

Abstract
Thermostable direct hemolysin (TDH), a major virulence factor of Vibrio parahaemolyticus, induces cytotoxicity in cultured cells. However, the mechanism of TDH's cytotoxic effect including its target molecules on the plasma membrane of eukaryotic cells remains unclear. In this study, we identified the role of lipid rafts, cholesterol- and sphingolipid-enriched microdomains, in TDH cytotoxicity. Treatment of cells with methyl-β-cyclodextrin (MβCD), a raft-disrupting agent, inhibited TDH cytotoxicity. TDH was associated with detergent-resistant membranes (DRMs), and MβCD eliminated this association. In contrast, there was no such association between a nontoxic TDH mutant and DRMs. The disruption of lipid rafts neither affected hemolysis nor inhibited Ca2+ influx into HeLa cells induced by TDH. These findings indicate that the cytotoxicity but not the hemolytic activity of TDH is dependent on lipid rafts. The exogenous and endogenous depletion of cellular sphingomyelin also prevented TDH cytotoxicity, but a direct interaction between TDH and sphingomyelin was not detected with either a lipid overlay assay or a liposome absorption test. Treatment with sphingomyelinase (SMase) at 100 mU/ml disrupted the association of TDH with DRMs but did not affect the localization of lipid raft marker proteins (caveolin-1 and flotillin-1) with DRMs. These results suggest that sphingomyelin is important for the association of TDH with lipid rafts but is not a molecular target of TDH. We hypothesize that TDH may target a certain group of rafts that are sensitive to SMase at a certain concentration, which does not affect other types of rafts.
Vibrio parahaemolyticus is a Gram-negative marine bacterium that is a major pathogen of food-borne gastroenteritis associated with seafood consumption (3, 15, 22). Most clinical isolates of V. parahaemolyticus show hemolysis on Wagatsuma blood agar, known as the Kanagawa phenomenon (KP), which has been recognized as being closely associated with the pathogenic trait of V. parahaemolyticus for humans (31, 41). Thermostable direct hemolysin (TDH), the factor responsible for KP, consists of 165 amino acids and forms a tetrameric structure under aqueous conditions (10). TDH is considered to be one of the major virulence factors of V. parahaemolyticus and exerts a variety of biological activities such as hemolytic activity, cytotoxicity, cardiotoxicity, and enterotoxicity (15, 32, 37). Previously reported animal experiments showed that the deletion of tdh lowers the pathogenicity of V. parahaemolyticus strains (36, 38).
The hemolytic activity of TDH has been well characterized and discussed. TDH forms pores of approximately 2 nm on erythrocyte membranes and causes colloidal osmotic lysis and is therefore considered to function as a pore-forming toxin (14, 27). The sensitivities to TDH differ among erythrocytes from different animal species, and TDH can cause the hemolysis of erythrocytes from human, rabbit, and sheep but not horse (15). While previous investigations indicated that GT1-ganglioside is a functional receptor for TDH on erythrocytes (45, 46), other studies reported contradictory findings (54, 55). In addition, TDH was found to be capable of exerting a cytotoxic effect on glycosphingolipid-deficient GM95 cells (G. Tang and T. Iida, unpublished data), which indicates that GT1-ganglioside is not the only functional receptor for TDH on cultured cells. In clinical courses of V. parahaemolyticus infection, cytotoxicity caused by TDH may be important to destroy intestinal epithelial cells and cause bloody, mucous stool (15). So far, several reports have highlighted the mode of action of TDH on cultured cells (5, 33, 34, 39, 48, 49), for example, that TDH induces Ca2+ influx into cells (34, 48) and modulates cytoskeletal organization (5). However, the mechanism of action of TDH upon the plasma membrane of target cells and its functional receptor(s) remains unclear.
The current view of the plasma membrane is that it is heterogeneous rather than a homogeneous “sea of phospholipids,” which was the classical concept (13, 26, 43). The plasma membrane contains specialized cholesterol- and sphingolipid-enriched microdomains, which are known as “lipid rafts.” Since lipid rafts are biochemically characterized by their insolubility by nonionic detergents, e.g., Triton X-100 at 4°C, the isolation of lipid rafts as detergent-resistant membranes (DRMs) is commonly used for their analysis (16, 29). Lipid rafts have also been implicated in important biological events such as signal transduction, protein sorting, and membrane trafficking (19, 43, 44). Moreover, these microdomains are reportedly subverted by various infectious agents like pathogens and toxins to facilitate their infectious processes such as binding, internalization, and signaling (30, 40). In particular, various bacterial pore-forming toxins including aerolysin and cholesterol-dependent cytolysins were previously reported to optimize their oligomerization via the concentration in lipid rafts (1, 25), which prompted us to address the relationship between lipid rafts and the toxicity of TDH because of the pore-forming nature of TDH.
In the study presented here, we assessed the role of lipid rafts in TDH cytotoxicity and hemolysis. Our data indicate that the cytotoxicity of TDH requires an association with lipid rafts, which is disrupted by the depletion of cholesterol or sphingomyelin.
MATERIALS AND METHODS
Antibodies and reagents.
Methyl-β-cyclodextrin (MβCD), sphingomyelin, cholesterol, and sphingomyelinase (SMase) from Bacillus cereus were purchased from Sigma (St. Louis, MO); antibodies to caveolin-1 (Cav-1) and flotillin-1 (Flt-1) were purchased from BD Biosciences (Franklin Lakes, NJ); anti-transferrin receptor (TfR) was purchased from Zymed (South San Francisco, CA); and anti-lysenin antibodies were purchased from Peptide Institute Inc. (Osaka, Japan).
Preparation of toxins.
TDH and R7 were prepared by introducing plasmid pKK223-3 harboring the gene for TDH or R7 into Escherichia coli JM109 by means of transformation. Expressed proteins were purified by a series of column chromatography steps described previously (49). Lysenin was purchased from Peptide Institute Inc.
Cytotoxicity assay.
Cytotoxicity was evaluated as previously described (24). HeLa, Rat-1, and CHO-K1 cells were obtained as described elsewhere previously (49). LA-1, LY-A/hCERT, LY-B, and LY-B/cLCB1 cells were provided by the Cell Bank, Riken BioResource Center (Tsukuba, Japan), through the National Bio-Resource Project of the MEXT, Japan. HeLa cells were exposed to TDH at 20 μg/ml for 3 h. Rat-1 cells, which are highly sensitive to TDH, and CHO-K1 cells, which are less sensitive to TDH, were exposed to TDH at 10 μg/ml for 30 min and at 100 μg/ml for 3 h, respectively. After centrifugation at 300 × g for 10 min, the supernatants were collected, and the lactate dehydrogenase (LDH) released into the supernatant was measured with the Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) according to the manufacturer's instructions. The LDH release (percent cytotoxicity) was calculated by using the following equation: (optical density at 492 nm [OD492] for experimental release − OD492 for spontaneous release)/(OD492 for maximum release − OD492 for spontaneous release) × 100. The spontaneous release was the amount of LDH released from the cytoplasm of untreated cells, whereas the maximum release was the amount of LDH released from toxin-untreated cells that were totally lysed with the lysis buffer included in the Cytotox 96 nonradioactive cytotoxicity assay kits (Promega).
Isolation of DRMs by sucrose gradient centrifugation and analysis of DRMs of the gradient.
DRMs were isolated as previously described (2, 8, 42, 43). Briefly, cells were washed with phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM KH2PO4, and 2 mM KH2PO4 at pH 7.4) 3 times and lysed with 1.75 ml of 1% Triton X-100 in Tris-buffered saline (TBS) (10 mM Tris-HCl at pH 7.4 and 150 mM NaCl) for 30 min on ice. After centrifugation at 800 × g for 10 min, the postnuclear supernatant was mixed with an equal volume of 80% sucrose in TBS. The mixture was then placed at the bottom of the ultracentrifugation tube and successively overlaid with 6 ml of 30% sucrose in TBS and 3 ml of 5% sucrose in TBS. After centrifugation at 35,000 rpm for 18 h at 4°C in an SW41 rotor (Beckman Coulter, Fullerton, CA), 1- or 2-ml fractions were collected from the top of the tube. Proteins of each fraction were precipitated with 10% trichloroacetic acid (TCA) and analyzed by means of sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. For immunoblotting, proteins of the gels were transferred onto nitrocellulose membranes, and TDH and R7 were detected with an anti-TDH monoclonal antibody (50).
Hemolysis assay.
Human erythrocytes from healthy volunteers were exposed to MβCD at 37°C for 1 h. After three washings with PBS, erythrocytes suspended in PBS (4%, vol/vol) were incubated with 10 μg/ml TDH at 37°C for 30 min. PBS and Triton X-100 (1%, vol/vol, final concentration) were used as negative and positive controls, respectively. Following centrifugation at 300 × g for 10 min, the supernatants were collected, and their optical density was measured at 570 nm to determine the percentage of hemoglobin released.
Ca2+ influx.
Intracellular Ca2+ levels of HeLa cells were monitored by incubating the HeLa cells with Fluo 4-AM (5 μM) for 30 min. After washing with Dulbecco's modified Eagle medium, the cells were then incubated with TDH at 37°C for 30 min, Fluo 4-AM was excited at 485 nm, emission was detected at 538 nm, and Ca2+ influx was expressed as ΔF(F30 min − F0 min)/F0 min.
Depletion of cellular sphingomyelin.
For the exogenous depletion of sphingomyelin, HeLa cells were treated with SMase for 1 h at 37°C. For the endogenous depletion of sphingomyelin, we used LY-A, LY-A/hCERT, LY-B, and LY-B/cLCB1 cells as previously described (11).
Preparation of liposomes.
First, sphingomyelin or cholesterol was dissolved in chloroform at 10 mM. Both lipid solutions were then mixed in a glass tube at a molecular ratio of 1:1. After the solvent had evaporated, the lipids were hydrated in PBS for 1 h, and the suspension was vortexed and sonicated. This was followed by the preparation of liposomes. For inhibitory experiments, TDH or lysenin was incubated with the prepared liposomes at 0 to 10 mM at room temperature for 30 min, after which cells or erythrocytes were exposed to the mixture. Cytotoxicity and hemolysis were determined as described above.
TLC.
Lipids were extracted by using the Bligh and Dyer method (4). Extracted lipids were spotted onto silica gel 60 F244 (catalog number 07589; Merck KGaA, Darmstadt, Germany) and separated by thin-layer chromatography (TLC) using a solvent system consisting of chloroform-methanol-water (65:25:4, vol/vol/vol). After drying of the TLC plates, the lipids were detected by means of Coomassie brilliant blue staining (35).
Lipid overlay assay.
The lipid overlay assay was performed as previously described (18). Briefly, aliquots of sphingomyelin or cholesterol (10 or 50 nmol) were spotted onto silica gel 60 (catalog number 16835; Merck). The plates were soaked in 0.4% polyisobutylmethacrylate in a chloroform-hexane (1:5) solution for 5 s and dried, followed by washing with PBS for 5 min and blocking with 1% fatty acid-free bovine serum albumin (BSA) in PBS for 1 h. The plates were then incubated with 100 μg/ml TDH or 1 μg/ml lysenin for 1.5 h. After washing with PBS, the plates were probed first with an anti-TDH or anti-lysenin polyclonal antibody and then with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G antibody. The ECL system (GE Healthcare UK Ltd., Buckinghamshire, England) was used for detection.
Fluorescent microscope analysis.
HeLa cells were incubated with 40 μg/ml TDH-Myc and with 5 μg/ml Alexa 546-conjugated cholera toxin B subunit (CTxB) or 5 μg/ml Alexa 546-conjugated transferrin (Molecular Probes, Eugene, OR) at 37°C for 30 min. After washing with PBS, the cells were fixed with 3% (wt/vol) paraformaldehyde for 15 min, washed with PBS, incubated with anti-c-Myc antibody (Cell Signaling Technology, Danvers, MA), and then incubated with Alexa 488-conjugated anti-mouse antibody (Molecular Probes). A Nikon (Tokyo, Japan) TE2000-U microscope was used for fluorescent microscopy.
Statistical analysis.
Statistical significance was determined by using the t test. A P value of <0.05 was considered statistically significant.
RESULTS
MβCD inhibits TDH cytotoxicity.
To examine whether lipid rafts are involved in the cytotoxicity induced by TDH, the effect of MβCD on TDH cytotoxicity to HeLa cells was tested, and it was found that the pretreatment of the cells with MβCD significantly inhibited such cytotoxicity (Fig. 1A). We also tested other cell lines, that is, Rat-1 cells, which are highly sensitive, and Chinese hamster ovary K1 (CHO-K1) cells, which are somewhat resistant to TDH (49). As shown in Fig. 1B and C, both cell lines pretreated with MβCD became resistant to the cytotoxic effect of TDH. Since MβCD disrupts lipid rafts by the depletion of cholesterol, we also examined whether TDH interacts directly with cholesterol, but the addition of an excess amount of cholesterol to the culture medium did not affect TDH cytotoxicity. Moreover, a lipid overlay assay showed no detectable interaction between TDH and cholesterol (data not shown). These results suggest that cholesterol depletion inhibits TDH cytotoxicity but that TDH does not interact directly with cholesterol.
FIG. 1.

Effect of methyl-β-cyclodextrin (MβCD) on cytotoxicity of TDH. MβCD inhibited TDH cytotoxicity for HeLa, Rat-1, and CHO-K1 cells. Each cell line was exposed to TDH at 20 μg/ml for 3 h for HeLa cells, at 10 μg/ml for 30 min for Rat-1 cells, and at 100 μg/ml for 3 h for CHO-K1 cells. Cytotoxicity was evaluated by determining the quantity of lactate dehydrogenase released. PBS was used as a negative control (0%), and the lysis buffer included in Cytotox 96 nonradioactive cytotoxicity assay kits (Promega) was used as a positive control (100%). Data are presented as the means for triplicate experiments. Error bars represent standard deviations (SD). Asterisks indicate significant differences from the results obtained with nontreated cells (P < 0.05).
Association of TDH with lipid rafts.
To investigate the role of lipid rafts in TDH cytotoxicity, we next assessed whether TDH is associated with lipid rafts. For the isolation of DRMs, TDH-treated HeLa cells were lysed with 1% Triton X-100 on ice, and the lysate was fractionated by sucrose gradient ultracentrifugation. As expected, the lipid raft marker proteins Cav-1 and Flt-1 were found in low-density DRM fractions, but the nonraft protein TfR was present in non-DRM fractions (Fig. 2A). TDH was detected in both DRM and non-DRM fractions. Next, the effect of MβCD on the association of TDH with lipid rafts was examined. After the pretreatment of HeLa cells with MβCD, they were incubated with TDH, and the Triton-insoluble fractions were separated. The majority of Cav-1 and Flt-1 was redistributed in non-DRM fractions, confirming the disruption of lipid rafts as a result of the treatment with MβCD. TDH was not detected in Triton-insoluble fractions but was completely localized in non-DRM fractions (Fig. 2B). These results indicate that TDH is, at least partly, associated with lipid rafts.
FIG. 2.

Association of TDH with lipid rafts. (A) HeLa cells were incubated with 10 μg/ml TDH at 37°C for 30 min. After the preparation of DRMs, the locations of TDH, caveolin-1 (Cav-1), flotillin-1 (Flt-1), and transferrin receptor (TfR) were analyzed by immunoblotting. (B) MβCD-pretreated HeLa cells were treated with 10 μg/ml TDH at 37°C for 30 min, and prepared DRMs were analyzed by immunoblotting against TDH, Cav-1, Flt-1, and TfR. (C) HeLa cells were incubated with 10 μg/ml R7 at 37°C for 30 min. DRMs were prepared and then analyzed by immunoblotting.
A mutant toxin of TDH is not associated with lipid rafts.
R7 is a mutant of TDH with a single amino acid substitution of serine for glycine 62 (47). R7 was isolated by in vitro mutagenesis and found not to induce hemolytic activity, cytotoxicity, or Ca2+ influx in erythrocytes but to competitively inhibit the binding of wild-type TDH to erythrocytes and cultured cells (49). To examine whether R7 is associated with lipid rafts, DRMs were isolated from R7-treated cells, and the location of R7 was analyzed. The result showed that, unlike TDH (Fig. 2A), R7 was not found in DRM fractions (Fig. 2C). This indicates that the mutant toxin of TDH is not associated with lipid rafts.
Effect of MβCD on TDH-induced Ca2+ influx into cultured cells.
As TDH was previously reported to induce Ca2+ influx into cultured cells (34, 48), we next examined whether MβCD affects Ca2+ influx into cultured cells induced by TDH. The Ca2+ indicator Fluo 4-AM was loaded onto HeLa cells, followed by the addition of TDH. After incubation for 30 min, intracellular Ca2+ levels were monitored (Fig. 3A). Similarly, LDH release from HeLa cells, an indicator of cell death, was evaluated 30 min after the addition of TDH (Fig. 3B). It was found that MβCD could not inhibit the elevation of intracellular Ca2+ levels induced by TDH (Fig. 3A), while LDH release was effectively blocked by MβCD (Fig. 3B). Taken together, these results suggest that although lipid rafts are essential for the cytotoxicity generated by TDH, TDH forms Ca2+-permeable pores on cellular membranes even without the involvement of lipid rafts.
FIG. 3.

Effect of MβCD on Ca2+ influx facilitated by TDH. (A) Ca2+ influx into HeLa cells facilitated by TDH or R7. HeLa cells were pretreated with 4 mM MβCD or not for 1 h, followed by Fluo 4-AM loading for 30 min. Cells were incubated with TDH or R7 for 30 min, and intracellular Ca2+ levels of cells were determined. Black squares, TDH treated; white triangles, MβCD pretreated and TDH treated; white squares, R7 treated. (B) Cytotoxicity of TDH or R7 for HeLa cells for 30 min. Black squares indicate that HeLa cells were treated with TDH, white triangles indicate that HeLa cells were pretreated with 4 mM MβCD for 1 h and treated with TDH, and white squares indicate that HeLa cells were treated with R7. Data are from three experiments ± SD.
MβCD has no effect on the hemolytic activity of TDH.
Next, we examined whether MβCD inhibits hemolysis induced by TDH. In contrast to cytotoxicity, hemolysis induced by TDH was not inhibited by MβCD (0 to 8 mM) (Fig. 4A). To exclude the possibility that the concentration of MβCD used in this study was not sufficient to disrupt the lipid rafts of erythrocytes, the effect of MβCD on location of the raft marker protein Flt-1 was examined. Although Flt-1 was found in DRM fractions from the erythrocytes that were not treated with MβCD, the location of Flt-1 was altered in the nonraft fractions when erythrocytes were treated with 4 mM MβCD (Fig. 4B). This demonstrated that the concentration of MβCD used in this study was sufficient for the disruption of lipid rafts and indicated that, unlike for the cytotoxicity of TDH, lipid rafts are not required for its hemolytic activity.
FIG. 4.
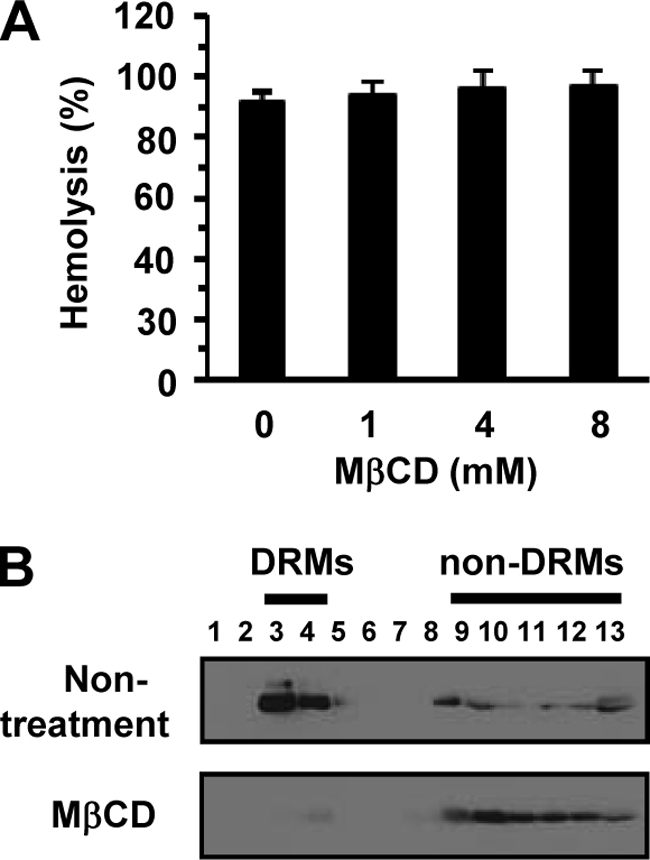
Disruption of lipid rafts in erythrocytes does not influence hemolysis caused by TDH. (A) Erythrocytes were pretreated with MβCD (0 to 8 mM) for 1 h at 37°C and washed with PBS. Nontreated and MβCD-treated erythrocytes were subsequently exposed to 10 μg/ml TDH for 30 min at 37°C. Hemolysis was determined by measuring the release of hemoglobin. Data are shown as means and SD from three independent experiments. (B) Erythrocytes were not treated with MβCD or treated with 4 mM MβCD for 1 h at 37°C and then lysed with 1% Triton X-100 for 30 min on ice. DRMs were prepared and analyzed by immunoblotting against Flt-1.
Depletion of cellular sphingomyelin inhibits TDH cytotoxicity.
Lipid rafts are microdomains enriched with cholesterol and sphingolipids. Although glycosphingolipids are important members of the sphingolipids, TDH is cytotoxic to glycosphingolipid-deficient mutant cells, GM95, which indicates that glycosphingolipids are not essential for the cytotoxicity of TDH. Sphingomyelin, another important member of the sphingolipids, is also a key component of lipid rafts (7). To investigate whether sphingomyelin is involved in the cytotoxicity of TDH, we evaluated the effect of TDH cytotoxicity on HeLa cells whose sphingomyelin was depleted. The HeLa cells were treated with exogenous sphingomyelinase (SMase) and then exposed to TDH. Interestingly, SMase reduced the cytotoxicity of TDH in a dose-dependent manner (Fig. 5A). Also, the SMase-induced depletion of sphingomyelin was confirmed by means of thin-layer chromatography (TLC) (Fig. 5B). Next, we tested the effect of the endogenous depletion of sphingomyelin, for which we used two types of cell lines, LY-A and LY-B, derived from CHO-K1 cells and deficient in sphingomyelin synthesis (6, 7, 11, 12). LY-A is deficient in the intracellular trafficking of ceramide, which accounts for its deficiency in sphingomyelin synthesis (6, 12). LY-B lacks a component of serine palmitoyltransferase, which catalyzes the first step of sphingolipid biosynthesis (11). These cells were exposed to TDH, followed by an assessment of cytotoxicity, which showed that LY-A and LY-B were more resistant to TDH than their parental cells (Fig. 5C). The partial cytotoxicity of TDH for LY-A and LY-B cells is related to the residual contents of sphingomyelin in LY-A and LY-B cells, which are ∼40% and ∼15%, respectively, of the levels in their parental cells (7). This led us to conclude that the cytotoxicity of TDH was abrogated by both endogenous and exogenous depletions of cellular sphingomyelin.
FIG. 5.

Depletion of sphingomyelin eliminates TDH cytotoxicity. (A) HeLa cells were pretreated with SMase for 1 h at 37°C to deplete cellular sphingomyelin. After washing with PBS, cells were incubated with 20 μg/ml TDH for 3 h. Means and SD from three experiments are shown. *, P < 0.01. (B) Thin-layer chromatography (TLC) analysis of the content of cellular sphingomyelin. Lane 1, 1 μg of sphingomyelin; lane 2, lipids extracted from nontreated HeLa cells; lane 3, lipids from HeLa cells treated with 50 mU/ml SMase for 1 h. PE, phosphatidylethanolamine; PC, phosphatidylcholine; SM, sphingomyelin. (C) CHO-K1 cells; LY-A cells; LY-A/hCERT cells, which expressed human CERT and restored the trafficking of ceramide; LY-B cells; and LY-B/cLCB1 cells, which complemented the serine palmitoyltransferase deficiency, were cultured in Nutridoma-BO medium for 2 days to deplete sphingomyelin of mutant cells. Cells were incubated with 200 μg/ml TDH for 3 h, and cytotoxicity was determined. Data are from three experiments ± SD. *, P < 0.01. SMase was not cytotoxic even at the highest concentration used in this study (100 mU/ml) (data not shown).
TDH does not interact directly with sphingomyelin.
The fact that the depletion of sphingomyelin inhibited the cytotoxicity of TDH raised the possibility that sphingomyelin is a target molecule for TDH. To investigate this possibility, we examined whether sphingomyelin/cholesterol liposomes inhibit the cytotoxicity of TDH. As shown in Fig. 6A and C, sphingomyelin/cholesterol liposomes significantly inhibited the cytotoxicity and hemolytic activity of lysenin, which is a sphingomyelin-binding toxin produced by the earthworm Eisenia foetida (53), indicating that the liposome preparations were appropriate for this inhibition test. In contrast, the cytotoxicity and hemolytic activity of TDH were not inhibited by the sphingomyelin/cholesterol liposomes, as shown in Fig. 6B and D. In addition, we performed a lipid overlay assay to evaluate the direct interaction between TDH and sphingomyelin. Sphingomyelin was spotted onto TLC plates, which were then probed with TDH or lysenin, and no direct interaction between TDH and sphingomyelin was detected (Fig. 6E), demonstrating that sphingomyelin is not a direct target molecule for TDH.
FIG. 6.

There is no direct interaction between TDH and sphingomyelin. (A to D) Liposomes including sphingomyelin and cholesterol were incubated with TDH or lysenin for 30 min at room temperature, and the toxin-liposome mixtures were added to HeLa cells or erythrocytes. (A and B) HeLa cells were incubated with liposomes and 1 μg/ml lysenin for 3 h (A) or 20 μg/ml TDH for 3 h (B). (C and D) Erythrocytes were exposed to liposomes and 500 ng/ml lysenin for 30 min (C) or 10 μg/ml TDH for 30 min (D). Data are shown as means and SD from three independent experiments. *, P < 0.01. (E) Binding of TDH or lysenin to sphingomyelin. Spotting of sphingomyelin (0, 10, and 50 nmol) onto the TLC plates was followed by lipid overlay assays.
Depletion of sphingomyelin results in a loss of association of TDH with lipid rafts.
Next, we examined the effect of sphingomyelin depletion on the association of TDH with lipid rafts. HeLa cells were treated with 100 mU/ml SMase and exposed to TDH, followed by the isolation of DRMs. In SMase-treated cells, Cav-1 and Flt-1 remained in the DRM fractions. However, TDH disappeared from DRM fractions and was found only in non-DRM fractions (Fig. 7). This finding indicated that the depletion of sphingomyelin inhibited the association of TDH with lipid rafts, which may therefore result in the loss of cytotoxicity due to TDH.
FIG. 7.
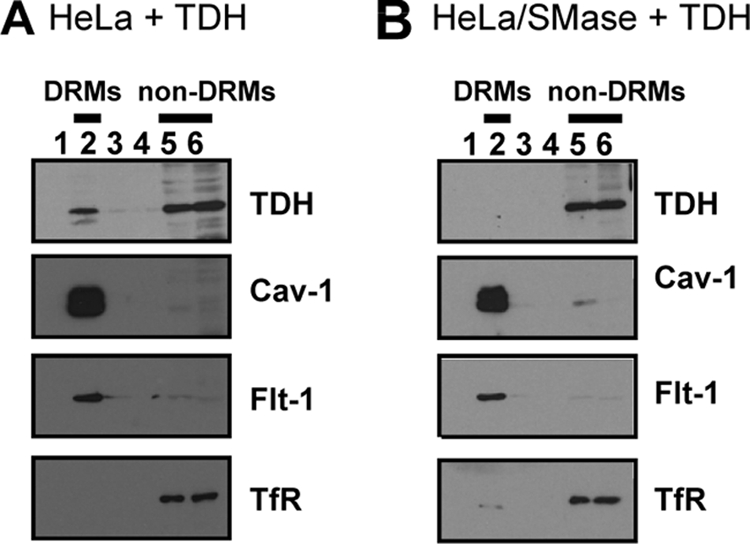
Depletion of sphingomyelin affects association of TDH with lipid rafts. HeLa cells were incubated with 100 mU/ml SMase or no SMase for 1 h and then exposed to 10 μg/ml TDH for 30 min. DRMs were prepared and analyzed by immunoblotting against TDH, Cav-1, Flt-1, and TfR. (A) Nontreated; (B) SMase treated.
Localization of c-Myc-tagged TDH.
Finally, fluorescent microscopy was used for the imaging of TDH on cells. We constructed TDH tagged with c-Myc at its C terminus (TDH-Myc) and used it for an immunofluorescence study. HeLa cells were treated with TDH-Myc and with the Alexa 546-conjugated cholera toxin B subunit (CTxB), which is a commonly used raft probe, or with Alexa 546-conjugated transferrin (Tfn), which stains cellular TfR, a nonraft marker. The distribution pattern of TDH-Myc was similar to that of CTxB (Fig. 8A) but not that of Tfn (Fig. 8B). However, the fact that the localization of TDH-Myc was not completely merged with that of CTxB suggests that although TDH-Myc was shown to be associated with lipid rafts, the lipid rafts recognized by TDH differ, at least partly, from the GM1-lipid rafts recognized by CTxB.
FIG. 8.

Distribution of TDH on cells by fluorescent microscopy. HeLa cells were incubated with TDH-Myc in the presence of Alexa 546-CTxB (A) or Alexa 546-Tfn (B) and fixed. The cells were then probed with anti-c-Myc antibody and subsequently stained with Alexa 488-conjugated anti-mouse antibody. Images were obtained by fluorescent microscopy. Arrowheads indicate nonspecific signals that were observed with anti-c-Myc antibody even in the absence of TDH-Myc (data not shown).
DISCUSSION
Although TDH is recognized as a major virulence factor of V. parahaemolyticus, the mechanism of its cytotoxicity remains unclear. In this study, we examined the role of lipid rafts in TDH cytotoxicity and found that (i) TDH is associated with lipid rafts, while a nontoxic TDH mutant is not, and (ii) MβCD eliminates TDH cytotoxicity and its association with lipid rafts. Based on these findings, we concluded that the cytotoxicity of TDH is dependent on lipid rafts.
TDH is a pore-forming toxin that forms 2-nm pores on the membrane of erythrocytes. Although many bacterial pore-forming toxins form sodium dodecyl sulfate (SDS)-resistant oligomers (51), to our knowledge, it has never been reported that TDH can form such a stable oligomer. On the other hand, it was previously reported that TDH forms a tetrameric structure in aqueous solution (10). The two-dimensional images showed a square shape of a tetramer of TDH and suggested the presence of a pore with a width of about 2 nm in the center of the tetramer. This size is consistent with the size of pores on erythrocytes observed previously (14). The pore in the center of the tetramer of TDH might account for the hemolytic activity of the toxin, and it seems to support our present finding that hemolysis by TDH is independent of lipid rafts. In the case of cultured cells, on the other hand, our finding demonstrated that the pores formed by TDH are also thought to be enough for Ca2+ influx into cultured cells but not for cytotoxicity (Fig. 3). It was previously reported that when cultured cells are pretreated with streptolysin O, a bacterial pore-forming toxin, at a low and sublethal dose, the cells can repair the injured plasma membrane (17, 52). Another study found that in response to aerolysin, another pore-forming toxin, caspase-1 activates lipid biogenesis, which could result in the repair of damage to the membrane and consequently in cell survival (9). We therefore hypothesize that pore formation by TDH in the nonraft membrane may not be sufficient for cell killing because nucleated cells may be able to repair their membrane damaged by TDH. In contrast, cytotoxic action occurs only when TDH is associated with lipid rafts. Because of the localization of TDH in lipid rafts, an irreversible membrane disruption that cannot be restored by the cells may occur and thus lead to cell death. Recently, various bacterial pore-forming toxins were reported to utilize lipid rafts (1, 25, 30). Lipid rafts are thought to serve as concentration devices for the optimization of oligomerization and pore formation by those pore-forming toxins on the plasma membrane. It is thus possible that TDH also facilitates its own cytotoxic effect as a result of concentration in lipid rafts. The corresponding model is presented in Fig. 9.
FIG. 9.

Model of hemolysis and cytotoxicity induced by TDH. In erythrocytes, TDH forms pores in the nonraft region on the membrane of erythrocytes in the presence or absence of lipid rafts. The pores are enough for the colloidal osmotic lysis of erythrocytes. In nucleated cells, TDH forms pores in the nonraft region on the plasma membrane in the presence or absence of lipid rafts. When lipid rafts are disrupted, nucleated cells may repair the pores, resulting in cell survival. When lipid rafts are intact, TDH can be associated with these rafts, and concentrated TDH may cause irreversible membrane disruption or other cytotoxic events in lipid rafts. The damage to the membrane that cannot be repaired by cells results in cell death.
When cells were treated with exogenous SMase, TDH was found not to be associated with DRMs, while raft marker proteins (Cav-1 and Flt-1) were still localized in DRM fractions (Fig. 7B). This prompted us to speculate that TDH may be associated with a certain type of lipid raft, which is disrupted at a certain concentration of SMase. Although the flotation of DRMs is generally used for analyses of lipid rafts, it has been pointed out frequently that DRMs are not the same as lipid rafts in living cells (28). Figure 8 shows the distribution of TDH-Myc determined by means of indirect immunofluorescence microscopy, which indicates that the distribution of TDH-Myc is similar but not identical to that of CTxB. This suggests that TDH-associated rafts may differ from GM1-lipid rafts. Recent biophysical approaches such as fluorescence resonance energy transfer and atomic force microscopy have shown that lipid rafts of living cells are more dynamic than the past model (13, 26, 43). Moreover, recent studies using the sphingomyelin-specific binding toxin lysenin have demonstrated that sphingomyelin-rich domains are distinct from GM1-rich domains in the cytoplasmic membrane (20, 21, 23). Thus, the heterogeneity of lipid rafts is steadily uncovered. TDH-associated rafts seem not to be identical to GM1-rich domains that are recognized by CTxB, as indicated by fluorescent microscopy. Although the fluorescent microscopic analysis in our study was not enough to clearly demonstrate that TDH associates with specific types of lipid rafts, it offered the possibility that TDH may serve as a potent probe for a certain group of lipid rafts in plasma membranes.
In conclusion, the results of our study demonstrate that the cytotoxic effect of TDH on nucleated cells is dependent on lipid rafts, while the hemolytic activity of TDH is not. It thus seems that the mechanism of cytotoxicity by TDH is different from that of hemolysis by TDH. Sphingomyelin was found to be important for the association of TDH with lipid rafts, although TDH does not interact directly with sphingomyelin. These results provided the possibility that TDH may target a certain group of rafts that are sensitive to SMase at a certain concentration that does not affect other types of rafts, which would support the notion of the heterogeneity of lipid rafts. It is also expected that TDH may prove to be a novel probe for the differentiation of the heterogeneity of lipid rafts.
Acknowledgments
We thank T. Miura and T. Taguchi for their helpful discussion, M. Rokuda for his technical assistance, and R. Dryselius for critical reading of the manuscript.
This study was supported by the Research for the Future Program funded by the Japan Society for the Promotion of Science and by Grants-in-Aid for Scientific Research on Priority Areas Applied Genomics and Matrix of Infection Phenomena, for Scientific Research and for Young Scientists (B), from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Editor: S. M. Payne
Footnotes
Published ahead of print on 23 November 2009.
REFERENCES
- 1.Abrami, L., M. Fivaz, and F. G. van der Goot. 2000. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 8:168-172. [DOI] [PubMed] [Google Scholar]
- 2.Abrami, L., S. Liu, P. Cosson, S. H. Leppla, and F. G. van der Goot. 2003. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 160:321-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blake, P. A., R. E. Weaver, and D. G. Hollis. 1980. Diseases of humans (other than cholera) caused by vibrios. Annu. Rev. Microbiol. 34:341-367. [DOI] [PubMed] [Google Scholar]
- 4.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911-917. [DOI] [PubMed] [Google Scholar]
- 5.Fabbri, A., L. Falzano, C. Frank, G. Donelli, P. Matarrese, F. Raimondi, A. Fasano, and C. Fiorentini. 1999. Vibrio parahaemolyticus thermostable direct hemolysin modulates cytoskeletal organization and calcium homeostasis in intestinal cultured cells. Infect. Immun. 67:1139-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukasawa, M., M. Nishijima, and K. Hanada. 1999. Genetic evidence for ATP-dependent endoplasmic reticulum-to-Golgi apparatus trafficking of ceramide for sphingomyelin synthesis in Chinese hamster ovary cells. J. Cell Biol. 144:673-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukasawa, M., M. Nishijima, H. Itabe, T. Takano, and K. Hanada. 2000. Reduction of sphingomyelin level without accumulation of ceramide in Chinese hamster ovary cells affects detergent-resistant membrane domains and enhances cellular cholesterol efflux to methyl-β-cyclodextrin. J. Biol. Chem. 275:34028-34034. [DOI] [PubMed] [Google Scholar]
- 8.Gabel, B. R., C. Elwell, S. C. D. van Ijzendoorn, and J. N. Engel. 2004. Lipid raft-mediated entry is not required for Chlamydia trachomatis infection of cultured epithelial cells. Infect. Immun. 72:7367-7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurcel, L., L. Abrami, S. Girardin, J. Tschopp, and F. G. van der Goot. 2006. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126:1135-1145. [DOI] [PubMed] [Google Scholar]
- 10.Hamada, D., T. Higurashi, K. Mayanagi, T. Miyata, T. Fukui, T. Iida, T. Honda, and I. Yanagihara. 2007. Tetrameric structure of thermostable direct hemolysin from Vibrio parahaemolyticus revealed by ultracentrifugation, small-angle X-ray scattering and electron microscopy. J. Mol. Biol. 365:187-195. [DOI] [PubMed] [Google Scholar]
- 11.Hanada, K., T. Hara, M. Fukasawa, A. Yamaji, M. Umeda, and M. Nishijima. 1998. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 273:33787-33794. [DOI] [PubMed] [Google Scholar]
- 12.Hanada, K., K. Kumagai, S. Yasuda, Y. Miura, M. Kawano, M. Fukasawa, and M. Nishijima. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426:803-809. [DOI] [PubMed] [Google Scholar]
- 13.Hancock, J. F. 2006. Lipid rafts: contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol. 7:456-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda, T., Y. Ni, T. Miwatani, T. Adachi, and J. Kim. 1992. The thermostable direct hemolysin of Vibrio parahaemolyticus is a pore-forming toxin. Can. J. Microbiol. 38:1175-1180. [DOI] [PubMed] [Google Scholar]
- 15.Honda, T., and T. Iida. 1993. The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct hemolysin and related hemolysins. Rev. Med. Microbiol. 4:106-113. [Google Scholar]
- 16.Hooper, N. M. 1999. Detergent-insoluble glycosphingolipid/cholesterol-rich microdomains, lipid rafts and caveolae. Mol. Membr. Biol. 16:145-156. [DOI] [PubMed] [Google Scholar]
- 17.Idone, V., C. Tam, J. W. Goss, D. Toomre, M. Pypaert, and N. W. Andrews. 2008. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180:905-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igarashi, K., K. Asai, M. Kaneda, M. Umeda, and K. Inoue. 1995. Specific binding of a synthetic peptide derived from an antibody complementarity determining region to phosphatidylserine. J. Biochem. (Tokyo) 117:452-457. [DOI] [PubMed] [Google Scholar]
- 19.Ikonen, E. 2001. Roles of lipid rafts in membrane transport. Curr. Opin. Cell Biol. 13:470-477. [DOI] [PubMed] [Google Scholar]
- 20.Ishitsuka, R., A. Yamaji-Hasegasa, A. Makino, Y. Hirabayashi, and T. Kobayashi. 2004. A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys. J. 86:296-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishitsuka, R., S. B. Saitoh, and T. Kobayashi. 2005. Imaging lipid rafts. J. Biochem. (Tokyo) 137:249-257. [DOI] [PubMed] [Google Scholar]
- 22.Janda, J. M., C. Powers, R. G. Bryant, and S. L. Abbott. 1988. Current perspectives on the epidemiology and pathogenesis of clinically significant Vibrio spp. Clin. Microbiol. Rev. 1:245-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiyokawa, E., T. Baba, N. Otsuka, A. Makino, S. Ohno, and T. Kobayashi. 2005. Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 280:24072-24084. [DOI] [PubMed] [Google Scholar]
- 24.Kodama, T., M. Rokuda, K. S. Park, V. V. Cantarelli, S. Matsuda, T. Iida, and T. Honda. 2007. Identification and characterization of VopT, a novel ADP-ribosyltransferase effector protein secreted via the Vibrio parahaemolyticus type III secretion system 2. Cell. Microbiol. 9:2598-2609. [DOI] [PubMed] [Google Scholar]
- 25.Lafont, F., L. Abrami, and F. G. van der Goot. 2004. Bacterial subversion of lipid rafts. Curr. Opin. Microbiol. 7:4-10. [DOI] [PubMed] [Google Scholar]
- 26.Lai, E. C. 2003. Lipid rafts make for slippery platforms. J. Cell Biol. 162:365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang, P. A., S. Kaiser, S. Myssina, C. Birka, C. Weinstock, H. Northoff, T. Wieder, F. Lang, and S. M. Huber. 2004. Effect of Vibrio parahaemolyticus haemolysin on human erythrocytes. Cell. Microbiol. 6:391-400. [DOI] [PubMed] [Google Scholar]
- 28.Lichtenberg, D., F. M. Goñi, and H. Heerklotz. 2005. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem. Sci. 30:430-436. [DOI] [PubMed] [Google Scholar]
- 29.London, E., and D. A. Brown. 2000. Insolubility of lipids in Triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta 1508:182-195. [DOI] [PubMed] [Google Scholar]
- 30.Mañes, S., G. del Real, and C. Martinez-A. 2003. Pathogens: raft hijackers. Nat. Rev. Immunol. 3:557-568. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto, Y., T. Kato, Y. Obara, S. Akiyama, K. Takizawa, and S. Yamai. 1969. In vitro hemolytic characteristic of Vibrio parahaemolyticus: its close correlation with human pathogenicity. J. Bacteriol. 100:1147-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyamoto, Y., Y. Obara, T. Nikkawa, S. Yamai, T. Kato, Y. Yamada, and M. Ohashi. 1980. Simplified purification and biophysicochemical characteristics of Kanagawa phenomenon-associated hemolysin of Vibrio parahaemolyticus. Infect. Immun. 28:567-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naim, R., I. Yanagihara, T. Iida, and T. Honda. 2001. Vibrio parahaemolyticus thermostable direct hemolysin can induce an apoptotic cell death in Rat-1 cells from inside and outside of the cells. FEMS Micriobiol. Lett. 195:237-244. [DOI] [PubMed] [Google Scholar]
- 34.Naim, R., T. Iida, A. Takahashi, and T. Honda. 2001. Monodansylcadaverine inhibits cytotoxicity of Vibrio parahaemolyticus thermostable direct hemolysin on cultured rat embryonic fibroblast cells. FEMS Micriobiol. Lett. 196:99-105. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura, K., and S. Handa. 1984. Coomassie brilliant blue staining of lipids on thin-layer plates. Anal. Biochem. 142:406-410. [DOI] [PubMed] [Google Scholar]
- 36.Nishibuchi, M., A. Fasano, R. G. Russell, and J. B. Kaper. 1992. Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect. Immun. 60:3539-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishibuchi, M., and J. B. Kaper. 1995. Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect. Immun. 63:2093-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park, K. S., T. Ono, M. Rokuda, M. H. Jang, T. Iida, and T. Honda. 2004. Cytotoxicity and enterotoxicity of the thermostable direct hemolysin-deletion mutants of Vibrio parahaemolyticus. Microbiol. Immunol. 48:313-318. [DOI] [PubMed] [Google Scholar]
- 39.Raimondi, F., J. P. Kao, C. Fiorentini, A. Fabbri, G. Donelli, N. Gasparini, A. Rubino, and A. Fasano. 2000. Enterotoxicity and cytotoxicity of Vibrio parahaemolyticus thermostable direct hemolysin in in vitro systems. Infect. Immun. 68:3180-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberger, C. M., J. H. Brumell, and B. B. Finlay. 2000. Microbial pathogenesis: lipid rafts as pathogen portals. Curr. Biol. 10:R823-R825. [DOI] [PubMed] [Google Scholar]
- 41.Sakazaki, T., K. Tamura, T. Kato, Y. Obata, and S. Yamai. 1968. Studies on the enteropathogenic, facultatively halophilic bacterium, Vibrio parahaemolyticus. III. Enteropathogenicity. Jpn. J. Med. Sci. Biol. 21:325-331. [DOI] [PubMed] [Google Scholar]
- 42.Schraw, W., Y. Li, M. S. McClain, F. G. van der Goot, and T. L. Cover. 2002. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 277:34642-34650. [DOI] [PubMed] [Google Scholar]
- 43.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature 387:569-572. [DOI] [PubMed] [Google Scholar]
- 44.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31-39. [DOI] [PubMed] [Google Scholar]
- 45.Takeda, Y., T. Takeda, T. Honda, J. Sakurai, N. Ohtomo, and T. Miwatani. 1975. Inhibition of hemolytic activity of the thermostable direct hemolysin of Vibrio parahaemolyticus by ganglioside. Infect. Immun. 12:931-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda, Y., T. Takeda, T. Honda, and T. Miwatani. 1976. Inactivation of the biological activities of the thermostable direct hemolysin of Vibrio parahaemolyticus by ganglioside GT1. Infect. Immun. 14:1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang, G. Q., T. Iida, K. Yamamoto, and T. Honda. 1994. A mutant toxin of Vibrio parahaemolyticus thermostable direct hemolysin which has lost hemolytic activity but retains ability to bind to erythrocytes. Infect. Immun. 62:3299-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang, G. Q., T. Iida, K. Yamamoto, and T. Honda. 1995. Ca(2+)-independent cytotoxicity of Vibrio parahaemolyticus thermostable direct hemolysin (TDH) on Intestine 407, a cell line derived from human embryonic intestine. FEMS Microbiol. Lett. 134:233-238. [DOI] [PubMed] [Google Scholar]
- 49.Tang, G. Q., T. Iida, H. Inoue, M. Yutsudo, K. Yamamoto, and T. Honda. 1997. A mutant cell line resistant to Vibrio parahaemolyticus thermostable direct hemolysin (TDH): its potential in identification of putative receptor for TDH. Biochim. Biophys. Acta 1360:277-282. [DOI] [PubMed] [Google Scholar]
- 50.Tang, G., T. Iida, K. Yamamoto, and T. Honda. 1997. Analysis of functional domains of Vibrio parahaemolyticus thermostable direct hemolysin using monoclonal antibodies. FEMS Microbiol. Lett. 150:289-296. [DOI] [PubMed] [Google Scholar]
- 51.Tweten, R. K. 2005. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Imuun. 73:6199-6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walev, I., S. C. Bhakdi, F. Hofmann, N. Djonder, A. Valeva, K. Aktories, and S. Bhakdi. 2000. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. U. S. A. 98:3185-3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamaji, A., Y. Sekizawa, K. Emoto, H. Sakuraba, K. Inoue, H. Kobayashi, and M. Umeda. 1998. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 273:5300-5306. [DOI] [PubMed] [Google Scholar]
- 54.Yoh, M., T. Honda, and T. Miwatani. 1988. Comparison of hemolysins of Vibrio cholerae non-O1 and Vibrio hollisae with thermostable direct hemolysin of Vibrio parahaemolyticus. Can. J. Microbiol. 34:1321-1324. [DOI] [PubMed] [Google Scholar]
- 55.Yoh, M., N. Morinaga, M. Noda, and T. Honda. 1995. The binding of Vibrio parahaemolyticus 125I-labeled thermostable direct hemolysin to erythrocytes. Toxicon 33:651-657. [DOI] [PubMed] [Google Scholar]