

Abstract
We previously showed that cytotoxic necrotizing factor 1 (CNF1) contributes to Escherichia coli K1 invasion of human brain microvascular endothelial cells (HBMEC) and interacts with the receptor on the surface of HBMEC. CNF1 is the cytoplasmic protein, and it remains incompletely understood how CNF1 is secreted across the inner and outer membranes in E. coli K1. In order to investigate the genetic determinants for secretion of CNF1 in E. coli K1, we performed Tn5 mutagenesis screening by applying β-lactamase as a reporter to monitor secretion of CNF1. We identified a Tn5 mutant that exhibited no β-lactamase activity in the culture supernatant and in which the mutated gene encodes a ferredoxin gene (fdx). In the fdx deletion mutant, there was no evidence of translocation of CNF1 into HBMEC. Western blot analysis of the fdx deletion mutant revealed that ferredoxin is involved in translocation of CNF1 across the cytoplasmic membrane. The fdx mutant exhibited significantly decreased invasion of HBMEC, similar to the decreased HBMEC invasion observed with the CNF1 mutant. The failures to secrete CNF1 and invade HBMEC of the fdx mutant were restored to the levels of the parent strain by complementation with fdx. These findings demonstrate for the first time that ferredoxin is involved in secretion of CNF1 across the inner membrane in meningitis-causing E. coli K1.
Neonatal Escherichia coli meningitis is associated with high mortality and morbidity, and a major contributing factor is our incomplete knowledge on the pathogenesis of E. coli meningitis (15, 16). Most cases of neonatal E. coli meningitis develop as a result of hematogenous spread (8, 14), but it is incompletely understood how circulating bacteria cross the blood-brain barrier and cause meningitis.
We have shown that cytotoxic necrotizing factor 1 (CNF1) contributes to E. coli K1 invasion of human brain microvascular endothelial cells (HBMEC) and penetration into the central nerve system (CNS) via the interaction with its receptor, 37 laminin receptor precursor (37LRP)/67 laminin receptor (67LR) (4, 12, 13). CNF1 is a cytoplasmic protein, and its secretion is a strategy utilized by meningitis-causing E. coli K1 to invade the blood-brain barrier (12). CNF1 is the paradigm of the RhoGTPase-activating bacterial toxins (2, 19). The CNF1 secretion pathway, however, remains incompletely understood. No typical signal peptide is found in the CNF1 sequence. A previous study by Kouokam et al. showed that CNF1 is tightly associated with outer membrane vesicles (18). Outer membrane vesicles from a number of bacterial species have been found to contain virulence factors, exhibit immunomodulatory effects, and adhere to and intoxicate host cells (20).
In order to study the genetic determinants for secretion of CNF1 in meningitis-causing E. coli K1, we designed a Tn5 mutational screening strategy by applying TEM β-lactamase as the reporter. Using this approach, we identified a mutant which was defective in CNF1 secretion into HBMEC, and this mutant is characterized in this report.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids are shown in Table 1. E. coli K1 strain RS218 (O18:K1:H7) is a cerebrospinal fluid isolate from a neonate with meningitis (12). E. coli K-12 strain DH5α was used as the host for plasmids, and EC100D pir116+ (Epicentre Biotechnologies, Madison, WI) was the host for the R6kγ origin plasmid. E. coli strains were routinely grown at 37°C in Luria broth. Where appropriate, the medium was supplemented with ampicillin (100 μg/ml), spectinomycin (100 μg/ml), tetracycline (10 μg/ml), or chloramphenicol (20 μg/ml).
TABLE 1.
Strains and plasmids used in the current study
| Strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| E. coli strains | ||
| RS218 | O18:K1:H7, isolated from cerebrospinal fluid of neonate with E. coli meningitis | 12 |
| EC100D | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ−rpsL nupG pir+ (DHFR) | Epicentre Biotechnologies |
| DH5α | F′ φ80dlacZ ΔM15Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+)phoA supE44 λ−thi-1 | Lab stock |
| Δcnf1 mutant | RS218 (O18:K1:H7) cnf1 deletion mutant | 12 |
| Δfdx mutant | RS218 (O18:K1:H7) fdx deletion mutant | This study |
| CΔfdx mutant | RS218 (O18:K1:H7) fdx deletion mutant complemented with fdx under control of arabinose promoter; complementation achieved by Tn7 site-specific insertion into second benign site in chromosome | This study |
| Plasmids | ||
| pBC-KS | Cloning vector with chloramphenicol resistance gene | Stratagene |
| pBAD-Myc/His | Arabinose promoter expression vector, ampicillin resistance | Invitrogen |
| pCX340 | PBR322 derivative, cloning vector used to fuse CNF1 to the mature form of TEM-1 β-lactamase, tetracycline resistance | 3 |
| pCX311 | Negative control, fusion of MBP to TEM-1, tetracycline resistance | 3 |
| pCXN | CNF1 coding region cloned into KpnI site of pCX340, tetracycline resistance | This study |
| pKD3 | Containing chloramphenicol resistance gene, R6kγ replication origin | 6 |
| pKD47 | Derivative of pKD46 (5); the only modification is that blaM in pKD46 is replaced by the spectinomycin resistance gene | This study |
| pGRG36 | Tn7 insertion vector, ampicillin resistance, temperature sensitive | 21 |
| pFBI | Contains β-lactamase coding region (signal peptide genetically deleted); spectinomycin resistance; R6kγ replication origin | This study |
| pFBI-CNF1 | CNF1 translationally fused with β-lactamase in pFBI | This study |
| pNFB | DNA fragment downstream of cnf1 in RS218 genome was PCR amplified and cloned in pFBI-CNF1 | This study |
| pNBC | Chloramphenicol resistance gene was inserted right after β-lactamase in pNFB | This study |
| pSR | Tn5 vector; spectinomycin resistance; R6kγ replication origin | This study |
| pGRGM | Multiple cloning site of pGRG36 ligated into PvuII site of pBC-KS; chloramphenicol resistance | This study |
| pGAP | AraC and arabinose promoter (pBAD) cloned into AvrII and XhoI site of pGRGM | This study |
| pGAP-fdx | Coding region of fdx cloned into NdeI and NotI site of pGAP | This study |
| pG-fdx | DNA fragment containing fdx obtained from pGAP-fdx by digestion with AvrII and PacI; ligated into same sites of pGRG36 | This study |
Construction of CNF1-Bla hybrid in the chromosome of RS218.
To integrate the TEM-1 blaM mature form DNA in frame with CNF1 into strain RS218 genomic DNA, blaM together with its upstream multiple cloning sites was cloned from pCX340 into pRS (a derivative of pSR, and the difference is that in pRS the R6kγ replication origin is upstream of the spectinomycin resistance gene), yielding pFBI (fuse Bla in frame). Then, cnf1 was amplified from RS218 genomic DNA with primers (Cnf1-s3 and Cnf1-a [Table 2]) and cloned into the KpnI site of plasmid pFBI, which gave pFBI-CNF1. After construction of pFBI-CNF1, the cnf1 downstream DNA fragment was amplified from RS218 genomic DNA with primers (NC-a3 and NC-s3 [Table 2]), digested with XbaI and NcoI, and cloned into pFBI-CNF1, and the resulting plasmid was designated as pNFB. The chloramphenicol resistance gene (obtained by digesting plasmid pKD3 with XbaI) (6) was then cloned into the XbaI site of pNFB, yielding pNBC.
TABLE 2.
Primers used in the study
| Primer | Sequence (5′-3′)a |
|---|---|
| NCHKs | CGACCTGTCCTGGTGATGC |
| Cnf1-s3 | GCGCGGTACCATGGGTAACCAATGGCAA |
| Cnf1-a | GGATCCGGTACCAAATTTTTTTGAAATACCTTCA |
| NC-s3 | GGCGTCTAGATTTTGATTCGGGAAATTATT |
| NC-a3 | GGCGCCATGGACTCTGCCCGATGATTTTC |
| NN-s | GTTGAAGTACTGGCTGTGGTT |
| NFB-CKS | GCTACTGAGGAAGAAGCATGGAA |
| NFB-CKA | TCGCAGGTGAGCCGAAACT |
| GRGM-f | TTTCACTTATCTGGTTGGTCG |
| GRGM-r | CGAGGCTTGTCAGTACATCA |
| AraCP-s | CCGGCCTAGGCTGATTCGTTACCAATTATGAC |
| AraCP-a | CCGGCTCGAGCATGGTTAATTCCTCCTGTTA |
| fdx-KOF | TTCGCCAATTTCGCGGCTATCCGTCCACTTAAGTCCCATACTAACCTCTGGTGTAGGCTGGAGCTGCTTC |
| fdx-KOR | CCAGTCGGTTCGTCGTGCGCTGAAAGGCCATTCCGTGGACGAGGTTTAATCATATGAATATCCTCCTTAG |
| FdxCKF | AGGATTTTCTCGTTGGATG |
| FdxCKR | AAGACTCAATGAGCTATGCC |
| fdx-a | CGCGCGGCCGCACTTAAGTCCCATACTAACCTC |
| fdx-s | GCGCGCATATGCCAAAGATTGTTATTTTG |
| Spc-SeqR | GCCTTGCTGTTCTTCTACGG |
| Tn7-ckf | ACGGTCGGGAACTGGAAC |
| Tn7-ckr | TGACCAGCCGCGTAACCT |
Restriction sites for cloning are underlined.
Primers (NN-s and NC-a [Table 2]) were used to amplify the DNA fragment from pNBC, and PCR products were digested with DpnI and gel purified. The PCR products were then electroporated into competent cells of strain RS218 containing pKD47 (a derivative of pKD46, with blaM in pKD46 replaced by spectinomycin resistance gene), allowing recombination to occur in the presence of arabinose. The temperature-sensitive pKD47 was cured by incubation at 37°C with agitation. The integration of blaM after cnf1 into the chromosome of strain RS218 was verified by PCR using primers (NFB-CKF and NFB-CKR [Table 2]), and the resulting strain was designated as strain NBC.
Transposome formation and transposition mutagenesis.
Transposon DNA was released by digesting plasmid pSR (Fig. 1C, modified from the pMini-Tn5 cycler [9]) with PvuII (New England Biolabs, Beverly, MA) and then gel purified (QIAquick gel extraction kit; Qiagen, Valencia, CA). Transposon DNA (Fig. 1C; up to 50 μg/ml) was incubated with 10 μg/ml hyperactive Tn5 transposase (Epicentre Technologies) for 1 h at 37°C in a 20-μl reaction volume. Transposomes (1 μl) were electroporated into competent NBC cells. Transposon insertion mutants were selected with spectinomycin.
FIG. 1.

Identification of the fdx gene as a genetic requirement for CNF1 secretion. (A) Schematic representation of the chromosomal structure of strains RS218 and NBC. In strain NBC, the cnf1-blaM translational chromosomal fusion was made by insertion of the bla gene after the cnf1 gene. (B) Strains RS218, NBC, and transposon mutant NBC-14H2 were grown overnight in brain heart infusion medium (static at 37°C). Bacterial culture supernatant was obtained by centrifugation at 5,000 × g for 10 min, and specific β-lactamase activity was determined based on the absorbance at 486 nm. The represented Bla activity data (means ± standard deviations) represent the results from three experiments in triplicate. (C) Transposon insertion within the fdx gene in the mutant strain NBC-14H2.
β-Lactamase activity assay.
Bacteria were grown in 96-well plates at 37°C overnight without agitation and then centrifuged at 3,200 rpm for 10 min, and 95 μl of supernatant from each clone was added to 5 μl of nitrocefin stock solution (Calbiochem, Gibbstown, NJ), which was incubated at room temperature for up to 24 h to allow the red color to develop. Nitrocefin is a chromogenic β-lactamase substrate that undergoes a distinctive color change from yellow (λmax, 390 nm at pH 7.0) to red (λmax, 486 nm at pH 7.0) as the amide bond in the β-lactam ring is hydrolyzed by β-lactamase. Nitrocefin stock solution (2 mM; Calbiochem) was prepared by dissolving 10.3 mg of nitrocefin in 0.5 ml of dimethyl sulfoxide and then adding 25 mM HEPES buffer (pH 7.3) to a final volume of 10 ml. Bla activity was read as positive if the color change to red occurred. Spectrophotometric assays for Bla using nitrocefin were also carried out by measuring changes in absorbance at 486 nm.
Genomic DNA isolation and sequencing.
Genomic DNA was isolated from individual Tn5 mutants as described previously (25). Chromosomal DNA was quantified with the a Quant-iT dsDNA BR assay kit (Invitrogen, Carlsbad, CA). Twelve microliters of genomic DNA (0.5 μg/μl) and 12 μl of sequencing primer SR-Seq (8 μM) were sent to the DNA Synthesis and Sequencing Facility (Johns Hopkins University School of Medicine) for sequencing.
fdx gene deletion and complementation.
To delete the fdx gene, a chloramphenicol resistance cassette was amplified from pKD3 (6) using primers fdx-KOF and fdx-KOR (Table 2). The PCR product was inserted into the chromosome by Lambda Red-mediated allele replacement (6). The correct insertion was verified by PCR.
For gene complementation, we applied Tn7 site-specific gene insertion into the second benign site in the chromosome of the mutant as described previously (21). Since the fdx gene is within the isc operon and does not have its own promoter, we used an arabinose promoter to initiate the transcription of the fdx gene. We amplified the multiple cloning site of pGRG36 (21) into the PvuII site of pBC-KS (the primers used for this purpose were GRGM-f and GRGM-r [Table 2]), yielding plasmid pGRGM. Next, araC and the arabinose promoter were obtained by PCR amplification from plasmid pBAD/Myc-His (Invitrogen) with primers AraPs and AraPa (Table 2) and then ligated into AvrII and XhoI sites of pGRGM, and the resulting plasmid was designated as pGAP. The coding region of the fdx gene was amplified from the genomic DNA of strain RS218 with primers fdx-s and fdx-a (Table 2) and ligated into the NdeI and NotI site of pGAP, yielding plasmid pGAP-fdx. The DNA fragment containing araC, the arabinose promoter, and the fdx gene was obtained by digesting pGAP-fdx with restriction enzymes AvrII and PacI and was subsequently ligated into the same sites of pGRG36. Finally, the ligation product was electroporated into the fdx mutant, and transformants were selected on LB plates containing ampicillin at 32°C. The transformant was streaked once on an LB plate containing ampicillin to ensure that the bacteria carried the plasmid and then grown without antibiotic selection in LB at 32°C, and 0.1% arabinose was added to induce expression of TnsABCD (Tn7 transposition machinery). Subsequently, the transformants were incubated at 42°C to prevent replication of the plasmid, and the insertion of Tn7 in the attachment site was verified by PCR with primers Tn7-ckf and Tn7-ckr (Table 2).
Assessment of CNF1 translocation into HBMEC.
HBMEC were cultured in clear-bottom 96-well plates (Becton Dickinson, Franklin Lakes, NJ) at 20,000 cells per well in experimental medium (M199-Ham F-12 [1:1] containing 5% fetal bovine serum, 2 mM glutamine, and 1 mM pyruvate) and incubated at 37°C in 5% CO2. Bacterial strains were grown overnight in brain heart infusion broth at 37°C, and the expression of the CNF1-Bla fusion from pCXN was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). On the day of infection, HBMEC were preloaded with CCF4/AM dye (final concentration, 1 μM; Invitrogen) as described previously (3, 23) and incubated with bacteria. The nonfluorescent esterified CCF4/AM substrate, upon entry into HBMEC, is rapidly converted to fluorescent green CCF4 by cellular esterases. Translocation of CNF1-Bla induces catalytic cleavage of the CCF4 β-lactam ring, which produces an easily detectable change in CCF4 fluorescence from green to blue emission (3, 28). After 45 min of infection, the translocation of the CNF1-Bla hybrid into HBMEC was observed under a Nikon fluorescence microscope.
Cell fractionation.
Cytoplasmic and periplasmic fractions were obtained by the method of osmotic shock as described previously (27). Periplasmic suspension was filtered through a 0.22-μm filter to remove any residual bacterial cells and then precipitated by the Na-deoxycholate-trichloroacetic acid method (5).
Western blot assays.
Cell fractions were separated by SDS-PAGE, and then protein samples were transferred to a nitrocellulose membrane. The blots were blocked with 5% skim milk in Tris-buffered saline (TBS; 25 mM Tris, pH 7.4, 150 mM NaCl) for 60 min at 22°C. The membrane was incubated for 2 h at 22°C with primary antibody. Primary antibodies used in this study were anti-CNF1 monoclonal antibody (DD1) (22), alkaline phosphatase (PhoA) monoclonal antibody (Millipore), and β-galactosidase (β-Gal) antiserum (Millipore), and disulfide oxidoreductase (DsbA) antiserum was also used (1). The membrane was washed with 0.5% Tween 20 in TBS and subsequently incubated for 60 min at room temperature with horseradish peroxidase-linked secondary antibodies. The membrane was developed with an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech). The E. coli proteins located in the cytoplasm (e.g., β-Gal protein) and periplasm (e.g., PhoA and DsbA) were used as markers for cytoplasmic and periplasmic fractions, respectively.
E. coli invasion assays in HBMEC.
E. coli invasion assays were performed in HBMEC as previously described (13). Briefly, confluent cultures of HBMEC (grown in 24-well plates) were incubated with 107 CFU of E. coli (multiplicity of infection of 100) in experimental medium. Plates were incubated for 90 min at 37°C in 5% CO2 to allow invasion to occur. The number of intracellular bacteria was determined by culturing on blood agar plates after extracellular bacteria were killed by incubation of the HBMEC monolayers with experimental medium containing gentamicin (100 μg/ml) for 1 h. Assays were performed in triplicate and repeated at least three times. Results are expressed as relative invasion frequencies (percent invasion compared to that of the parent strain, RS218).
RESULTS
Screening of genetic determinants for secretion of CNF1.
To investigate the secretion of CNF1 in meningitis-causing E. coli K1, we applied β-lactamase (Bla) as the reporter gene, which was translationally fused to the C terminal of the cnf1 gene in the chromosome of strain RS218. The resulting strain was designated as strain NBC (CNF1-Bla-CAT) (Fig. 1A). In the NBC strain, Bla's secretion is entirely dependent on CNF1's secretion machinery. We were able to visualize the color change of nitrocefin in the culture supernatant of NBC (yellow to red), which was induced by Bla activity, compared to no detectable color change with the parent strain, RS218. These findings demonstrated that CNF1 secretion can be detected by Bla activity in strain NBC, and strain NBC is suitable for screening of genes involved in CNF1 secretion in E. coli K1 strain RS218.
We next performed mini-Tn5 in vitro mutagenesis and constructed a mutant library of strain NBC. Those mutants with the transposon being inserted within the cnf1-bla coding region or the promoter region of cnf1 were excluded by PCR (with primers NCHKs and NC-a [Table 2]). For β-lactamase assays, the NBC strain was used as a positive control, while the wild-type strain RS218 was used as a negative control. We identified a mutant (NBC-14H2) that exhibited negative Bla activity based on visual color change and the spectrometric reading, and the characterization of this mutant is the purpose of this report.
Ferredoxin is essential for CNF1 secretion.
The transposon mutant was significantly defective in secretion of CNF1 into the culture medium, based on the β-lactamase activity (Fig. 1B). We determined the location of the transposon insertion by direct DNA sequencing of the mutant's genomic DNA. The insertion was shown to occur within the fdx gene, encoding ferredoxin, which is located within the isc operon, downstream of the hscA and hscB genes (Fig. 1C). Genes located within isc operon have been shown to be involved in iron sulfur protein assembly and may operate in vivo as a complex (26). However, the function of ferredoxin has not been biochemically determined.
We deleted the fdx gene from the RS218 genome. The growth rates of the fdx mutant were similar to those of the parent strain, RS218, and both strains had similar numbers of bacteria after 24 h of incubation (Fig. 2). We subsequently analyzed its capability for translocating CNF1 into HBMEC by using a CNF1-Bla fusion protein expressed from the plasmid pCXN. E. coli transformants harboring pCXN were preinduced with 1 mM IPTG and then added to HBMEC preloaded with CCF4/AM dye. After 45 min of incubation, the CNF1 translocation into HBMEC was visualized under fluorescence microscopy. As expected, the wild-type strain successfully translocated CNF1-Bla hybrid protein into HBMEC, as shown by the emission of blue fluorescence, while the fdx mutant failed to do so (Fig. 3). The failure to translocate the CNF1-Bla fusion in the fdx mutant was restored in the complemented strain CΔfdx (Fig. 3).
FIG. 2.
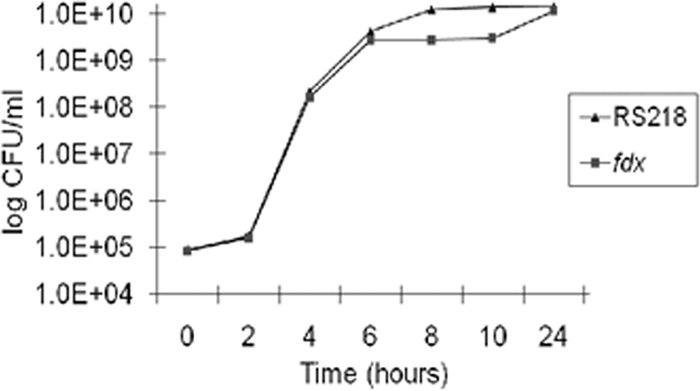
Growth curves of the parent strain RS218 and its fdx mutant in brain heart infusion (BHI) medium. Overnight cultures of RS218 and the fdx mutant were diluted 1:10,000 into fresh BHI medium, and bacteria were cultured at 37°C with shaking for up to 24 h. The CFU were determined at different time intervals as indicated in the figure. The experiment was repeated three times.
FIG. 3.

Analysis of translocation of CNF1 into HBMEC. HBMEC were preloaded with the BlaM substrate CCF4/AM and then infected with E. coli strains bearing different plasmids as indicated on the left. Plasmid pCX311 expresses a maltose-binding protein-Bla fusion and was used as the negative control, and pCXN expresses a CNF1-Bla fusion. RS218 is the wild-type strain, Δfdx is a fdx deletion mutant, and CΔfdx is a complemented strain of the fdx deletion mutant with fdx. For strain CΔfdx, 0.1% arabinose was added to promote the transcription of the complemented fdx gene. The software ImageJ was used to merge the green and blue channels (24).
Bacterial lysates of the Δfdx/pCXN construct containing the CNF1-Bla fusion protein, induced by 1 mM IPTG, however, was capable of translocation into HBMEC, as shown in a CCF4/AM assay (data not shown). These findings demonstrate that the CNF1-Bla fusion expressed in strain Δfdx is functionally active, and the failure to translocate CNF1 into HBMEC in the fdx mutant is due to the lack of its secretion.
Ferredoxin is required for secretion of CNF1 at the step of crossing the cytoplasmic membrane.
We next examined the secretion of CNF1 across the cytoplasmic membrane by comparing the location of CNF1 expression in cytoplasmic and periplasmic fractions derived from the parent strain, RS218, and its fdx mutant. As shown in Fig. 4A, the cytoplasmic fraction contained β-Gal protein but was devoid of periplasmic proteins (PhoA and DsbA). In contrast, the periplasmic fraction contained PhoA and DsbA but was devoid of β-Gal, suggesting that our cell fractions exhibited the expected protein profiles. As shown by Western blotting (Fig. 4A), the presence of CNF1 in both the cytoplasmic and periplasmic fractions was demonstrated in the parent strain, RS218. In contrast, CNF1 expression was evident in the cytoplasm but was not detectable in the periplasmic fraction in the fdx mutant (Fig. 4A). The absence of CNF1 in the periplasmic fraction of the fdx mutant was, however, restored by complementation of the fdx mutant with the fdx gene (strain CΔfdx). These findings suggest that CNF1 secretion was blocked at the step of crossing the inner membrane in the Δfdx mutant.
FIG. 4.
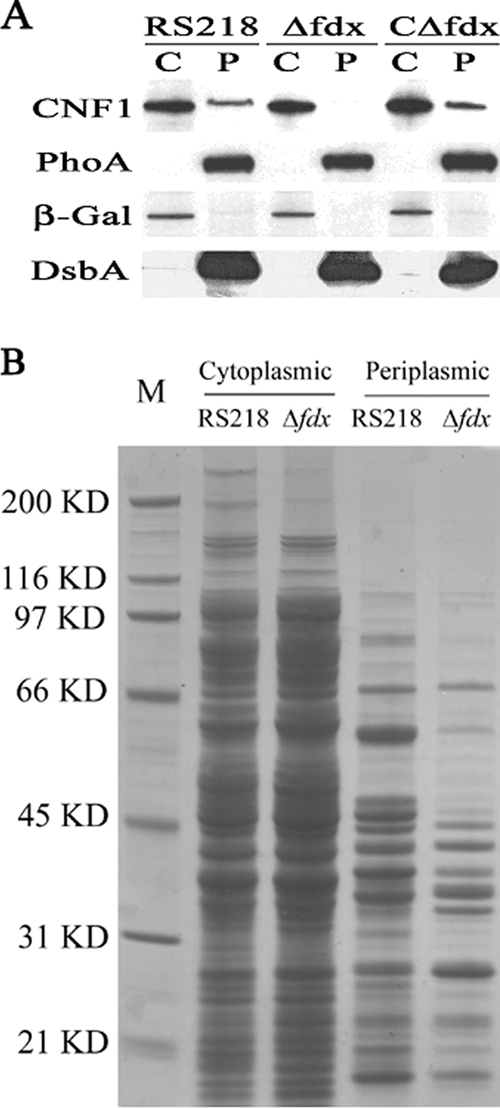
(A) CNF1 subcellular localization and secretion in E. coli RS218. Western blot analysis was carried out with the cytoplasmic (C) and periplasmic (P) fractions from RS218, Δfdx, and CΔfdx (with 0.1% arabinose to promote the transcription of complemented fdx gene). The amount of cytoplasmic protein loaded was 40 μg, and the amount of periplasmic protein loaded was equal to the total periplasmic protein that was collected from 3 ×109 bacteria (the number of bacteria was estimated from the optical density at 620 nm [OD620]). CNF1, PhoA, β-Gal, and DsbA were detected by their respective specific antibodies, as described in Materials and Methods. (B) SDS-PAGE analysis of the protein profile in the cytoplasmic and periplasmic fractions prepared from RS218 and Δfdx as indicated in the figure. M, molecular marker; size positions are indicated along with the masses on the left. The amount of cytoplasmic protein loaded was 15 μg. The loaded periplasmic protein was equal to the total periplasmic protein that was collected from 1010 bacteria cells (the number of bacteria was estimated from the OD620).
To determine whether or not the failure to secrete CNF1 across the inner membrane is unique to CNF1 in the fdx mutant, we examined and compared the patterns of cytoplasmic and periplasmic proteins between strain RS218 and its fdx mutant. SDS-PAGE and Coomassie blue staining revealed that the fdx mutant exhibited somewhat different patterns of the periplasmic proteins compared with those of RS218, while the patterns of the cytoplasmic proteins were similar between strain RS218 and the fdx mutant (Fig. 4B). Taken together, these findings suggest that ferredoxin may be involved in the secretion of CNF1 and several other proteins across the inner membrane in E. coli K1.
Ferredoxin promotes E. coli K1 invasion of HBMEC.
We previously showed that CNF1 contributes to E. coli K1 invasion of HBMEC (12). Because CNF1 secretion was impaired in the fdx mutant, our next experiment was to examine the HBMEC invasion abilities of the fdx mutant and its complemented strain, compared to those of the wild-type strain, RS218, and the CNF1 mutant. The in vitro HBMEC assays revealed that the fdx mutant was significantly defective in invasion of HBMEC compared to the parent strain (Fig. 5), while the invasion frequency was restored to the level of the parent strain by complementation with fdx. To initiate the transcription of the fdx gene in the complemented strain CΔfdx, 0.1% arabinose was added to both brain heart infusion medium and HBMEC invasion assay medium, but arabinose did not affect HBMEC invasion frequencies except for strain CΔfdx. As expected, the CNF1 mutant was significantly defective in invasion of HBMEC, which is consistent with our previous data (12).
FIG. 5.
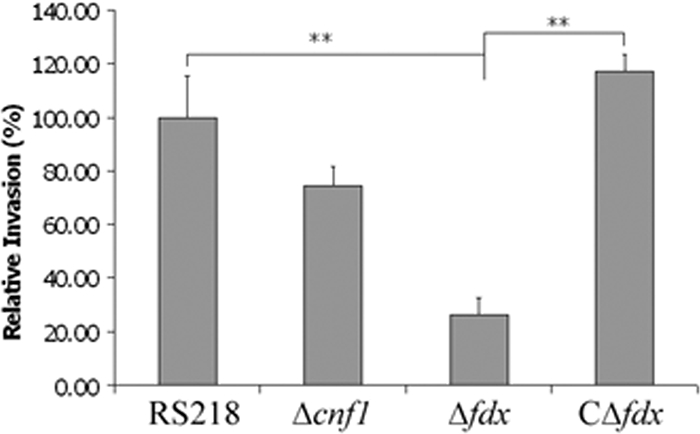
The fdx deletion mutant of E. coli strain RS218 exhibits significantly decreased invasion of HBMEC. To determine whether ferredoxin plays a role in E. coli invasion of HBMEC, invasion assays were performed using the fdx deletion mutant and the complemented strain CΔfdx. In strain CΔfdx, the fdx gene is under the control of the pBAD promoter. **, P < 0.01. The data (means ± standard deviations) represent assay results with 0.1% arabinose.
DISCUSSION
We previously showed that CNF1, a cytoplasmic protein, contributes to E. coli K1 invasion of HBMEC and penetration into the CNS, and we identified the HBMEC receptor for CNF1, 37LRP/67LR (4, 12, 13), but it remains incompletely understood how CNF1 is transported across the inner and outer membranes in E. coli K1. Recent studies have shown that CNF1 from uropathogenic E. coli strains J96 and CP9 is transported to the culture supernatant in a complex with outer membrane vesicles (7, 18).
In order to investigate the secretion and/or translocation of CNF1 from the cytoplasm of E. coli K1, we constructed a recombinant E. coli strain fused with β-lactamase in the C terminus of the cnf1 gene in the chromosome of RS218 (strain NBC). In the NBC strain, Bla's secretion is entirely dependent upon CNF1's secretion machinery, and we initially used Bla activity to monitor CNF1 secretion from the cytoplasm. From screening of the Tn5 library of strain NBC, we identified the mutant that exhibited no detectable Bla activity in the culture supernatant and did not have the transposon inserted into the cnf1-bla coding region or the promoter region of cnf1.
We subsequently identified that the transposon insertion occurred within the fdx gene, and a CNF1 translocation assay with the fdx deletion mutant demonstrated that ferredoxin is essential for CNF1 secretion into HBMEC. Moreover, we showed that ferredoxin was involved in secretion of CNF1 across the inner membrane of strain RS218. This was shown by our demonstrations that (i) deletion of fdx resulted in a failure to detect the presence of CNF1 in the periplasmic fraction of strain RS218, while the known periplasmic proteins, PhoA and DsbA, were present in the periplasmic fraction, and (ii) complementation of the fdx mutant with fdx restored the secretion of CNF1 in the periplasmic fraction. These findings demonstrate that ferredoxin affects the secretion of CNF1 across the inner membrane in E. coli K1 strain RS218.
More importantly, the fdx deletion mutant exhibited significantly decreased invasion of HBMEC compared to the parent strain RS218, and this invasion defect was restored to the level of the parent strain by complementation with fdx. CNF1 has been reported to contribute to E. coli K1 invasion of HBMEC, as shown by significantly decreased HBMEC invasion of the CNF1 mutant compared to the parent strain RS218 (4, 12, 13), and it is likely that the HBMEC invasion defect of the fdx mutant is related to the failure to secrete CNF1. Of interest, the decreased invasion frequency was significantly greater with the fdx mutant than with the CNF1 mutant, e.g., 26% versus 74% relative invasion frequency, respectively, compared to the invasion frequency of the parent strain RS218, suggesting that ferredoxin may affect secretion of CNF1 as well as other E. coli K1 determinants involved in invasion of HBMEC. Our comparison of periplasmic protein profiles showed different patterns of proteins between the parent strain and the fdx mutant, and it is tempting to speculate that fdx may also involve secretion of other E. coli K1 determinants involved in HBMEC invasion. Additional studies are needed to clarify this issue.
Ferredoxins are small iron-sulfur proteins that mediate electron transfer and have either a [4Fe-4S], [3Fe-3S], or [2Fe-2S] clusterwhose reduction potential is highly negative (−300 mV or lower) (10, 11). E. coli ferredoxin is an adrenodoxin-type [2Fe-2S] ferredoxin, and it does not mediate electron transport in the NADP photoreduction system of spinach and is incapable of replacing the Pseudomonas putida ferredoxin in camphor hydroxylation (10, 17). The genetic localization of the fdx gene suggests that it may be involved in biogenesis of Fe-S proteins (26). However, the exact physiological role of E. coli ferredoxin has not yet been genetically or biochemically determined. The electronic versatility of Fe-S clusters make it possible that ferredoxin may be involved in transmitting energy that is required for CNF1 secretion. There might also be other yet-undetermined mechanisms for the involvement of ferredoxin in CNF1 secretion.
In summary, we have demonstrated for the first time that ferredoxin affects the secretion of CNF1 across the inner membrane in meningitis-causing E. coli K1, and the fdx mutant was defective in secretion of CNF1 into HBMEC as well as invasion of HBMEC. Studies are needed to understand how ferredoxin affects the CNF1 secretion and also whether ferredoxin affects secretion of other virulence factors that are involved in E. coli K1 invasion of HBMEC.
Acknowledgments
This work was supported in part by an NIH grant (NS26310-22).
We thank Alison O'Brien for providing the CNF1 monoclonal antibody, K. Ito for providing DsbA antiserum, Nancy L. Craig for providing the pGRG36 plasmid, Fred Heffron for providing the plasmid pMini-Tn5 cycler, and Eric Oswald for providing plasmids pCX311 and pCX340. We also thank George Niemann and Bo Ma for suggestions and discussions.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 16 November 2009.
REFERENCES
- 1.Akiyama, Y., S. Kamitani, N. Kusukawa, and K. Ito. 1992. In vitro catalysis of oxidative folding of disulfide-donded proteins by the Escherichia coli dsbA (ppfA) gene product. J. Biol. Chem. 267:22440-22445. [PubMed] [Google Scholar]
- 2.Boquet, P. 2001. The cytotoxic necrotizing factor 1 (CNF1) from Esherichia coli. Toxicon 39:1673-1680. [DOI] [PubMed] [Google Scholar]
- 3.Charpentier, X., and E. Oswald. 2004. Identification of the secretion and translocation domain of the Enteropathogenic and Enterohemorrhagic Escherichia coli effector Cif, using TEM-1 β-lactamase as a new fluorescence-based reporter. J. Bacteriol. 186:5486-5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung, J. W., S. J. Hong, K. J. Kim, D. Goti, M. F. Stins, S. Shin, V. L. Dawson, T. M. Dawson, and K. S. Kim. 2003. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 278:16857-16862. [DOI] [PubMed] [Google Scholar]
- 5.Cold Spring Harb. Protoc. 2006. doi: 10.1101/pdb.prot4258. [DOI]
- 6.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis, J. M., H. M. Carvalho, S. B. Rasmussen, and A. D. O'Brien. 2006. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 74:4401-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietzman, D. E., G. W. Fischer, and F. D. Schoenknecht. 1974. Neonatal Escherichia coli septicemia: bacterial counts in blood. J. Pediatr. 85:128-130. [DOI] [PubMed] [Google Scholar]
- 9.Geddes, K., M. Worley, G. Niemann, and F. Heffron. 2005. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect. Immun. 73:6260-6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holden, H. M., B. L. Jacobson, J. K. Hurley, G. Tollin, B. H. Oh, L. Skjeldal, Y. K. Chae, H. Cheng, B. Xia, and J. L. Markley. 1994. Structure-function studies of [2Fe-2S] ferredoxins. J. Bioenerg. Biomembr. 26:67-88. [DOI] [PubMed] [Google Scholar]
- 11.Kakuta, Y., T. Horio, Y. Takahashi, and K. Fukuyama. 2001. Crystal structure of Escherichia coli Fdx, an adrenodoxin-type ferredoxin involved in the assembly of iron-sulfur clusters. Biochemistry 40:11007-11012. [DOI] [PubMed] [Google Scholar]
- 12.Khan, N. A., Y. Wang, K. J. Kim, J. W. Chung, C. A. Wass, and K. S. Kim. 2002. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 277:15607-15612. [DOI] [PubMed] [Google Scholar]
- 13.Kim, K. J., J. W. Chung, and K. S. Kim. 2005. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 280:1360-1368. [DOI] [PubMed] [Google Scholar]
- 14.Kim, K. S., H. Itabashi, P. Gemski, J. Sadoff, R. L. Warren, and A. S. Cross. 1992. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Invest. 90:897-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, K. S. 2003. Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat. Rev. Neurosci. 4:376-385. [DOI] [PubMed] [Google Scholar]
- 16.Kim, K. S. 2008. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 6:625-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knoell, H. E., and J. Knappe. 1974. Escherichia coli ferredoxin, an iron-sulfur protein of the adrenodoxin type. Eur. J. Biochem. 50:245-252. [DOI] [PubMed] [Google Scholar]
- 18.Kouokam, J. C., S. N. Wai, M. Fällman, U. Dobrindt, J. Hacker, and B. E. Uhlin. 2006. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect. Immun. 74:2022-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemonier, M., L. Landraud, and E. Lemichez. 2007. Rho GTPase-activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol. Rev. 31:515-534. [DOI] [PubMed] [Google Scholar]
- 20.Mashburn-Warren, L. M., and M. Whiteley. 2006. Special delivery: vesicle trafficking in prokaryotes. Mol. Microbiol. 61:839-846. [DOI] [PubMed] [Google Scholar]
- 21.McKenzie, G. J., and N. L. Craig. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meysick, K. C., M. Mills, and A. D. O'Brien. 2001. Epitope mapping of monoclonal antibodies capable of neutralizing cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli. Infect. Immun. 69:2066-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills, E., K. Baruch, X. Charpentier, S. Kobi, and I. Rosenshine. 2008. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe 3:104-113. [DOI] [PubMed] [Google Scholar]
- 24.Rasband, W. S. 2009. Image J, 1997-2009. National Institutes of Health, Bethesda, MD. http://rsb.info.nih.gov/ij/.
- 25.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Takahashi, Y., and M. Nakamura. 1999. Functional assignment of the ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster involved in the assembly of Fe-S clusters in Escherichia coli. J. Biochem. 126:917-926. [DOI] [PubMed] [Google Scholar]
- 27.Wai, S. N., B. Lindmark, T. Söderblom, A. Takade, M. Westermark, J. Oscarsson, J. Jass, A. Richter-Dahlfors, Y. Mizunoe, and B. E. Uhlin. 2003. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 115:25-35. [DOI] [PubMed] [Google Scholar]
- 28.Zlokarnik, G., P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer, and R. Y. Tsien. 1998. Quantitation of transcription and clonal selection of single living cells with β-lactamase as reporter. Science 279:84-88. [DOI] [PubMed] [Google Scholar]