.) with the indicated dose of bortezomib diluted in 200 μl 0.9% NaCl containing 10 mg/ml mannitol. Control animals received the same volume of diluent without any drug. All procedures were performed in accordance with the Animal Care and Use guidelines of The Scripps Research Institute and Yale University.
Intracellular HBV DNA analysis.
DNA was prepared from snap-frozen liver tissue as previously described (9). Briefly, tissue was homogenized in DNA lysis buffer (50 mM Tris, 20 mM EDTA, 1% sodium dodecyl sulfate [SDS]), and the lysates were incubated at least 16 h at 37°C with 25 μg of proteinase K/ml. The lysates were sequentially extracted with phenol (pH 8.0), a 1:1 phenol (pH 8.0)-chloroform mixture, and chloroform. The DNA was then precipitated with an equal volume of isopropanol and washed in 80% ethanol. For Southern hybridizations, 20 μg of DNA was digested with HindIII, separated by agarose gel (1.4%) electrophoresis, and transferred to a nylon membrane utilizing standard protocols. Membranes were hybridized with a 32P-labeled DNA probe consisting of the entire HBV (strain ayw) genome.
Intracellular HBV RNA analysis.
For total-RNA preparation, tissue was homogenized in GTC solution (4.2 M guanidine isothiocyanate, 25 mM sodium citrate [pH 7.3], 0.5% sarcosyl) containing 100 mM β-mercaptoethanol (4). The pH of the lysate was adjusted to 4.0 with 0.1 volume 2 M sodium acetate (pH 4.0); the lysates were extracted twice with a 2.5:1 mixture of phenol (pH 4.0)-chloroform; and RNA was precipitated with an equal volume of isopropanol. The RNA was resuspended in diethylpyrocarbonate-treated H2O and was further extracted with a 1:1 mixture of phenol (pH 8.0)-chloroform and chloroform. The RNA was once again precipitated with an equal volume of isopropanol, washed in 80% ethanol, and dissolved in 55 μl H2O. For Northern hybridizations, 10 to 20 μg of total RNA was electrophoresed through a 1% agarose-formaldehyde RNA gel and was then transferred to a nylon membrane using standard protocols. Membranes were hybridized with a radiolabeled HBV DNA probe as described above or with a 32P-labeled DNA probe consisting of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) open reading frame.
Real-time reverse-transcription PCR (RT-PCR).
One microgram of total RNA prepared from snap-frozen mouse liver tissue was reverse transcribed with random hexamers using the TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Real-time PCR was performed using an Applied Biosystems 7500 real-time PCR system as follows. Reaction mixtures contained 100 ng reverse-transcribed RNA, 12.5 μl SYBR green reaction mix (Applied Biosystems), and 200 nM sense and antisense primers in a final volume of 25-μl. The primers used for PCR were as follows: GAPDH, 5′-TCT GGA AAG CTG TGC CGT G-3′ (sense) and 5′-CCA GTG AGC TTC CCG TTC AG-3′ (antisense); ISG15, 5′-CAA TGG CCT GGG ACC TAA A-3′ (sense) and 5′-CTT CTT CAG TTC TGA CAC CGT CAT-3′ (antisense); Usp18, 5′-CTC CGG CTT GTG TAG ACT CT-3′ (sense) and 5′-GGG ACT GGC GGG ACT-3′ (antisense); TGTP, 5′-CAG CCC ACA AGC GTC A-3′ (sense) and 5′-TGG AAT GGT GGC TAA TGC TC-3′ (antisense). After an initial incubation at 95°C for 5 min, PCR amplification was performed by cycling 50 times for 1 min at 95°C, followed by 1 min at 60°C. Relative gene expression was quantified by the ΔΔCT method using the 7500 System Sequence Detection software (Applied Biosystems).
RNase protection assay.
The RNase protection assay for intrahepatic cytokine expression was performed exactly as described previously (8, 11).
HBcAg.
Liver tissue was homogenized in 2× SDS sample buffer, and expression of liver HBV core antigen (HBcAg) was determined by Western blotting using an HBcAg-specific antibody (Dako, Carpinteria CA). Protein loading was controlled by immunoblotting the same membrane with an antibody specific for β-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
Serum HBV DNA analysis.
Mouse serum (5 μl) was diluted 1:100 in TNE buffer (10 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA) and applied to a nitrocellulose membrane using a Hybri-dot manifold (Bethesda Research Labs, Inc.). The membrane was denatured in 0.2 M NaOH-1.5 M NaCl for 10 min and then neutralized in 0.2 M Tris-HCl-1.5 M NaCl for 5 min. Nucleic acids were cross-linked on the membrane by UV irradiation and were hybridized with a radiolabeled HBV DNA probe as described above. Virion-associated DNA was visualized with a phosphorimager (Fujifilm, Tokyo, Japan) and quantified with ImageGauge software, version 4.22 (Fujifilm).
Histology.
Liver tissue was fixed in 10% zinc-buffered formalin, processed by routine methods, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Sections of liver were evaluated for expression of HBV protein using a NeoMarkers HBcAg-specific monoclonal antibody (1:300; LabVision, Fremont, CA) and the chromogen diaminobenzidine tetrahydrochloride by routine immunohistochemistry methods (Research Histology, Department of Pathology, Yale University School of Medicine).
sALT activity assay.
The level of serum alanine aminotransferase (sALT) was determined with Infinity ALT reagent (Thermo Electron, Louisville, CO) using a Spectramax spectrophotometer (Molecular Devices, Sunnyvale, CA) and enzyme standards (Verichem Laboratories, Providence, RI).
Proteasome activity assay.
Proteasomes were purified from homogenized mouse liver tissue, and activity was measured using the fluorogenic proteasome substrate (N-succinyl)-Leu-Leu-Val-Tyr-(amino-4-methylcoumarin) as previously described (30).
Statistical analysis.
Unless otherwise noted in the figure legends, experiments were performed using groups of four mice. This sample size is sufficient to detect an 80% reduction in HBV DNA levels using Student's t test with a significance level (alpha) of 0.05 and a power (1 − beta) of 0.80, assuming standard deviations equivalent to 50% of the mean DNA levels (22). A two-sample Student t test using the two-tailed distribution was used to determine significant differences in serum HBV DNA and antigen levels. P values of <0.05 were considered statistically significant.
RESULTS
Bortezomib inhibits HBV replication in transgenic mice.
A single dose of 1 mg/kg bortezomib inhibits proteasome activity in mice by 80 to 90% in multiple organs, including the liver (1, 16, 20). This dose of bortezomib was shown to inhibit proteasome activity maximally 6 h after injection, with activity returning to baseline levels approximately 40 h later (20). Because the activity of the drug may differ in different mouse strains, we first confirmed that this dose of bortezomib also inhibited proteasome activity in the livers of HBV transgenic mice (Fig. 1A). We then injected HBV 1.3.32 transgenic mice i.v. with a single dose of saline or 1 mg/kg bortezomib, and we monitored HBV replication by Southern blot analysis of liver HBV DNA replication intermediates 1 day after administration of the drug. Prior to injection, the groups of mice were matched for serum HBeAg levels such that no significant difference in expression was noted between the two groups (Fig. 1B). In contrast to HBeAg expression prior to injection, we found significant reductions in levels of HBV DNA replication intermediates in the liver 1 day following bortezomib administration (Fig. 1C).
FIG. 1.

Bortezomib inhibits HBV replication in transgenic mice. (A) Proteasome activity in HBV transgenic mouse livers (n = 3) 24 h after i.v. injection with saline (filled squares) or Velcade (filled triangles). Results of a 40-min assay using 2.5 μg of purified protein (left) and a 2-h assay using 1 μg of purified protein (right) are shown. Filled circles indicate reactions for which no protein was added. RFU, relative fluorescence units. (B) Relative HBeAg expression in three groups of four mice prior to administration of saline or 1 mg/kg bortezomib. CPM, counts per minute. (C) Quantification by Southern blot phosphorimager analysis of liver HBV DNA replication in the same animals 1 day following administration of the drug. In the box plots, the horizontal lines indicate the mean, the shaded boxes indicate ±1 standard error of the mean, the vertical lines indicate the minimum and maximum values, and P values indicate the results of Student's t test. DLU, digital light units.
Time course and dose response of HBV inhibition.
We next monitored HBV replication at 1, 2, 3, and 6 days after administration of bortezomib. Levels of HBV DNA replication intermediates in the liver were reduced within 24 h after injection of the drug and remained suppressed for at least the next 2 to 3 days before returning to near-baseline levels by day 6 (Fig. 2A). However, there was no significant change in the level of HBV 3.5- and 2.1-kb mRNA expression compared to that of GAPDH or the steady-state level of total HBcAg protein in the liver (Fig. 2A). Under these conditions, we also observed only transient mild liver damage as measured by sALT levels, which peaked at 180 U/liter at day 2 postinjection and returned to normal levels by day 6 (Fig. 2A). Therefore, bortezomib inhibits HBV replication by a mechanism that does not reduce HBV gene expression or result in the destruction of a large number of hepatocytes.
FIG. 2.

Time course and dose response of HBV inhibition. (A) Groups of age-, sex-, and serum HBeAg-matched transgenic mice (3 mice per group) were injected intravenously with a single dose of 1 mg/kg bortezomib and were then analyzed 1, 2, 3, or 6 days postinjection. (B) Groups of age-, sex-, and serum HBeAg-matched transgenic mice (3 to 4 mice per group) were injected intravenously with a single dose of 0.2 mg/kg, 1 mg/kg, or 5 mg/kg bortezomib and were then analyzed 24 h postinjection. HBV replication in the liver was measured by Southern blot (SB) analysis of relaxed-circle (RC) and single-stranded (SS) DNA replication forms and was compared to levels in control animals that did not receive the drug. The expression of 3.5- and 2.1-kb HBV mRNA from total liver RNA was examined by Northern blot (NB) analysis and was compared to the expression of the GAPDH housekeeping gene. HBcAg levels were measured by Western blot (WB) analysis and were compared to β-actin expression. The level of serum alanine aminotransferase (sALT) was determined in order to assess hepatocellular toxicity.
We then determined the dose of bortezomib required to inhibit HBV replication in the transgenic mice. As we found in the preceding time course experiment, levels of HBV DNA replication intermediates in the liver were reduced 24 h after a single i.v. injection of 1 mg/kg bortezomib (Fig. 2B). However, virus replication was no longer blocked when a 5-fold-lower dose (0.2 mg/kg) was administered. We also observed no reduction in the level of HBV replication when a 5-fold-higher dose was used (5 mg/kg) but instead found a slight increase in the level of HBV DNA replication intermediates that was neither statistically significant (P = 0.16) nor reproducible in a second experiment (data not shown). Again, expression of HBV 3.5- and 2.1-kb mRNAs and the steady-state level of total HBcAg were not significantly changed in the liver at all doses of the drug tested (Fig. 2B). Evidence of mild hepatocellular damage, as measured by sALT activity, was present only at the highest (5-mg/kg) dose of the drug (Fig. 2B). The animals that received 5 mg/kg bortezomib also showed visible signs of stress or illness at this dose, including ruffled fur, labored breathing, and hunched posture (data not shown). These animals were therefore euthanized, and longer treatments at the higher dose were not tested.
Bortezomib treatment reduces levels of serum HBV DNA but not those of secreted antigens.
To determine if bortezomib administration altered the release of secreted viral antigens, serum HBeAg and HBsAg levels were measured by ELISA. In general, the drug only modestly changed the amounts of these antigens in mice that were assayed 1, 2, 3, and 6 days after injection with 1 mg/kg bortezomib (Fig. 3A) or 24 h after a single dose of 0.2, 1, or 5 mg/kg bortezomib (Fig. 3C). HBeAg levels in animals given 5 mg/kg bortezomib were reduced approximately 50%, and this reduction was statistically significant (P = 0.04). There was also a consistent trend of increased HBsAg levels in mice 24 h after treatment with 1 mg/kg bortezomib, but this difference failed to achieve significance in two independent experiments (P = 0.23, P = 0.07). Thus, consistent with the fact that bortezomib did not change HBV 3.5- and 2.1-kb RNA or HBcAg protein levels in the liver, with some minor exceptions, the production and release of the secreted viral antigens were also generally not significantly affected by the drug.
FIG. 3.

Bortezomib reduces serum HBV DNA levels but not HBeAg and HBsAg secretion. (A and B) HBeAg, HBsAg, and HBV DNA levels in mouse serum 1, 2, 3, and 6 days after intravenous injection of 1 mg/kg bortezomib (3 mice per group). (C and D) HBeAg, HBsAg, and HBV DNA levels in mouse serum 24 h after a single dose of 0.2, 1, or 5 mg/kg bortezomib (4 mice per group). Data are displayed as expression levels relative to those for control mice that did not receive the drug. Error bars represent standard errors of the means, and P values indicate the results of Student's t test.
In contrast to the results for secreted viral antigens, mice given a single dose of 1 mg/kg bortezomib had serum HBV DNA levels that were maximally reduced to 8% of untreated levels at 3 days posttreatment (P = 0.03) (Fig. 3B). There was also a slight increase in HBV DNA levels in the sera of mice treated with 5 mg/kg bortezomib for 24 h (Fig. 3D) that was consistent with the increased levels of liver HBV replication intermediates (Fig. 2B), but this difference also failed to achieve statistical significance (P = 0.22). Therefore, the reduction in serum HBV DNA levels in mice treated with bortezomib correlated with the decrease in levels of HBV DNA replication intermediates in the liver.
Histological analysis of bortezomib-treated mice.
We also determined if bortezomib treatment induced inflammation or other histological abnormalities in the livers of HBV transgenic mice. Zinc-formalin-fixed liver tissues isolated from representative mice from the time course (Fig. 2A) and dose-response (Fig. 2B) experiments were sectioned and stained with hematoxylin-eosin. At all time points and at all doses of the drug examined, there was no evidence of significant hepatitis or other pathology in the liver (Fig. 4), consistent with the relatively modest ALT elevation in the bortezomib-treated mice. Furthermore, consistent with the similar HBcAg levels measured by Western blotting (Fig. 2), there were no significant differences in the number or distribution of HBcAg-positive hepatocytes between mice injected with saline and mice injected with bortezomib (Fig. 5). Thus, the reduced levels of HBV replication cannot be explained by a large loss of HBV-producing hepatocytes.
FIG. 4.

Representative liver histology for bortezomib-treated mice. (A to D) No significant pathological changes were observed in sections of liver from mice injected intravenously with a single dose of bortezomib (1 mg/kg) and analyzed at day 1 (B), 2 (C), or 6 (D) postinjection relative to liver sections from control mice (A). (E to H) There were also no significant hepatic changes observed between control mice (E) and mice injected intravenously with a single dose of bortezomib at 0.2 mg/kg (F), 1.0 mg/kg (G), or 5.0 mg/kg (H) and then analyzed 24 h postinjection. Bars, 50 μm.
FIG. 5.
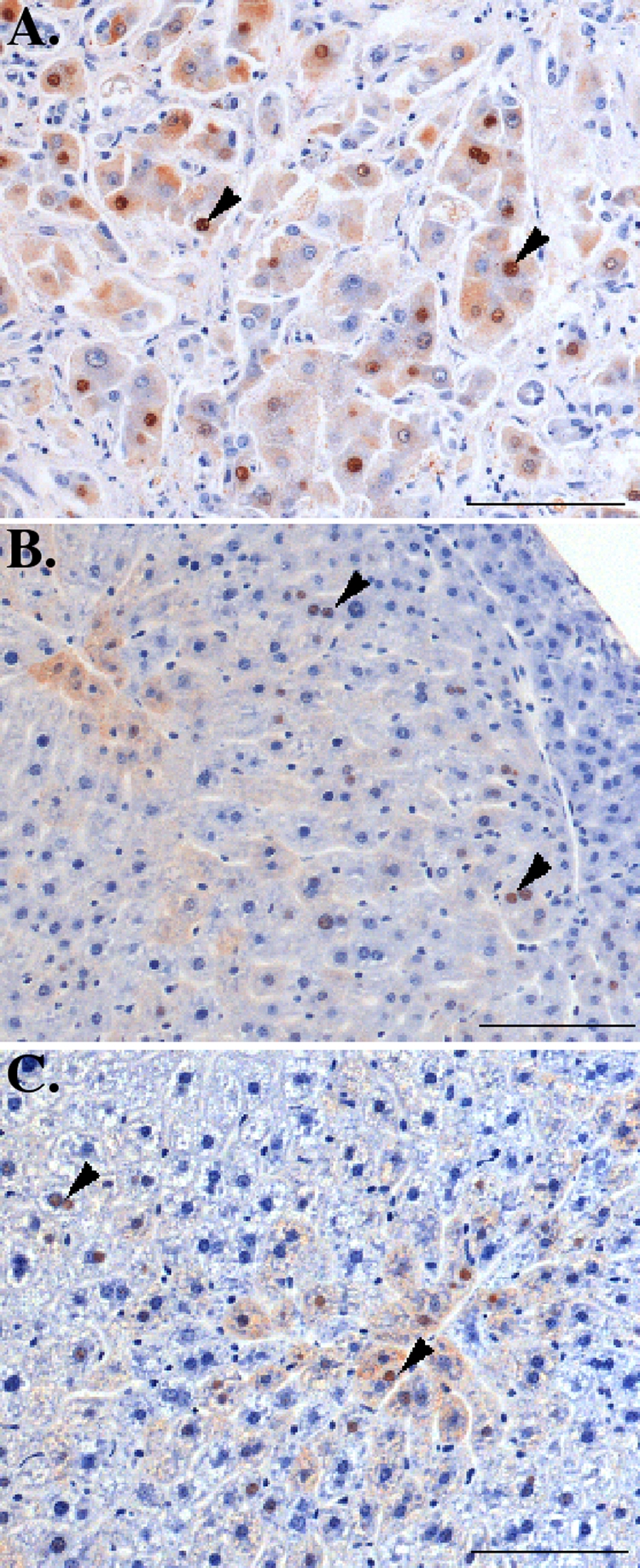
Representative liver immunohistochemistry for HBcAg expression in bortezomib-treated mice. (A) A control section of human liver from an HBV-positive patient revealed numerous HBV-positive hepatocytes with strong cytoplasmic and nuclear staining (arrowheads). (B and C) There was no significant difference in the number and distribution of HBV-positive hepatocytes (arrowheads) in HBV-infected mice 1 day after intravenous injection of saline (B) or 1 mg/kg bortezomib (C). Bars, 100 μm.
Inhibition of HBV replication by bortezomib does not occur through an intrahepatic inflammatory cytokine response.
Because IFN-α/β and IFN-γ also inhibit HBV replication without reducing viral mRNA expression or hepatocyte viability in the transgenic mice (8), we next determined if bortezomib administration induced intrahepatic IFN production by examining the expression of IFN-regulated genes in the liver. Quantitative RT-PCR was performed using total RNA isolated from the liver to monitor the expression of three representative IFN-inducible genes: ISG15, Usp18, and TGTP. The expression of ISG15 and Usp18 is preferentially induced by IFN-α/β, while IFN-γ preferentially induces TGTP (38). In mice from the time course experiment (Fig. 2A), the expression of these genes was not induced by bortezomib; instead, their expression levels were transiently decreased 24 h after drug injection (Fig. 6A). In addition, we also examined the intrahepatic expression of other inflammatory cytokines (including tumor necrosis factor alpha [TNF-α], TNF-β, interleukin-1α [IL-1α], IL-1β, and IFN-γ) by an RNase protection assay, and we found that the expression of these genes was not increased by bortezomib administration (Fig. 6B). As observed in the preceding time course analysis, the expression of IFN-stimulated genes also was not induced in the livers of mice from the dose-response experiment but rather was decreased (Fig. 6C). Thus, the bortezomib-mediated inhibition of HBV replication does not appear to occur through indirect intrahepatic induction of inflammatory cytokines. This conclusion is consistent with the absence of liver inflammation seen in the histological analysis (Fig. 4).
FIG. 6.

Bortezomib does not induce an IFN response in the liver. (A and C) Intrahepatic expression of the representative IFN-inducible genes ISG15, Usp18, and TGTP was measured by quantitative real-time RT-PCR (qRT-PCR) in mice either 1, 2, 3, or 6 days after intravenous injection of 1 mg/kg bortezomib (A) or 24 h after a single dose of 0.2, 1.0, or 5.0 mg/kg bortezomib (C). Results for each individual mouse are shown. Results are displayed as fold changes compared to one control mouse and are normalized to GAPDH expression. (B) RNase protection assay (RPA) of cytokine mRNA expression in the livers of individual mice 1, 2, 3, and 6 days after intravenous injection of 1 mg/kg bortezomib.
DISCUSSION
We found that bortezomib administration effectively reduced the levels of HBV DNA replication intermediates in the livers of HBV transgenic mice and the levels of HBV DNA in the sera. Furthermore, this block occurred at a step downstream of HBV mRNA and protein expression. The inhibition of HBV replication by bortezomib in the transgenic mice was surprising, since we have previously found that although proteasome inhibition blocks HBV release in cell culture, it does not affect the levels of intracellular DNA replication intermediates (7, 31). Although the reason for the difference between cell culture and mice is unclear, it is possible that the proteasome-sensitive aspect of the HBV replication cycle is more accurately reflected in the liver in vivo. For example, the inhibitory effect may be due to a gene(s) that is expressed in a strictly differentiation specific manner, and therefore this effect may be regulated differently in the liver and in cell culture. Alternatively, the antiviral mechanism may depend on a mediator produced by an extrahepatic tissue, and it would therefore be apparent only in vivo. However, we cannot rule out the possibility that the antiviral effect on HBV is restricted to mice, or is limited to the specific transgenic mouse line (1.3.32) used for these experiments. Additional studies of different HBV transgenic mouse lines, chimpanzees, and/or humans would be needed to determine whether this antiviral activity extends to chronic infections in other organisms.
In addition to measuring HBV DNA levels in the liver and serum, we also assessed the levels of secreted viral antigens (HBeAg, HBsAg) in the serum after bortezomib treatment. Consistent with the fact that we did not observe a change in the expression of the 3.5- and 2.1-kb HBV mRNAs in the liver, we also found little to no change in the levels of serum HBe and HBs antigens at the 1-mg/kg dose. The slight increase in HBsAg levels is consistent with previous results obtained in cell culture (7) and may reflect stabilization of the protein by proteasome inhibition.
We observed that the inhibition of HBV replication was sensitive to the dose of bortezomib administered to the animal. The dose at which we observed the inhibition of HBV (1 mg/kg) has been shown previously to inhibit proteasome activity in multiple mouse tissues (including the liver) by approximately 85% (1, 16, 20). Not surprisingly, a 5-fold-lower dose of bortezomib did not affect baseline levels of HBV replication. Somewhat unexpected was the observation that a 5-fold-higher dose also did not reduce virus replication. There are a number of possible explanations for this result, including systemic and/or local effects of the drug at the higher dose that mask the effects of proteasome inhibition on HBV replication. Alternatively, different levels of proteasome inhibition may differentially affect positive or negative factors for virus replication. A mechanism such as this has precedence in other drugs. For example, celecoxib loses anti-inflammatory activity in rats at high doses, because at high concentrations it induces, rather than inhibits, NF-κB activation (25).
Although the precise mechanism of HBV inhibition is not known at this time, there are a number of possibilities. First, it is possible that the ubiquitin-proteasome pathway influences some aspect of pregenomic RNA (pgRNA) encapsidation that is disrupted by proteasome inhibition. Interestingly, HBV RNA encapsidation and Pol priming require the activity of cellular chaperones (12, 13), and the activity of these proteins is intimately linked with the ubiquitin-proteasome pathway (44). Second, hepadnaviral Pol proteins have a short half-life (40, 41), and we have found that HBV Pol expression is enhanced by proteasome inhibition in cell culture (unpublished data). Therefore, proteasome inhibition may perturb the level of functional Pol available to mediate HBV DNA synthesis. Third, it is possible that proteasome inhibition interferes with HBx function, although this may be an unlikely explanation, since 1.3.32 transgenic mice express low levels of HBx mRNA and contain no detectable levels of HBx protein (9). Finally, it should also be noted that we cannot rule out the possibility that bortezomib inhibits HBV replication through a proteasome-independent mechanism.
Interestingly, IFN-α/β and IFN-γ also potently block HBV replication in transgenic mouse hepatocytes at a similar step in the virus life cycle (36, 37). Thus, one potential explanation for our finding is that bortezomib indirectly inhibits HBV replication by inducing an inflammatory cytokine response in the liver. However, we were unable to detect induction of IFN-α/β- or IFN-γ-activated genes, increased inflammatory cytokine mRNA expression, or inflammation in the livers of bortezomib-treated mice. Therefore, while it is difficult to rule out entirely, inhibition of HBV in vivo by bortezomib does not appear to be due to the induction of an IFN response in the liver.
Several studies have demonstrated that inhibitors of cellular proteins exploited by viral pathogens represent a viable strategy for antiviral therapy. For example, inhibitors of Abl-family tyrosine kinases and the ErbB-1 kinase block poxvirus replication in vivo (28, 39). Similarly, downregulation of the Raf/MEK/extracellular signal-regulated kinase (ERK) signaling cascade with a MEK inhibitor results in impaired propagation of influenza virus (19, 27). Furthermore, α-glucosidase inhibitors exhibit antiviral activity against several enveloped viruses, including HBV and dengue virus (3, 23). We have shown that proteasome inhibition suppresses HBV replication in transgenic mice. Overall, these results support recent evidence of the interaction between the HBV replication cycle and the ubiquitin-proteasome pathway, and they imply that viral or cellular proteins required for replication may be regulated by the proteasome. Our results further suggest the possibility that inhibitors of the proteasome or other proteasome-dependent cellular pathways required for HBV replication may be therapeutically useful in the treatment of chronic infection.
Acknowledgments
This work was supported by grant CA40489 (to F.V.C.) and Research Scholar Development Award K22 AI64757 (to M.D.R.) from the N.I.H. M.L.G. was supported by a predoctoral fellowship from the Ford Foundation.
Footnotes
Published ahead of print on 30 November 2009.
REFERENCES
- 1.Adams, J., V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash, and P. J. Elliott. 1999. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 59:2615-2622. [PubMed] [Google Scholar]
- 2.Allen, M. I., M. Deslauriers, C. W. Andrews, G. A. Tipples, K. A. Walters, D. L. Tyrrell, N. Brown, and L. D. Condreay. 1998. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 27:1670-1677. [DOI] [PubMed] [Google Scholar]
- 3.Chang, J., L. Wang, D. Ma, X. Qu, H. Guo, X. Xu, P. M. Mason, N. Bourne, R. Moriarty, B. Gu, J. T. Guo, and T. M. Block. 2009. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob. Agents Chemother. 53:1501-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-choloroform extraction. Anal. Biochem. 162:156-159. [DOI] [PubMed] [Google Scholar]
- 5.Chotiyaputta, W., and A. S. Lok. 2009. Hepatitis B virus variants. Nat. Rev. Gastroenterol. Hepatol. 6:453-462. [DOI] [PubMed] [Google Scholar]
- 6.Fätkenheuer, G., M. Nelson, A. Lazzarin, I. Konourina, A. I. Hoepelman, H. Lampiris, B. Hirschel, P. Tebas, F. Raffi, B. Trottier, N. Bellos, M. Saag, D. A. Cooper, M. Westby, M. Tawadrous, J. F. Sullivan, C. Ridgway, M. W. Dunne, S. Felstead, H. Mayer, and E. van der Ryst. 2008. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 359:1442-1455. [DOI] [PubMed] [Google Scholar]
- 7.Garcia, M. L., R. Byfield, and M. D. Robek. 2009. Hepatitis B virus replication and release are independent of core lysine ubiquitination. J. Virol. 83:4923-4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guidotti, L. G., T. Ishikawa, M. V. Hobbs, B. Matzke, R. Schreiber, and F. V. Chisari. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25-36. [DOI] [PubMed] [Google Scholar]
- 9.Guidotti, L. G., B. Matzke, H. Schaller, and F. V. Chisari. 1995. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gulick, R. M., J. Lalezari, J. Goodrich, N. Clumeck, E. DeJesus, A. Horban, J. Nadler, B. Clotet, A. Karlsson, M. Wohlfeiler, J. B. Montana, M. McHale, J. Sullivan, C. Ridgway, S. Felstead, M. W. Dunne, E. van der Ryst, and H. Mayer. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hobbs, M. V., W. O. Weigle, D. J. Noonan, B. E. Torbett, R. J. McEvilly, R. J. Koch, G. J. Cardenas, and D. N. Ernst. 1993. Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J. Immunol. 150:3602-3614. [PubMed] [Google Scholar]
- 12.Hu, J., and C. Seeger. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu, J., D. O. Toft, and C. Seeger. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, Z., Z. Zhang, E. Doo, O. Coux, A. L. Goldberg, and T. J. Liang. 1999. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol. 73:7231-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, J., J. Kwong, E. C. Sun, and T. J. Liang. 1996. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J. Virol. 70:5582-5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeBlanc, R., L. P. Catley, T. Hideshima, S. Lentzsch, C. S. Mitsiades, N. Mitsiades, D. Neuberg, O. Goloubeva, C. S. Pien, J. Adams, D. Gupta, P. G. Richardson, N. C. Munshi, and K. C. Anderson. 2002. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 62:4996-5000. [PubMed] [Google Scholar]
- 17.Liu, B., P. T. Sarkis, K. Luo, Y. Yu, and X. F. Yu. 2005. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 79:9579-9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lorenzo, M. E., J. U. Jung, and H. L. Ploegh. 2002. Kaposi's sarcoma-associated herpesvirus K3 utilizes the ubiquitin-proteasome system in routing class major histocompatibility complexes to late endocytic compartments. J. Virol. 76:5522-5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ludwig, S., T. Wolff, C. Ehrhardt, W. J. Wurzer, J. Reinhardt, O. Planz, and S. Pleschka. 2004. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 561:37-43. [DOI] [PubMed] [Google Scholar]
- 20.Luker, G. D., C. M. Pica, J. Song, K. E. Luker, and D. Piwnica-Worms. 2003. Imaging 26S proteasome activity and inhibition in living mice. Nat. Med. 9:969-973. [DOI] [PubMed] [Google Scholar]
- 21.Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444-11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald, J. H. 2009. Handbook of biological statistics, 2nd ed., p. 110-114. Sparky House Publishing, Baltimore, MD.
- 23.Mehta, A., N. Zitzmann, P. M. Rudd, T. M. Block, and R. A. Dwek. 1998. Alpha-glucosidase inhibitors as potential broad based anti-viral agents. FEBS Lett. 430:17-22. [DOI] [PubMed] [Google Scholar]
- 24.Michael, D., and M. Oren. 2003. The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 13:49-58. [DOI] [PubMed] [Google Scholar]
- 25.Niederberger, E., I. Tegeder, G. Vetter, A. Schmidtko, H. Schmidt, C. Euchenhofer, L. Brautigam, S. Grosch, and G. Geisslinger. 2001. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-κB. FASEB J. 15:1622-1624. [DOI] [PubMed] [Google Scholar]
- 26.Palombella, V. J., O. J. Rando, A. L. Goldberg, and T. Maniatis. 1994. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 78:773-785. [DOI] [PubMed] [Google Scholar]
- 27.Pleschka, S., T. Wolff, C. Ehrhardt, G. Hobom, O. Planz, U. R. Rapp, and S. Ludwig. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301-305. [DOI] [PubMed] [Google Scholar]
- 28.Reeves, P. M., B. Bommarius, S. Lebeis, S. McNulty, J. Christensen, A. Swimm, A. Chahroudi, R. Chavan, M. B. Feinberg, D. Veach, W. Bornmann, M. Sherman, and D. Kalman. 2005. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat. Med. 11:731-739. [DOI] [PubMed] [Google Scholar]
- 29.Richardson, P. G., B. Barlogie, J. Berenson, S. Singhal, S. Jagannath, D. Irwin, S. V. Rajkumar, G. Srkalovic, M. Alsina, R. Alexanian, D. Siegel, R. Z. Orlowski, D. Kuter, S. A. Limentani, S. Lee, T. Hideshima, D. L. Esseltine, M. Kauffman, J. Adams, D. P. Schenkein, and K. C. Anderson. 2003. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348:2609-2617. [DOI] [PubMed] [Google Scholar]
- 30.Robek, M. D., M. L. Garcia, B. S. Boyd, and F. V. Chisari. 2007. Role of immunoproteasome catalytic subunits in the immune response to hepatitis B virus. J. Virol. 81:483-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robek, M. D., S. F. Wieland, and F. V. Chisari. 2002. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J. Virol. 76:3570-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rock, K. L., C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang, and A. L. Goldberg. 1994. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761-771. [DOI] [PubMed] [Google Scholar]
- 33.Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129-1136. [DOI] [PubMed] [Google Scholar]
- 34.Schubert, U., D. E. Ott, E. N. Chertova, R. Welker, U. Tessmer, M. F. Princiotta, J. R. Bennink, H. G. Krausslich, and J. W. Yewdell. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. U. S. A. 97:13057-13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stohwasser, R., H. G. Holzhutter, U. Lehmann, P. Henklein, and P. M. Kloetzel. 2003. Hepatitis B virus HBx peptide 116-138 and proteasome activator PA28 compete for binding to the proteasome α4/MC6 subunit. Biol. Chem. 384:39-49. [DOI] [PubMed] [Google Scholar]
- 36.Wieland, S. F., A. Eustaquio, C. Whitten-Bauer, B. Boyd, and F. V. Chisari. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 102:9913-9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wieland, S. F., L. G. Guidotti, and F. V. Chisari. 2000. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. 74:4165-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wieland, S. F., R. G. Vega, R. Muller, C. F. Evans, B. Hilbush, L. G. Guidotti, J. G. Sutcliff, P. G. Schultz, and F. V. Chisari. 2003. Searching for interferon-induced genes that inhibit hepatitis B virus replication in transgenic mouse hepatocytes. J. Virol. 77:1227-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang, H., S. K. Kim, M. Kim, P. A. Reche, T. J. Morehead, I. K. Damon, R. M. Welsh, and E. L. Reinherz. 2005. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 115:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao, E., H. Schaller, and J. E. Tavis. 2003. The duck hepatitis B virus polymerase and core proteins accumulate in different patterns from their common mRNA. Virology 311:81-88. [DOI] [PubMed] [Google Scholar]
- 41.Yao, E., and J. E. Tavis. 2003. Kinetics of synthesis and turnover of the duck hepatitis B virus reverse transcriptase. J. Biol. Chem. 278:1201-1205. [DOI] [PubMed] [Google Scholar]
- 42.Yewdell, J. 2002. To DRiP or not to DRiP: generating peptide ligands for MHC class I molecules from biosynthesized proteins. Mol. Immunol. 39:139-146. [DOI] [PubMed] [Google Scholar]
- 43.Yewdell, J. W., L. C. Anton, and J. R. Bennink. 1996. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 157:1823-1826. [PubMed] [Google Scholar]
- 44.Young, J. C., V. R. Agashe, K. Siegers, and F. U. Hartl. 2004. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 5:781-791. [DOI] [PubMed] [Google Scholar]
- 45.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 46.Zhang, Z., U. Protzer, Z. Hu, J. Jacob, and T. J. Liang. 2004. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 78:4566-4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Z., N. Torii, A. Furusaka, N. Malayaman, Z. Hu, and T. J. Liang. 2000. Structural and functional characterization of interaction between hepatitis B virus X protein and the proteasome complex. J. Biol. Chem. 275:15157-15165. [DOI] [PubMed] [Google Scholar]