

Abstract
As resistance determinants, KPC β-lactamases demonstrate a wide substrate spectrum that includes carbapenems, oxyimino-cephalosporins, and cephamycins. In addition, clinical strains harboring KPC-type β-lactamases are often identified as resistant to standard β-lactam-β-lactamase inhibitor combinations in susceptibility testing. The KPC-2 carbapenemase presents a significant clinical challenge, as the mechanistic bases for KPC-2-associated phenotypes remain elusive. Here, we demonstrate resistance by KPC-2 to β-lactamase inhibitors by determining that clavulanic acid, sulbactam, and tazobactam are hydrolyzed by KPC-2 with partition ratios (kcat/kinact ratios, where kinact is the rate constant of enzyme inactivation) of 2,500, 1,000, and 500, respectively. Methylidene penems that contain an sp2-hybridized C3 carboxylate and a bicyclic R1 side chain (dihydropyrazolo[1,5-c][1,3]thiazole [penem 1] and dihydropyrazolo[5,1-c][1,4]thiazine [penem 2]) are potent inhibitors: Km of penem 1, 0.06 ± 0.01 μM, and Km of penem 2, 0.006 ± 0.001 μM. We also demonstrate that penems 1 and 2 are mechanism-based inactivators, having partition ratios (kcat/kinact ratios) of 250 and 50, respectively. To understand the mechanism of inhibition by these penems, we generated molecular representations of both inhibitors in the active site of KPC-2. These models (i) suggest that penem 1 and penem 2 interact differently with active site residues, with the carbonyl of penem 2 being positioned outside the oxyanion hole and in a less favorable position for hydrolysis than that of penem 1, and (ii) support the kinetic observations that penem 2 is the better inhibitor (kinact/Km = 6.5 ± 0.6 μM−1 s−1). We conclude that KPC-2 is unique among class A β-lactamases in being able to readily hydrolyze clavulanic acid, sulbactam, and tazobactam. In contrast, penem-type β-lactamase inhibitors, by exhibiting unique active site chemistry, may serve as an important scaffold for future development and offer an attractive alternative to our current β-lactamase inhibitors.
In Klebsiella pneumoniae, β-lactam resistance is mediated predominantly by class A SHV, TEM, and CTX-M β-lactamases (7, 35). Single amino acid substitutions in the SHV and TEM β-lactamases can drastically alter the substrate profiles of the enzymes and confer resistance to extended-spectrum cephalosporins and β-lactamase inhibitors (5, 12, 34, 36). β-Lactamases with altered substrate profiles (i.e., extended-spectrum or inhibitor-resistant β-lactamases) have significantly challenged the clinician's approach to the treatment of serious infectious diseases (36). Thus, the search for effective mechanism-based inhibitors of novel β-lactamases merits significant effort (8, 9, 32).
First identified in K. pneumoniae, KPC class A β-lactamases threaten the use of all current β-lactam antibiotics (57). These β-lactamase enzymes are present in an increasing number of bacterial genera, becoming the major carbapenemase expressed by Gram-negative pathogens (e.g., Enterobacter spp., Escherichia coli, Citrobacter freundii, Pseudomonas spp., Serratia marcescens, Proteus mirabilis, and Salmonella enterica) in the United States (3, 10, 11, 16, 17, 25, 37, 45, 49, 53, 59). Moreover, KPC β-lactamases are becoming geographically widespread (having been detected, e.g., in the United States, China, France, Colombia, Greece, Sweden, Norway, Argentina, the United Kingdom, Israel, Brazil, Puerto Rico, Canada, Ireland, Trinidad and Tobago, Poland, Italy, and Finland) (1, 2, 15, 23, 24, 29-31, 33, 38, 39, 42, 50, 51, 53, 55, 57). Evidence suggests that many K. pneumoniae strains in the United States harboring KPCs are genetically related (19).
Why are KPC β-lactamases so problematic? KPC-2 has an overall structure similar to those of other class A enzymes, and interestingly, this β-lactamase has only 50% protein sequence conservation compared to CTX-M-1, 39% compared to SHV-1, and 35% compared to TEM-1. KPC-2 is more like other class A carbapenemases, having 55% identity to NmcA and Imi-1, 63% identity to Sfc-1, and 57% identity to Sme-1. The KPC-2 β-lactamase possesses a large and shallow active site, allowing it to accommodate “bulkier” β-lactams (26). As a result of these structural characteristics, KPC-2 is regarded as a versatile β-lactamase (37); it is a penicillinase, carbapenemase, and cephamycinase and an extended-spectrum β-lactamase (57, 58). Microbiologists and clinicians have observed that many blaKPC-2-containing strains are resistant to β-lactam-β-lactamase inhibitor combinations (6, 19, 50, 54, 55, 59). According to Clinical and Laboratory Standards Institute (CLSI) breakpoints, blaKPC-2-carrying clinical strains for which the MICs of amoxicillin-clavulanic acid are ≥32/16 mg/liter and those of piperacillin-tazobactam are ≥128/4 mg/liter are resistant (14, 58). These observations led us to examine the kinetic properties of the KPC-2 β-lactamase tested against commercially available and novel inhibitors.
A β-lactamase inhibitor demonstrating an affinity in the nanomolar range for KPC-2 and other class A carbapenemases would be an important addition to our therapeutic armamentarium. Thus, we wondered if penem inhibitors that possess an sp2-hybridized C3 carboxylate (a property resembling a characteristic of carbapenems), a complex and reactive R1 side chain, and inactivation chemistry different from that of clavulanic acid could be exploited to inhibit KPC enzymes (41). The methylidene inhibitors penem 1 and penem 2 have dihydropyrazolo[1,5-c][1,3]thiazole and dihydropyrazolo[5,1-c][1,4]thiazine moieties, respectively (see Fig. 1). These penems demonstrate similar levels of in vivo efficacy in mice and have been shown to be effective inhibitors of several class A, C, and D β-lactamases (4, 43, 46-48, 52).
FIG. 1.

Chemical structures of the classical β-lactamase inhibitors, the novel penem β-lactamase inhibitors, cefotaxime, and imipenem.
In this paper, we show why K. pneumoniae containing blaKPC-2 and an E. coli laboratory strain harboring blaKPC-2 are not susceptible to the commercially available β-lactamase inhibitors. Our results demonstrated that clavulanic acid, sulbactam, and tazobactam are hydrolyzed by the KPC-2 β-lactamase. 6-Methylidene penems with complex fused bicyclic R1 side chains are better inhibitors because they possess greater affinity for the active site, have low Kms, and act as mechanism-based inactivators.
MATERIALS AND METHODS
Bacterial strains and plasmids.
K. pneumoniae possessing blaKPC-2 and E. coli containing blaKPC-2 in a pBR322-catI vector (pBR322-catI-blaKPC-2) were kind gifts from Fred Tenover of the Centers for Disease Control and Prevention (Atlanta, GA) (58). We verified the sequence of blaKPC-2 in the pBR322-catI vector. All DNA sequencing reactions were conducted by the Genomics Core Facility at Case Western Reserve University (Cleveland, OH). E. coli DH10B cells (Invitrogen, Carlsbad, CA) were used as a host strain for the pBR322-catI-blaKPC-2 plasmid.
Antibiotic susceptibility.
K. pneumoniae 1534 possessing blaKPC-2, E. coli DH10B, and E. coli DH10B expressing blaKPC-2 were phenotypically characterized using lysogeny broth agar dilution MICs according to CLSI guidelines (13). MICs of antibiotics were determined using a Steers replicator that delivered 10-μl samples containing 104 CFU per spot. For determination of the β-lactamase inhibitor MICs, ampicillin was maintained at a constant concentration of 50 mg/liter and clavulanic acid and sulbactam concentrations were increased (43). In testing of piperacillin-tazobactam, the combination was used at an 8:1 ratio. Finally, penem 1 and penem 2 were maintained at 4 mg/liter while the concentrations of piperacillin, imipenem, and cefotaxime were increased.
Sulbactam was a gift from Thomas Gootz, formerly of Pfizer (Groton, CT). Clavulanic acid was obtained from GlaxoSmithKline (Brentford, United Kingdom). Tazobactam was given to us by Wyeth Pharmaceuticals (Pearl River, NY). Penem 1 and penem 2 were kindly provided by Tarek Mansour of Wyeth Pharmaceuticals (Pearl River, NY). The details of the chemical synthesis of penems 1 and 2 are summarized by Venkatesan et al. (47). Imipenem was purchased from U.S. Pharmacopeia (Rockville, MD). Piperacillin and cefotaxime were purchased from Sigma (St. Louis, MO). The chemical structures of compounds used in this study are shown in Fig. 1.
β-Lactamase purification.
The KPC-2 β-lactamase was purified from E. coli DH10B cells carrying the pBR322-catI-blaKPC-2 plasmid. The cells were grown for 18 h in 500 ml of superoptimal broth (SOB) containing 20 mg/liter chloramphenicol (Sigma) at 37°C with shaking. Cells were pelleted by centrifugation at 5000 × g for 15 min. Cell pellets were frozen for 18 h at −20°C and resuspended in 50 mM Tris-Cl, pH 7.4, and periplasmic proteins were released using stringent periplasmic fractionation with 40 mg/liter lysozyme followed by the addition of 2.0 mM EDTA. The supernatant was further enriched for β-lactamase by using preparative isoelectric focusing as described previously (27, 43). A second purification step was performed using fast protein liquid chromatography (FPLC) with a size exclusion HiLoad 16/60 Superdex-75 column (GE Healthcare Life Sciences, Uppsala, Sweden) on the ÄKTA P-900 system (GE Healthcare Life Sciences). Proteins were eluted with 10 mM phosphate-buffered saline (PBS) at pH 7.4, and fractions were analyzed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein samples were resolved on an SDS-PAGE gel with 5% acrylamide for stacking and 12% acrylamide for separation and stained with Coomassie brilliant blue R250. A final purification step using anion-exchange chromatography with a HiTrap Q HP column (GE Healthcare Life Sciences) was needed. Proteins were eluted with a salt gradient using a mixture of 50 mM Tris-Cl at pH 8.8 and 1.0 M sodium chloride at pH 8.8. The purity of each preparation was again assessed by SDS-PAGE. Protein concentrations were determined with a protein assay using bovine serum albumin as a protein standard according to the protocol of the assay system manufacturer (Bio-Rad, Hercules, CA).
ESI-MS.
Electrospray ionization (ESI)-mass spectrometry (MS) analysis of the intact KPC-2 β-lactamase, purified from E. coli DH10B cells carrying the pBR322-catI-blaKPC-2 plasmid, was performed with a Q-STAR XL quadrupole time-of-flight mass spectrometer (Applied Biosystems, Foster City, CA) equipped with a nanospray source as described previously (44). β-Lactamase enzymes (E) were incubated with an inhibitor (I) for 15 min at room temperature in 10 mM PBS, pH 7.4. The I:E ratios were greater than the turnover number at 15 min (see below). Reactions were terminated by equilibration with 0.1% trifluoroacetic acid (TFA). All samples were desalted and concentrated using a C18 ZipTip according to the protocol of the manufacturer (Millipore, Billerica, MA). Eluted protein samples were diluted with 50% acetonitrile and 0.1% TFA to a concentration of 10 μM and infused at a rate of 0.5 μl per min, and data were collected for 2 min. Spectra were deconvoluted using the Applied Biosystems Analyst program.
Kinetics.
Steady-state kinetic parameters were determined using an 8453 diode array spectrophotometer (Agilent, Santa Clara, CA). Each assay was performed with 10 mM PBS, pH 7.4, at room temperature. In all assays, the enzyme was maintained at 10 nM while substrate concentrations were varied from 5 to 200 μM. To measure hydrolysis rates, we used the following extinction coefficients (Δɛ): nitrocefin (NCF) Δɛ, 17,400 M−1 cm−1 at 482 nm; piperacillin Δɛ, −820 M−1 cm−1 at 235 nm; cefotaxime Δɛ, −7,250 M−1 cm−1 at 262 nm; imipenem Δɛ, −9,000 M−1 cm−1 at 299 nm; and penem 1 Δɛ, −32,400 M−1 cm−1 at 290 nm. We also observed that in 10 mM PBS at room temperature, imipenem undergoes spontaneous hydrolysis. We subtracted the rate of this spontaneous hydrolysis from our measured velocity to determine the “true velocity” (v).
The kinetic parameters Vmax and Km were obtained by nonlinear least-squares fitting of the data (using a Michaelis-Menten equation) with Enzfitter (Biosoft Corporation, Ferguson, MO):
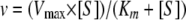 |
(1) |
where [S] is the concentration of substrate.
Kinetic assays were performed under steady-state conditions to determine the Kms of inhibitors for the enzyme according to a previously established model represented in equation 2 (21, 28). In every case, the enzyme concentration was maintained at 10 nM while inhibitor concentrations varied from 25 to 200 μM for clavulanic acid, 125 μM to 1.0 mM for sulbactam, 250 μM to 2.5 mM for tazobactam, 100 nM to 5 μM for penem 1, and 10 to 250 nM for penem 2. A final concentration of 100 μM NCF was used as the reporter substrate. The branched-pathway model followed by penems 1 and 2 can be represented by the following rate equation:
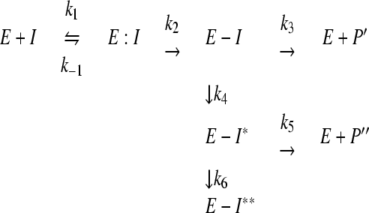 |
(2) |
In this model, formation of the noncovalent enzyme-inhibitor complex E:I is represented by the dissociation constant, K, which is equivalent to k−1/k1. k2 is the first-order rate constant for the acylation step, or the formation of acyl enzyme E-I. k3 is the rate constant for the hydrolysis of the E-I acyl enzyme. The rearrangement of E-I to yield E-I* is represented by the rate constant k4. The rate constant for the hydrolysis of the E-I* acyl enzyme corresponds to k5. Finally, the formation of a terminally inactivated acyl enzyme species, E-I**, is represented by rate constant k6. P′ and P″ are reaction products.
Inverse initial steady-state velocities (1/v0) were plotted against the inhibitor concentration ([I]) to obtain a straight line. As previously established, for brief periods of observation, the k5 step of equation 2 can be neglected (21, 28). Under these conditions, measuring the initial steady-state velocity immediately after mixing yielded a Km for the hydrolysis reaction, or the Michaelis constant for the inhibitors. The initial velocity (v0) measured after mixing is represented by equation 3.
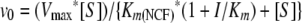 |
(3) |
where Km(NCF) is the Km of NCF.
Km(observed) was determined by dividing the value for the y intercept by the slope of the line. The data were corrected to account for the affinity of NCF for the β-lactamase:
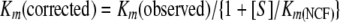 |
(4) |
The rate constant of enzyme inactivation, kinact, was measured directly by time-dependent inactivation of the enzyme in the presence of an inhibitor. To perform this determination, a fixed concentration of enzyme (10 nM), 100 μM NCF, and increasing concentrations of an inactivator (50 to 1,250 μM for clavulanic acid, 500 μM to 5.0 mM for sulbactam, 250 μM to 2.5 mM for tazobactam, 400 nM to 20 μM for penem 1, and 100 nM to 1.5 μM for penem 2) were used in each assay. The observed rate constant for inactivation (kobs) was determined by nonlinear least-squares fitting of the data using Origin 8.0 (Northampton, MA). Here, A0 is initial absorbance, vf is final velocity, and t is time:
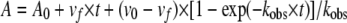 |
(5) |
Each kobs value was plotted versus the inhibitor concentration and fit to determine kinact. The kinetic parameter kinact was obtained with nonlinear least-squares fitting of the data using Enzfitter (Biosoft Corporation) and the hyperbolic equation below:
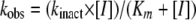 |
(6) |
kinact as presented in equation 2 corresponds to (k2 × k4)/(k2 + k3 + k4). The inhibitor efficiency expressed by the kinact/Km ratio is equivalent to (k2 × k4)/ (K × k3).
Partition ratios, or turnover numbers (tn = kcat/kinact or k3/k4), for KPC-2 were obtained by incubating 1.0 μM KPC-2 with increasing concentrations of an inhibitor (1.0 to 10.0 mM for clavulanic acid, 500 μM to 5.0 mM for sulbactam, 50 μM to 1.0 mM for tazobactam, 100 μM to 1.0 mM for penem 1, and 5 to 100 μM for penem 2) at room temperature for 15 min in 10 mM PBS, pH 7.4. The ratio of inhibitor to enzyme (I:E ratio) necessary to inhibit the hydrolysis of NCF by greater than 90% was determined.
Recovery from inhibition by penem 1 was measured by incubating 1.0 μM KPC-2 with 1.0 mM penem 1 for 18 h at room temperature. A spin concentrator with a 10,000-molecular-weight cutoff was used to remove the unbound inhibitor/product from the EI mixture. NCF (100 μM) hydrolysis by KPC-2 was monitored for 40 min. Hydrolysis of 25 μM penem 1 by 200, 50, and 25 nM KPC-2 at room temperature was measured at A290 over 30 min (λmax for penem 1, 290 nm).
UVD spectra.
UV difference (UVD) spectra were obtained for 25 μM penem 1, 25 μM penem 1 reacted with 0.25 μM KPC-2, and 25 μM penem 1 reacted with 50 mM sodium hydroxide (NaOH) in 10 mM PBS, pH 7.4. UVD spectra were measured from wavelengths of 200 to 400 nm on an Agilent 8453 diode array spectrophotometer for 1 min at room temperature.
Molecular modeling.
The crystal structure of KPC-2 (Protein Data Bank accession no. 2OV5) was used to generate molecular representations of the β-lactamase interactions with the two penem inhibitors. The applications Protein Reports and Utility Tools in Discovery Studio 2.1 molecular modeling software (Accelrys, San Diego, CA) were used to correct crystallographic disorder and prepare the protein for molecular modeling. Hydrogen atoms were added, the pH was set at 7.4, and the crystallographic waters were removed. The KPC-2 β-lactamase was immersed in a “water box” and was centered 7.0 Å away from any face of the box. Then the molecule was subjected to energy minimization in several steps using steepest-descent and conjugate gradient algorithms to reach the minimum convergence (0.02 after 10,000 iterations and a final potential energy of −370,864 kJ/mol). Energy minimizations and molecular dynamics simulations for the enzyme and complexes were performed using force-field parameters of CHARMm and a dielectric constant of 1.0. The particle mesh Ewald (PME) method was used to treat long-range electrostatics. Bonds that involved hydrogen atoms were constrained with the SHAKE algorithm.
The penem structures were constructed using Fragment Builder tools. The CHARMm force field was applied. The protein was solvated and minimized using a Standard Dynamics Cascade protocol of Discovery Studio 2.1. The hydrolyzed penems were automatically docked into the active site of the β-lactamase using the Flexible Docking module of Discovery Studio 2.1. The protocol allowed docking of penems into the flexible active site of KPC-2. Flexible docking using the ChiFlex algorithm (40) created side chains and penem conformations, while the LibDock algorithm (18) docked the low-energy penem conformations into the active site of KPC-2. In the presence of penems, the side chains of selected active site residues were refined using the ChiRotor algorithm (40), after which a final simulated annealing and energy minimization step for each ligand conformation was carried out using CDOCKER (56).
RESULTS AND DISCUSSION
Susceptibility testing.
In Table 1, we summarize the results of our susceptibility testing with β-lactams of the K. pneumoniae clinical isolate containing KPC-2, an E. coli transformant with blaKPC-2, and an E. coli DH10B control. As expected, for the K. pneumoniae clinical strain and the E. coli strain containing blaKPC-2, the MICs of all β-lactams tested were elevated (8 mg/liter for imipenem and 8,192 mg/liter for ampicillin). MICs of the β-lactam-β-lactamase inhibitor combinations for blaKPC-2-expressing strains were also high (ampicillin-clavulanic acid MIC, 50/32 mg/liter; ampicillin-sulbactam MIC, 50/512 mg/liter; and piperacillin-tazobactam MIC, 512/64 mg/liter). These observations are consistent with current reports of MICs for clinical strains (6, 19, 50, 54, 55, 59). We next assayed the activities of piperacillin, cefotaxime, and imipenem in combination with penem 1 or penem 2. The lowest MICs were obtained when either penem 1 or 2 was combined with piperacillin, cefotaxime, or imipenem. Notably, penem 1 and penem 2 lower the MICs of imipenem from 8 to 2 mg/liter for blaKPC-2-bearing strains. Interestingly, the most significant reduction for blaKPC-2-carrying strains was seen when cefotaxime was combined with penem 1 and penem 2. MICs were lowered from 8 and 16 to 0.25 and 0.5 mg/liter, a six-doubling-dilution difference, when combined with 4 mg/liter of penems 1 and 2.
TABLE 1.
MICs of β-lactams and combinations of β-lactams with commercially available β-lactamase inhibitors and penems 1 and 2
| Drug or combinationa | MIC (mg/liter) for: |
||
|---|---|---|---|
| K. pneumoniae 1534 blaKPC-2 | E. coli DH10B (pBR322) blaKPC-2 | E. coli DH10B | |
| Ampicillin | 8,192 | 4,096 | 1 |
| Ampicillin-clavulanic acid | 50/32 | 50/32 | 50/1 |
| Ampicillin-sulbactam | 50/512 | 50/512 | 50/1 |
| Piperacillin | 1,024 | 1,024 | 2 |
| Piperacillin-tazobactam | 512/64 | 512/64 | 4/0.5 |
| Piperacillin-penem 1 | 16/4 | 32/4 | 1/4 |
| Piperacillin-penem 2 | 32/4 | 64/4 | 4/4 |
| Cefotaxime | 16 | 8 | 0.06 |
| Cefotaxime-penem 1 | 0.25/4 | 0.25/4 | 0.06/4 |
| Cefotaxime-penem 2 | 0.25/4 | 0.5/4 | 0.06/4 |
| Imipenem | 8 | 8 | 0.125 |
| Imipenem-penem 1 | 2/4 | 2/4 | 0.125/4 |
| Imipenem-penem 2 | 2/4 | 2/4 | 0.25/4 |
In drug combinations, ampicillin was maintained at a constant concentration of 50 mg/liter and clavulanic acid and sulbactam concentrations were increased. For piperacillin-tazobactam, the two components were increased but maintained at a ratio of 8:1. Penem 1 and penem 2 were held at a constant concentration of 4 mg/liter while the concentrations of piperacillin, imipenem, and cefotaxime were increased.
Kinetics of KPC-2 with substrates and inhibitors.
KPC-2 was purified, and steady-state kinetic parameters were measured (Table 2). As demonstrated previously, KPC-2 hydrolyzes piperacillin, NCF, cefotaxime, and imipenem. Consistent with the MIC data, the catalytic efficiency (kcat/Km ratio) is lowest for cefotaxime, at 0.61 μM−1 s−1, and highest for NCF, at 6.4 μM−1 s−1, among the substrates tested (58).
TABLE 2.
KPC-2 kinetic parametersa
| Drug | Km (μM) | kcat(s−1) | kcat/Km ratio (μM−1 s−1) |
|---|---|---|---|
| Piperacillin | 16 ± 2 | 17 ± 6 | 1.1 ± 0.2 |
| NCF | 5 ± 1 | 30 ± 1 | 6.4 ± 0.2 |
| Cefotaxime | 120 ± 14 | 78 ± 5 | 0.6 ± 0.1 |
| Imipenem | 19 ± 1 | 19 ± 1 | 1.0 ± 0.1 |
Values are means ± standard deviations.
To measure the kinetic parameters of inactivation, we used NCF as the reporter substrate (Table 3). In our assays, clavulanic acid, tazobactam, and sulbactam were unable to effectively inhibit NCF hydrolysis by KPC-2 (kinact/Km ratio, <0.003 μM−1 s−1 for all three inhibitors). More importantly, we also showed that KPC-2 hydrolyzes over 2,500 molecules of clavulanic acid, 1,000 molecules of sulbactam, and 500 molecules of tazobactam in 15 min. After 24 h, there is at least 50% further recovery of KPC-2 activity from inhibition by sulbactam and tazobactam at drug concentrations equal to the tn measured at 15 min. By 72 h after the addition of the drug, KPC-2 partially recovers (∼5%) from inhibition by clavulanic acid.
TABLE 3.
KPC-2 inhibitor kinetics with the β-lactamase inhibitorsa
| Drug | Km (μM) | kinact (s−1) | kinact/Km ratio (μM−1 s−1) | tn (kcat/kinact) |
|---|---|---|---|---|
| Clavulanate | 8.4 ± 0.8 | 0.027 ± 0.001 | 0.0036 ± 0.0004 | ∼2,500 |
| Sulbactam | 135 ± 1 | 0.16 ± 0.01 | 0.0012 ± 0.0001 | ∼1,000 |
| Tazobactam | 78.5 ± 7.9 | 0.20 ± 0.01 | 0.0025 ± 0.0002 | ∼500 |
| Penem 1 | 0.06 ± 0.01 | 0.011 ± 0.001 | 0.18 ± 0.02 | ∼250 |
| Penem 2 | 0.006 ± 0.001 | 0.039 ± 0.002 | 6.5 ± 0.6 | ∼25 |
Values are means ± standard deviations.
In contrast, we observed that the kinact/Km ratios for penems 1 and 2 with the KPC-2 β-lactamase were 0.18 and 6.5 μM−1 s−1, respectively; these values were in accordance with the lower MIC determinations for penems 1 and 2. Despite significantly lower MICs of combinations with these penems than of combinations with other β-lactamase inhibitors, KPC-2 also hydrolyzed 250 molecules of penem 1 and 25 molecules of penem 2 in 15 min. In the case of penem 1, we were able to determine a kcat of 2.5 s−1. After 24 h, KPC-2 can recover from inhibition by penem 1 (∼20%) or penem 2 (∼30%) at inhibitor concentrations equal to the tn measured at 15 min. Recovery after 24 h at a 1,000:1 penem 1-to-KPC-2 ratio is represented in Fig. 2D.
FIG. 2.

(A) UVD spectra for 25 μM penem 1, a mixture of 25 μM penem 1 and 0.25 μM KPC-2, and a mixture of 25 μM penem 1 and 50 mM NaOH after 50 s of incubation. (B) UVD spectra for a mixture of 25 μM penem 1 and 0.25 μM KPC-2 monitored over periods of 5, 25, and 50 s. (C) Penem 1 hydrolysis by KPC-2 monitored at A290 using ratios of 125:1 (25 μM penem 1 and 200 nM KPC-2), 500:1 (25 μM penem 1 and 50 nM KPC-2), and 1,000:1 (25 μM penem 1 and 25 nM KPC-2). (D) Hydrolysis of NCF (100 μM) by 1.0 μM KPC-2 after inhibition by 1.0 mM penem 1 at a 1,000:1 inhibitor-to-enzyme ratio during 24 h of incubation at room temperature compared to hydrolysis by KPC-2 alone.
KPC-2, inhibitor inactivation products, and mechanism of inhibition.
Timed MS was used to assess the formation of KPC-2-β-lactamase inhibitor complexes. A molecular mass of 28,477 ± 3 Da (amu) was obtained for KPC-2 alone. In preparation for MS, KPC-2 was incubated for 15 min with clavulanic acid, sulbactam, tazobactam, penem 1, or penem 2 at a concentration greater than the tn. Notwithstanding the lower partition ratios (tn), we were not able to trap penem 1 or penem 2 inactivating KPC-2.
We next employed timed UVD spectra to gain further insight into the process of KPC-2 inhibition by penem 1 and penem 2. The UVD spectra at 50 s for penem 1 alone, penem 1 reacted with KPC-2, and penem 1 reacted with NaOH are represented in Fig. 2A. The significant change in A290 when KPC-2 was incubated with penem 1 suggests that penem 1 is hydrolyzed by KPC-2 (Fig. 2B). This interpretation is supported by the UVD changes that are observed after the base hydrolysis of penem 1 (A290 also decreases with time).
We next studied the hydrolysis of penem 1 by KPC-2 at A290 (Fig. 2C). Our results show that when the I:E ratio is >tn (i.e., >250:1), a new steady state is reached. We observed that the hydrolysis of penem 1 is biphasic, with rapid initial hydrolysis (E-I → E + P′; rate constant, k3) followed by a lower steady-state rate (E-I* → E + P″ rate constant, k5) (equation 2) after about 800 s. After 24 h at a high inhibitor-to-enzyme ratio (1,000:1), not all of penem 1 was hydrolyzed (data not shown). Remarkably, if excess penem 1 is removed, most of KPC-2's activity rapidly recovers from inhibition at a 1,000:1 ratio, with a slight lag (Fig. 2D). We also observed that there is an initial rate of hydrolysis which may be due to free enzyme (either enzyme that has not acylated or enzyme that has acylated and deacylated) (Fig. 2D). In addition, the slope of the line after the lag is lower than that for the control without penem 1, which is indicative of a terminally inactivated enzyme-inhibitor complex (E-I**; equation 2).
To begin to understand how penem 1 and penem 2 interact with KPC-2, we modeled the penems in the active site of KPC-2. We focused upon the penems because they were the best inhibitors among those tested, including clavulanate, sulbactam, and tazobactam. Based upon our work with SHV-1 and OXA-1, we conceptualized a mechanism in which the acyl enzyme proceeds to the linear imine that ultimately undergoes 7-endo-trig cyclization to yield a cyclic enamine, the 1,4-thiazepine derivative (2, 37). Here, we focus on the deacylated forms of penems 1 and 2 before formation of the postulated seven-membered 1,4-thiazepine ring (E + P′).
In Fig. 3, the molecular representation of penem 1 (orange) within the active site of KPC-2 is superimposed with the representation of penem 2 (purple) in the active site. When comparing the models of the major active site interactions with penem 1 and penem 2, we note several major differences. To begin with, the carbonyl oxygen atom of penem 1 is pointing toward the oxyanion hole, whereas the carbonyl oxygen atom of penem 2 is flipped and pointing away from the oxyanion hole. Next, we note that residues T237 and R220 have hydrogen bonding interactions with the C3 carboxylate of penem 1, whereas neither is close enough to the C3 carboxylate of penem 2 for hydrogen bonding interactions. Instead, the C3 carboxylate of penem 2 is close enough for hydrogen bonding with either K234 or T235. Lastly, we observe hydrophobic interactions with a potential for π-π stacking between the W105 ring and the bicyclic ring of penem 1. However, in the penem 2 model, W105 shifts away about 50° or 2.5 Å from the penem 2 molecule. Overall, our model indicates why the penems participate in interactions leading to lower Kms and higher kinact/Km ratios than those for the other inhibitors tested.
FIG. 3.
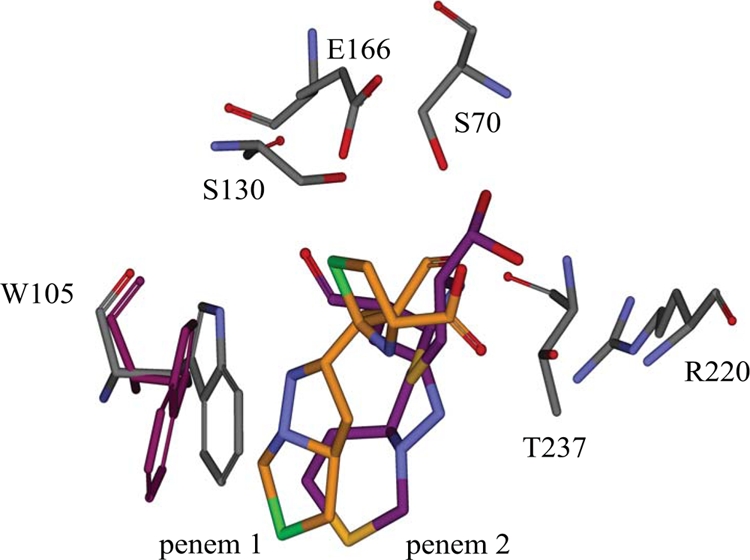
Superimposition of molecular representations of penem 1 (gray-orange) and penem 2 (gray-purple) within the KPC-2 active site. Atom colors: N (blue), O (red), S (green in penem 1, yellow in penem 2).
Conclusions.
Herein, we summarize the kinetic and biochemical correlates of resistance to inhibition of KPC-2 by clavulanic acid, sulbactam, and tazobactam and we explore the turnover of two novel penems. Three important conclusions arise from the findings of our study. First, we show why the commercially available β-lactamase inhibitors are ineffective against KPC-2. To our knowledge, this ability to readily hydrolyze clavulanic acid, sulbactam, and tazobactam is very uncommon in class A enzymes (22). This unprecedented observation partly explains why MICs of β-lactam-β-lactamase inhibitor combinations are so high. For clinical isolates, this situation is compounded by the presence of multiple β-lactamases (e.g., TEM and SHV, etc). Although penem 1 and penem 2 are hydrolyzed by KPC-2 while acting as mechanism-based inactivators, they potentially offer a better alternative than the commercial inhibitors for inhibition of KPC-producing strains. We suspect that unraveling the chemistry that drives the hydrolysis of the commercially available inhibitors and penems 1 and 2 through a branched kinetic mechanism (20, 21, 28) may serve to offer new approaches to inhibiting carbapenemases.
Second, we were intrigued by the synergy between cefotaxime and penem 1 or 2. We predict that this synergy is due to the lower catalytic efficiency of the KPC-2 β-lactamase for cefotaxime (kcat/Km ratio, 0.6 ± 0.1 μM−1 s−1) than for the other substrates tested. One wonders if combinations like this may potentially be exploited to inhibit KPC-producing bacteria in the future.
Third, penem 1 and penem 2 differ by only one carbon atom; thus, it was surprising that their turnover patterns were different. Using our model, we illustrate why these inhibitors are effective (i.e., position in the oxyanion hole) and why they may behave so differently.
We close with the thought that the future challenge in medicinal chemistry will be to anticipate the different catalytic pathways these versatile enzymes (i.e., β-lactamases) will follow as we continuously stress them with each new generation of β-lactams. We recall that both imipenem (thienamycin) and clavulanic acid were discovered in the late 1970s. It is sobering to contemplate that it has taken only 30 years to evolve a versatile β-lactamase whose carbapenem-resistant phenotype is matched by a very robust inhibitor-resistant profile. In fact, the latter may be the preeminent property of the KPC β-lactamase.
Acknowledgments
The Veterans Affairs Merit Review Program, the National Institutes of Health (grant no. RO1 AI063517-01), and the Geriatric Research, Education and Clinical Center, VISN 10, supported these studies.
We also thank Andrea Endimiani and Andrea M. Hujer for careful review of the manuscript.
Footnotes
Published ahead of print on 14 December 2009.
REFERENCES
- 1.Akpaka, P. E., W. H. Swanston, H. N. Ihemere, A. Correa, J. A. Torres, J. D. Tafur, M. C. Montealegre, J. P. Quinn, and M. V. Villegas. 2009. Emergence of KPC-producing Pseudomonas aeruginosa in Trinidad and Tobago. J. Clin. Microbiol. 47:2670-2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baraniak, A., R. Izdebski, M. Herda, J. Fiett, W. Hryniewicz, M. Gniadkowski, I. Kern-Zdanowicz, K. Filczak, and U. Lopaciuk. 2009. Emergence of Klebsiella pneumoniae ST258 with KPC-2 in Poland. Antimicrob. Agents Chemother. 53:4565-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett, J. W., M. L. Herrera, J. S. Lewis II, B. W. Wickes, and J. H. Jorgensen. 2009. KPC-2-producing Enterobacter cloacae and Pseudomonas putida coinfection in a liver transplant recipient. Antimicrob. Agents Chemother. 53:292-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bethel, C. R., A. M. Distler, M. W. Ruszczycky, M. P. Carey, P. R. Carey, A. M. Hujer, M. Taracila, M. S. Helfand, J. M. Thomson, M. Kalp, V. E. Anderson, D. A. Leonard, K. M. Hujer, T. Abe, A. M. Venkatesan, T. S. Mansour, and R. A. Bonomo. 2008. Inhibition of OXA-1 β-lactamase by penems. Antimicrob. Agents Chemother. 52:3135-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonomo, R. A., and L. B. Rice. 1999. Inhibitor resistant class A β-lactamases. Front. Biosci. 4:e34-e41. [DOI] [PubMed] [Google Scholar]
- 6.Bratu, S., M. Mooty, S. Nichani, D. Landman, C. Gullans, B. Pettinato, U. Karumudi, P. Tolaney, and J. Quale. 2005. Emergence of KPC-possessing Klebsiella pneumoniae in Brooklyn, New York: epidemiology and recommendations for detection. Antimicrob. Agents Chemother. 49:3018-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bush, K., G. A. Jacoby, and A. A. Medeiros. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39:1211-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buynak, J. D. 2004. The discovery and development of modified penicillin- and cephalosporin-derived β-lactamase inhibitors. Curr. Med. Chem. 11:1951-1964. [DOI] [PubMed] [Google Scholar]
- 9.Buynak, J. D. 2006. Understanding the longevity of the β-lactam antibiotics and of antibiotic/β-lactamase inhibitor combinations. Biochem. Pharmacol. 71:930-940. [DOI] [PubMed] [Google Scholar]
- 10.Cai, J. C., H. W. Zhou, R. Zhang, and G. X. Chen. 2008. Emergence of Serratia marcescens, Klebsiella pneumoniae, and Escherichia coli isolates possessing the plasmid-mediated carbapenem-hydrolyzing β-lactamase KPC-2 in intensive care units of a Chinese hospital. Antimicrob. Agents Chemother. 52:2014-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castanheira, M., H. S. Sader, L. M. Deshpande, T. R. Fritsche, and R. N. Jones. 2008. Antimicrobial activities of tigecycline and other broad-spectrum antimicrobials tested against serine carbapenemase- and metallo-β-lactamase-producing Enterobacteriaceae: report from the SENTRY Antimicrobial Surveillance Program. Antimicrob. Agents Chemother. 52:570-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaibi, E. B., D. Sirot, G. Paul, and R. Labia. 1999. Inhibitor-resistant TEM β-lactamases: phenotypic, genetic and biochemical characteristics. J. Antimicrob. Chemother. 43:447-458. [DOI] [PubMed] [Google Scholar]
- 13.CLSI. CLSI methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. CLSI M7-A1. Approved standard, 1st ed. Clinical and Laboratory Standards Institute, Wayne, PA.
- 14.CLSI. 2007. Performance standards for antimicrobial susceptibility testing; seventeenth informational supplement, M100-S17. Document no. 19087. Clinical and Laboratory Standards Institute, Wayne, PA.
- 15.Cuzon, G., T. Naas, M. C. Demachy, and P. Nordmann. 2008. Plasmid-mediated carbapenem-hydrolyzing β-lactamase KPC-2 in Klebsiella pneumoniae isolate from Greece. Antimicrob. Agents Chemother. 52:796-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshpande, L. M., R. N. Jones, T. R. Fritsche, and H. S. Sader. 2006. Occurrence and characterization of carbapenemase-producing Enterobacteriaceae: report from the SENTRY Antimicrobial Surveillance Program (2000-2004). Microb. Drug Resist. 12:223-230. [DOI] [PubMed] [Google Scholar]
- 17.Deshpande, L. M., P. R. Rhomberg, H. S. Sader, and R. N. Jones. 2006. Emergence of serine carbapenemases (KPC and SME) among clinical strains of Enterobacteriaceae isolated in the United States Medical Centers: report from the MYSTIC Program (1999-2005). Diagn. Microbiol. Infect. Dis. 56:367-372. [DOI] [PubMed] [Google Scholar]
- 18.Diller, D. J., and K. M. Merz, Jr. 2001. High throughput docking for library design and library prioritization. Proteins 43:113-124. [DOI] [PubMed] [Google Scholar]
- 19.Endimiani, A., A. M. Hujer, F. Perez, C. R. Bethel, K. M. Hujer, J. Kroeger, M. Oethinger, D. L. Paterson, M. D. Adams, M. R. Jacobs, D. J. Diekema, G. S. Hall, S. G. Jenkins, L. B. Rice, F. C. Tenover, and R. A. Bonomo. 2009. Characterization of blaKPC-containing Klebsiella pneumoniae isolates detected in different institutions in the Eastern USA. J. Antimicrob. Chemother. 63:427-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farmer, T. H., J. W. Page, D. J. Payne, and D. J. Knowles. 1994. Kinetic and physical studies of β-lactamase inhibition by a novel penem, BRL 42715. Biochem. J. 303(Pt. 3):825-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frere, J. M., C. Dormans, C. Duyckaerts, and J. De Graeve. 1982. Interaction of β-iodopenicillanate with the β-lactamases of Streptomyces albus G and Actinomadura R39. Biochem. J. 207:437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frere, J. M., C. Dormans, V. M. Lenzini, and C. Duyckaerts. 1982. Interaction of clavulanate with the β-lactamases of Streptomyces albus G and Actinomadura R39. Biochem. J. 207:429-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giani, T., M. M. D'Andrea, P. Pecile, L. Borgianni, P. Nicoletti, F. Tonelli, A. Bartoloni, and G. M. Rossolini. 2009. Emergence in Italy of Klebsiella pneumoniae sequence type 258 producing KPC-3 carbapenemase. J. Clin. Microbiol. 47:3793-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldfarb, D., S.-B. Harvey, K. Jessamine, P. Jessamine, B. Toye, and M. Desjardins. 2009. Detection of plasmid-mediated KPC-producing Klebsiella pneumoniae in Ottawa, Canada: evidence of intrahospital transmission. J. Clin. Microbiol. 47:1920-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hossain, A., M. J. Ferraro, R. M. Pino, R. B. Dew III, E. S. Moland, T. J. Lockhart, K. S. Thomson, R. V. Goering, and N. D. Hanson. 2004. Plasmid-mediated carbapenem-hydrolyzing enzyme KPC-2 in an Enterobacter sp. Antimicrob. Agents Chemother. 48:4438-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ke, W., C. R. Bethel, J. M. Thomson, R. A. Bonomo, and F. van den Akker. 2007. Crystal structure of KPC-2: insights into carbapenemase activity in class A β-lactamases. Biochemistry 46:5732-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, S., M. Thomas, D. M. Shlaes, S. D. Rudin, J. R. Knox, V. Anderson, and R. A. Bonomo. 1998. Kinetic analysis of an inhibitor-resistant variant of the OHIO-1 β-lactamase, an SHV-family class A enzyme. Biochem. J. 333(Pt. 2):395-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matagne, A., P. Ledent, D. Monnaie, A. Felici, M. Jamin, X. Raquet, M. Galleni, D. Klein, I. Francois, and J. M. Frere. 1995. Kinetic study of interaction between BRL 42715, β-lactamases, and d-alanyl-d-alanine peptidases. Antimicrob. Agents Chemother. 39:227-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naas, T., P. Nordmann, G. Vedel, and C. Poyart. 2005. Plasmid-mediated carbapenem-hydrolyzing β-lactamase KPC in a Klebsiella pneumoniae isolate from France. Antimicrob. Agents Chemother. 49:4423-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navon-Venezia, S., I. Chmelnitsky, A. Leavitt, M. J. Schwaber, D. Schwartz, and Y. Carmeli. 2006. Plasmid-mediated imipenem-hydrolyzing enzyme KPC-2 among multiple carbapenem-resistant Escherichia coli clones in Israel. Antimicrob. Agents Chemother. 50:3098-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osterblad, M., J. Kirveskari, S. Koskela, P. Tissari, K. Vuorenoja, A. J. Hakanen, M. Vaara, and J. Jalava. 2009. First isolations of KPC-2-carrying ST258 Klebsiella pneumoniae strains in Finland, June and August 2009. Euro Surveill. 14:pii=19349. [PubMed] [Google Scholar]
- 32.Page, M. G. 2000. β-Lactamase inhibitors. Drug Resist. Updat. 3:109-125. [DOI] [PubMed] [Google Scholar]
- 33.Pasteran, F. G., L. Otaegui, L. Guerriero, G. Radice, R. Maggiora, M. Rapoport, D. Faccone, A. Di Martino, and M. Galas. 2008. Klebsiella pneumoniae carbapenemase-2, Buenos Aires, Argentina. Emerg. Infect. Dis. 14:1178-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paterson, D. L., and R. A. Bonomo. 2005. Extended-spectrum β-lactamases: a clinical update. Clin. Microbiol. Rev. 18:657-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paterson, D. L., K. M. Hujer, A. M. Hujer, B. Yeiser, M. D. Bonomo, L. B. Rice, and R. A. Bonomo. 2003. Extended-spectrum β-lactamases in Klebsiella pneumoniae bloodstream isolates from seven countries: dominance and widespread prevalence of SHV- and CTX-M-type β-lactamases. Antimicrob. Agents Chemother. 47:3554-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez, F., A. Endimiani, K. M. Hujer, and R. A. Bonomo. 2007. The continuing challenge of ESBLs. Curr. Opin. Pharmacol. 7:459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Queenan, A. M., B. Foleno, C. Gownley, E. Wira, and K. Bush. 2004. Effects of inoculum and β-lactamase activity in AmpC- and extended-spectrum β-lactamase (ESBL)-producing Escherichia coli and Klebsiella pneumoniae clinical isolates tested by using NCCLS ESBL methodology. J. Clin. Microbiol. 42:269-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roche, C., M. Cotter, N. O'Connell, and B. Crowley. 2009. First identification of class A carbapenemase-producing Klebsiella pneumoniae in the Republic of Ireland. Euro Surveill. 14:pii=19163. [PubMed] [Google Scholar]
- 39.Samuelsen, O., U. Naseer, S. Tofteland, D. H. Skutlaberg, A. Onken, R. Hjetland, A. Sundsfjord, and C. G. Giske. 2009. Emergence of clonally related Klebsiella pneumoniae isolates of sequence type 258 producing plasmid-mediated KPC carbapenemase in Norway and Sweden. J. Antimicrob. Chemother. 63:654-658. [DOI] [PubMed] [Google Scholar]
- 40.Spassov, V. Z., L. Yan, and P. K. Flook. 2007. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: a side-chain prediction algorithm based on side-chain backbone interactions. Protein Sci. 16:494-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabei, K., X. Feng, A. M. Venkatesan, T. Abe, U. Hideki, T. S. Mansour, and M. M. Siegel. 2004. Mechanism of inactivation of β-lactamases by novel 6-methylidene penems elucidated using electrospray ionization mass spectrometry. J. Med. Chem. 47:3674-3688. [DOI] [PubMed] [Google Scholar]
- 42.Tegmark Wisell, K., S. Haeggman, L. Gezelius, O. Thompson, I. Gustafsson, T. Ripa, and B. Olsson-Liljequist. 2007. Identification of Klebsiella pneumoniae carbapenemase in Sweden. Euro Surveill. 12:E071220.3. [DOI] [PubMed] [Google Scholar]
- 43.Thomson, J. M., A. M. Distler, and R. A. Bonomo. 2007. Overcoming resistance to β-lactamase inhibitors: comparing sulbactam to novel inhibitors against clavulanate resistant SHV enzymes with substitutions at Ambler position 244. Biochemistry 46:11361-11368. [DOI] [PubMed] [Google Scholar]
- 44.Thomson, J. M., A. M. Distler, F. Prati, and R. A. Bonomo. 2006. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J. Biol. Chem. 281:26734-26744. [DOI] [PubMed] [Google Scholar]
- 45.Tibbetts, R., J. G. Frye, J. Marschall, D. Warren, and W. Dunne. 2008. Detection of KPC-2 in a clinical isolate of Proteus mirabilis and first reported description of carbapenemase resistance caused by a KPC β-lactamase in P. mirabilis. J. Clin. Microbiol. 46:3080-3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venkatesan, A. M., A. Agarwal, T. Abe, H. Ushirogochi, M. Ado, T. Tsuyoshi, O. Dos Santos, Z. Li, G. Francisco, Y. I. Lin, P. J. Petersen, Y. Yang, W. J. Weiss, D. M. Shlaes, and T. S. Mansour. 2008. 5,5,6-Fused tricycles bearing imidazole and pyrazole 6-methylidene penems as broad-spectrum inhibitors of β-lactamases. Bioorg. Med. Chem. 16:1890-1902. [DOI] [PubMed] [Google Scholar]
- 47.Venkatesan, A. M., A. Agarwal, T. Abe, H. Ushirogochi, I. Yamamura, M. Ado, T. Tsuyoshi, O. Dos Santos, Y. Gu, F. W. Sum, Z. Li, G. Francisco, Y. I. Lin, P. J. Petersen, Y. Yang, T. Kumagai, W. J. Weiss, D. M. Shlaes, J. R. Knox, and T. S. Mansour. 2006. Structure-activity relationship of 6-methylidene penems bearing 6,5 bicyclic heterocycles as broad-spectrum β-lactamase inhibitors: evidence for 1,4-thiazepine intermediates with C7 R stereochemistry by computational methods. J. Med. Chem. 49:4623-4637. [DOI] [PubMed] [Google Scholar]
- 48.Venkatesan, A. M., A. Agarwal, T. Abe, H. Ushirogochi, I. Yamamura, T. Kumagai, P. J. Petersen, W. J. Weiss, E. Lenoy, Y. Yang, D. M. Shlaes, J. L. Ryan, and T. S. Mansour. 2004. Novel imidazole substituted 6-methylidene-penems as broad-spectrum β-lactamase inhibitors. Bioorg. Med. Chem. 12:5807-5817. [DOI] [PubMed] [Google Scholar]
- 49.Villegas, M. V., K. Lolans, A. Correa, J. N. Kattan, J. A. Lopez, and J. P. Quinn. 2007. First identification of Pseudomonas aeruginosa isolates producing a KPC-type carbapenem-hydrolyzing β-lactamase. Antimicrob. Agents Chemother. 51:1553-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villegas, M. V., K. Lolans, A. Correa, C. J. Suarez, J. A. Lopez, M. Vallejo, and J. P. Quinn. 2006. First detection of the plasmid-mediated class A carbapenemase KPC-2 in clinical isolates of Klebsiella pneumoniae from South America. Antimicrob. Agents Chemother. 50:2880-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei, Z. Q., X. X. Du, Y. S. Yu, P. Shen, Y. G. Chen, and L. J. Li. 2007. Plasmid-mediated KPC-2 in a Klebsiella pneumoniae isolate from China. Antimicrob. Agents Chemother. 51:763-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss, W. J., P. J. Petersen, T. M. Murphy, L. Tardio, Y. Yang, P. A. Bradford, A. M. Venkatesan, T. Abe, T. Isoda, A. Mihira, H. Ushirogochi, T. Takasake, S. Projan, J. O'Connell, and T. S. Mansour. 2004. In vitro and in vivo activities of novel 6-methylidene penems as β-lactamase inhibitors. Antimicrob. Agents Chemother. 48:4589-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolter, D. J., N. Khalaf, I. E. Robledo, G. J. Vazquez, M. I. Sante, E. E. Aquino, R. V. Goering, and N. D. Hanson. 2009. Surveillance of carbapenem-resistant Pseudomonas aeruginosa isolates from Puerto Rican medical center hospitals: dissemination of KPC and IMP-18 β-lactamases. Antimicrob. Agents Chemother. 53:1660-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodford, N., P. M. Tierno, Jr., K. Young, L. Tysall, M. F. Palepou, E. Ward, R. E. Painter, D. F. Suber, D. Shungu, L. L. Silver, K. Inglima, J. Kornblum, and D. M. Livermore. 2004. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A β-lactamase, KPC-3, in a New York medical center. Antimicrob. Agents Chemother. 48:4793-4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woodford, N., J. Zhang, M. Warner, M. E. Kaufmann, J. Matos, A. Macdonald, D. Brudney, D. Sompolinsky, S. Navon-Venezia, and D. M. Livermore. 2008. Arrival of Klebsiella pneumoniae producing KPC carbapenemase in the United Kingdom. J. Antimicrob. Chemother. 62:1261-1264. [DOI] [PubMed] [Google Scholar]
- 56.Wu, G., D. H. Robertson, C. L. Brooks III, and M. Vieth. 2003. Detailed analysis of grid-based molecular docking: a case study of CDOCKER—a CHARMm-based MD docking algorithm. J. Comput. Chem. 24:1549-1562. [DOI] [PubMed] [Google Scholar]
- 57.Yigit, H., A. M. Queenan, G. J. Anderson, A. Domenech-Sanchez, J. W. Biddle, C. D. Steward, S. Alberti, K. Bush, and F. C. Tenover. 2001. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45:1151-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yigit, H., A. M. Queenan, J. K. Rasheed, J. W. Biddle, A. Domenech-Sanchez, S. Alberti, K. Bush, and F. C. Tenover. 2003. Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzing β-lactamase KPC-2. Antimicrob. Agents Chemother. 47:3881-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang, R., H. W. Zhou, J. C. Cai, and G. X. Chen. 2007. Plasmid-mediated carbapenem-hydrolysing β-lactamase KPC-2 in carbapenem-resistant Serratia marcescens isolates from Hangzhou, China. J. Antimicrob. Chemother. 59:574-576. [DOI] [PubMed] [Google Scholar]