

Abstract
Mutations of ephrin B1 in humans result in craniofrontonasal syndrome. Because little is known of the role and mechanism of action of ephrin B1 in bone, we examined the function of osteoblast-produced ephrin B1 in vivo and identified the molecular mechanism by which ephrin B1 reverse signaling regulates bone formation. Targeted deletion of the ephrin B1 gene in type 1α2 collagen-producing cells resulted in severe calvarial defects, decreased bone size, bone mineral density, and trabecular bone volume, caused by impairment in osterix expression and osteoblast differentiation. Coimmunoprecipitation of the TAZ complex with TAZ-specific antibody revealed a protein complex containing ephrin B1, PTPN13, NHERF1, and TAZ in bone marrow stromal (BMS) cells. Activation of ephrin B1 reverse signaling with soluble EphB2-Fc led to a time-dependent increase in TAZ dephosphorylation and shuttling from cytoplasm to nucleus. Treatment of BMS cells with exogenous EphB2-Fc resulted in a 4-fold increase in osterix expression as determined by Western blotting. Disruption of TAZ expression using specific lentivirus small hairpin RNA (shRNA) decreased TAZ mRNA by 80% and ephrin B1 reverse signaling-mediated increases in osterix mRNA by 75%. Knockdown of NHERF1 expression reduced basal levels of osterix expression by 90% and abolished ephrin B1-mediated induction of osterix expression. We conclude that locally produced ephrin B1 mediates its effects on osteoblast differentiation by a novel molecular mechanism in which activation of reverse signaling leads to dephosphorylation of TAZ and subsequent release of TAZ from the ephrin B1/NHERF1/TAZ complex to translocate to the nucleus to induce expression of the osterix gene and perhaps other osteoblast differentiation genes. Our findings provide strong evidence that ephrin B1 reverse signaling in osteoblasts is critical for BMS cell differentiation and bone formation.
Osteoporosis is a common disease characterized by an age-dependent decrease in bone mineral density (BMD) and a microarchitectural deterioration of bone tissue, with a consequent increase in the risk of developing fragility fractures of the hip, spine, and other skeletal sites (19). The decrease in bone mass occurs when the body fails to form enough new bone to replace the amount of old bone resorbed leading to reduced bone strength. There are two major known causes of osteoporosis: low peak BMD, which is typically achieved by around age 30, and high bone loss rate, which occurs particularly after menopause and during the natural process of aging. The accumulation of peak bone mass depends on bone growth during early skeletal development and the balance between osteoblastic bone formation and osteoclastic bone resorption during the postnatal growth period. Therefore, understanding the regulatory factors that govern bone development, bone size, bone mineralization, and bone quality during active growth periods as well as bone homeostasis during menopause and aging is essential for development of therapeutics to prevent osteoporosis.
Ephrin ligands and their receptors have been shown to play key roles in the growth and development of multiple tissues including the skeleton (14, 50, 53). There are two types of ephrin ligands and their receptors. Ephrin A's are membrane-anchored proteins, while ephrin B's are transmembrane proteins. In general, ephrin A's bind to ephrin A receptors (EphA) while ephrin B's interact with ephrin B receptors (EphB), with few exceptions (29). Ephrin B1 preferentially binds to EphB2 and -B3 receptors with high affinity and interacts with EphB1 and -B4 receptors with low affinity (29). It has been shown that both ephrin B1 and B2 and their receptors (EphB2, -B3, -B4, -B6, and -A4) are expressed in bone cells (58). However, only ephrin B1 and B2 are expressed in osteoclasts during osteoclast precursor differentiation, while ephrin B1 and B2 and their receptors are consistently coexpressed during osteoblast differentiation (58). The interaction of ephrin B1 and B2 with their multiple receptors via cell-cell contact leads to the activation of a bidirectional signal in which both the receptor-mediated forward signal and the ligand-mediated reverse signal activate downstream signaling cascades (13, 58). In the cells that coexpress both ephrin ligands and their receptors, the ephrin ligands and receptor proteins can be segregated into distinct membrane domains from which they signal biological effects via cell surface interactions (36). Although EphB4 forward signaling and ephrin B2 reverse signaling have been implicated in regulating osteoblastic bone formation and osteoclastic bone resorption processes (58), homozygotes for targeted null mutations of ephrin B2 or EphB4 receptor exhibit severe defects in angiogenesis of both arteries and veins and embryonic lethality (16, 18, 35). In contrast, total disruption of the ephrin B1 gene in mice results in perinatal lethality and defects in skeletal patterning, while mutations of ephrin B1 in humans have been found to cause craniofrontonasal syndrome (9, 13, 50, 51). Mutation of the cytoplasmic tail of ephrin B1, which allows its extracellular domain to interact with ephrin receptors, produces the same bone phenotypes as those of ephrin B1 knockout (KO) mice (13, 14). However, individual KO of EphB1, -B2, -B3, or -A4 receptor, the major receptors for ephrin B1, showed mild phenotypes of behavior or the nerve or digestive system, while KO of both EphB2 and EphB3 receptors resulted in embryonic lethality (15, 24, 26, 39, 52, 58). These experimental and genetic studies strongly suggest that ephrin B1-mediated reverse signaling via its cytoplasmic tail is essential in craniofacial development and the bone formation processes.
Recent studies of the structure and function of ephrin B1 have shown that a highly conserved cytoplasmic tail of ephrin B1 contains several tyrosine residues that can be phosphorylated when the extracellular domain of ephrin B1 contacts its multiple cognate EphB receptors (7, 13, 34). The C terminus of ephrin B1 also contains a conserved binding motif (YYKV) which PDZ proteins can recognize and to which they can bind (7, 33, 34, 49). Therefore, ephrin B1 functions as a receptorlike signaling molecule to transduce signals into the interior of the cell through tyrosine phosphorylation and interaction with PDZ domain-containing proteins including Pick1, GRIP, PTP-BL, and Par-6 in endothelial cells and Xenopus laevis oocytes (6, 7, 31, 33, 34, 40, 46, 49). However, whether any of these PDZ domain-containing proteins are also involved in ephrin B1 reverse signaling to regulate mesenchymal stem cell differentiation and bone formation is unknown.
Schroeder et al. have shown that a PDZ-containing protein, Na/H exchange regulatory factor 1 (NHERF1), also known as solute carrier family 9, isoform 3 regulator 1 (Slc9a3r1), is highly expressed in MC3T3-E1 osteoblasts (44). Targeted disruption of NHERF1 resulted in postnatal lethality often accompanied by bone fractures due to a 25 to 30% reduction in BMD that is believed to be due to increased phosphate transport by Npt2a in the kidneys (45). However, the severity of the skeletal phenotype in the NHERF1 KO mice cannot be explained by the sole function of NHERF1 of redistributing Npt2a because targeted disruption of Npt2a produced only a mild skeletal phenotype that improved over time (3). Thus, NHERF1 may be involved in regulating other biological events besides regulating phosphate transport by Npt2a. Because NHERF2 has been shown to bind to both the membrane receptors and the transcriptional coactivator with PDZ binding motif (TAZ) via its two PDZ domains (30) and because TAZ contains a well-conserved WW domain that can interact with transcription factors such as Runx2, Smad, and Pax3 (27, 28, 30), we hypothesized that ephrin B1 could also form a complex with TAZ through osteoblast-produced NHERF1, a homolog of NHERF2, and transmit extracellular signals into the nuclei of bone cells to regulate bone formation. To test this hypothesis, we generated conditional KO mice with disruption of ephrin B1 in type 1α2 collagen-producing cells for evaluation of skeletal phenotypes and explored the involvement of NHERF1 and TAZ in mediating the effects of the ephrin B1 ligand on osteoblast function.
MATERIALS AND METHODS
Plasmids, recombinant proteins, and antibodies.
The full-length cDNA of mouse EphB2 was amplified by PCR and cloned into the Acc651 and XhoI sites of pcDNA3.1 (Invitrogen; pcDNA-EphB2). Plasmid pcDNA3-ephrin B1 was provided by Philippe Soriano (Mount Sinai School of Medicine, New York). Plasmids pEGFP-TAZ and pEGFP-N-TAZ were generated by inserting mouse TAZ and truncated N-TAZ released from pcDNA-TAZ and pcDNA-N-TAZ (kindly provided by Cai Bin Cui at the University of North Carolina) into the corresponding HindIII and XbaI sites of the pEGFP-C1 vector (BD Bioscience) (11, 13). Retroviral vectors pMX-ephrinB2-IRES-EGFP and pMX-EphB4-IRES-EGFP were generous gifts from Koichi Matsuo at the Keio University (58). Recombinant proteins ephrin B1-Fc, ephrin B2-Fc, EphB2-Fc, and control Fc and antibodies against human IgG, NHERF1, histone H3, and β-actin were purchased from Sigma. Antibody specific to mouse NHERF2 was from Alpha Diagnostic International (San Antonio, TX). Antibodies against ephrin B1, ephrin B2, EphB2, and EphB4 were from R & D Systems. Anti-phospho-Tyr317 ephrin B was a product of PhosphoSolutions (Aurora, CO). Polyclonal antibody specific to PTPN13 was purchased from Santa Cruz. Polyclonal antibodies to TAZ and phospho-YAP (Ser127) were from Cell Signaling Technology, while monoclonal antibody specific to TAZ was from BD Pharmingen. Retroviral pLKO.1-shRNA clones were purchased from Sigma and Open Biosystems. The hairpin sequences (linker sequences are in lowercase) of targeting shRNA were as follows: mouse TAZ, CCTGCATTTCTGTGGCAGATActcgagTATCTGCCACAGAAATGCAGG; mouse NHERF1, GACCGAATTGTGGAGGTCAATctcgagATTGACCTCCACAATTCGGTC; enhanced green fluorescent protein (EGFP), TACAACAGCCACAACGTCTATctcgagATAGACGTTGTGGCTGTTGTA.
Mice.
The floxed ephrin B1 mice with a mixed background of C57BL/6J and 129S4 strains were kindly provided by Philippe Soriano (13). The Col1α2-iCre transgenic mice expressing improved Cre recombinase (iCre) were reported previously (17). Ephrin B1 conditional KO mice were generated by crossing an efnb1flox/flox female with a Cre transgenic male under the control of regulatory sequences of the col1α2 gene to generate iCre+, efnb1flox/y hemizygous males. The iCre+, efnb1flox/y hemizygous males were then bred with efnb1flox/flox homozygous females to generate ephrin B1 conditional KO and control wild-type (WT) mice (Fig. 1A). All mice were housed at the Jerry L. Pettis Memorial VA Medical Center Veterinary Medical Unit (Loma Linda, CA) under standard approved laboratory conditions with controlled illumination (14 h light, 10 h dark) and temperature (22°C) and unrestricted food and water. All of the procedures were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the Jerry L. Pettis Memorial VA Medical Center. Genotyping of the ephrin B1 gene and iCre transgene was monitored by PCR using DNA extracted from tail snips as reported previously (13, 17). The mouse sex-determining gene sry on the Y chromosome was amplified by PCR with specific primers to determine the gender of mouse fetuses (22).
FIG. 1.

Ephrin B1 deficiency in osteoblasts caused skull defects and reduced body size. (A) Breeding scheme for generation of ephrin B1 (efnb1) conditional KO and WT mice. (B) Loss of ephrin B1 expression in osteoblasts from ephrin B1 conditional KO mice measured by Western blotting. (C) Female embryos at E19.5. The arrow indicates the exencephaly. (D) Alizarin red and alcian blue staining of the skulls of the same embryos as in panel C. Arrows indicate skull defects. (E) Alizarin red and alcian blue staining of the same embryo skeletons as in panel C. Arrows indicate calvarial defects. (F) Body sizes of 3-week-old WT and KO mice. (G) Body lengths of WT and KO mice at 3 and 8 weeks of age (n = 14).
Skeleton staining.
Alizarin red and alcian blue staining was performed using established methods (37). Briefly, embryonic day 19.5 (E19.5) embryos were deskinned, eviscerated, and fixed in 95% ethanol for 5 days. The carcasses were fixed in acetone for 4 days and then stained for 3 days in a solution containing 0.1% alizarin red, 0.3% alcian blue, acetic acid, and 70% ethanol (1:1:1:17, vol/vol/vol/vol). The embryos were then transferred to a solution of 1% KOH in 20% glycerol until clear and then stored in glycerol.
Evaluation of bone phenotypes.
Total bone mineral content (BMC) and bone mineral density (BMD) were measured by dual-energy X-ray absorptiometry (DEXA) as described previously (20, 21). The volumetric BMD and geometric parameters at the middiaphysis of the femurs isolated from 8-week-old mice were determined by peripheral quantitative computed tomography (pQCT) as reported previously (21). Cortical and trabecular bone microarchitectures of the femurs isolated from 8-week-old mice were assessed using micro-computed tomography (μ-CT) (Inveon CT module; Siemens, Malvern, PA). The femurs isolated from 8-week-old mice were scanned by X-ray (80 peak kV [kVp]; anode current, 250 μA) with an axial length of 1,024 slides at 10 μm/slice and parallel length of 2,048 slices. The voxel size was 10 μm. Reconstruction analysis was performed with COBRA software (Exxim, Pleasanton, CA). The sections of 1 mm at the middiaphysis were analyzed for cortical measurements and a fixed section of 3.2 mm at the distal end was analyzed for trabecular measurements by using Amira software (Mercury Computer Systems, Inc., Chelmsford, MA). The bones analyzed were adjusted for length so that the regions of interest chosen for cortical and trabecular bone parameters were anatomically the same between the mutant bone and control littermates' bone.
Dynamic calcein labeling and histomorphometry.
Three-week-old mice were injected intraperitoneally with calcein 7 days (20 mg/kg of body weight) and 2 days prior to the expected day of euthanization in order to label mineralizing bone surfaces. Mouse femurs were fixed in 10% formalin overnight. The bones were washed, dehydrated, and embedded in methyl methacrylate resin for sectioning. Longitudinal sections of comparable anatomic positions of the femurs were analyzed by fluorescence microscopy. For analysis of cortical bone formation parameters, middiaphysis of left femurs were used as a sampling site. For evaluation of bone resorption parameters, the right femurs were partially demineralized, embedded in glycomethacrylate, and cut into sections. The sections were stained for tartrate-resistant acid phosphatase (TRAP), and the TRAP covered surface was measured. All bone histomorphometric parameters were measured as previously described (2, 42). Mineral apposition rate (MAR) and the bone formation rate (BFR)/bone surface (BS) ratio were calculated as described previously (41).
Serum assay.
The mouse serum level of C telopeptide was measured as described previously (47).
Primary cell culture.
Bone marrow stromal (BMS) cells derived from the femurs and the tibias of ephrin B1 conditional KO and corresponding control mice were cultured as reported previously (56). The cells were cultured in α minimal essential medium (αMEM) containing 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml) until 60 to 70% confluence prior to experiments.
Nodule assay and ALP staining.
BMS cells isolated from the femurs and the tibias of ephrin B1 conditional KO and corresponding control mice were grown to 80% confluence. The cells were then treated with a mineralization medium containing 10 mM β-glycerophosphate, 50 μg/ml ascorbic acid, and 5% FBS for 24 days. The cells were washed, fixed, and stained with 40 mM alizarin red (pH 4.2). The mineralized area was measured as described previously (55, 56). For alkaline phosphatase (ALP) staining, primary bone marrow stromal cells were grown to 70% confluence and treated with 2 μg/ml of clustered EphB2-Fc or control Fc in a differentiated medium for 6 days, followed by ALP staining and ALP activity assay (55, 56).
Transient transfection and viral transduction.
Transient transfection was carried out by using Lipofectamine reagent according to instructions of the manufacturer (Invitrogen). Production of murine leukemia virus (MLV) and lentivirus was achieved by transfection in Plat-E and 293T cells as described previously (38, 43). Forty-eight hours after transfection, culture supernatants containing retroviral particles were collected, spun at 2,000 × g for 10 min, and filtered through a 0.45-μm filter. Titers were determined by infecting 293T cells with serial dilutions and examining green fluorescent protein (GFP) expression of infected cells by flow cytometry 24 h after infection. Primary BMS cells or C3H10T1/2 cells were transduced by adding culture supernatant containing retroviral particles into the 6-well culture plates in the presence of 8 μg/ml of Polybrene.
RNA extraction and quantitative PCR.
RNA was extracted from primary cultures or bone marrow-free femurs and tibias of 3-week-old ephrin B1 KO mice and corresponding WT littermates as described previously (54, 56). An aliquot of RNA (2 μg) was reverse-transcribed into cDNA in 20 μl of reaction mixture by the oligo(dT)2-18 primer. Real-time PCR was carried out as reported previously (54, 56). Primers for peptidyl prolyl isomerase A (PPIA) were used to normalize the expression data of test genes. Sequences of the primers were as follows: osterix forward, 5′-TGGCGTCCTCTCTGCTTGA; osterix reverse, 5′-TCAGTGAGGGAAGGGTGGGT; PPIA forward, 5′-CCATGGCAAATGCTGGACCA; PPIA reverse, 5′-TCCTGGACCCAAAACGCTCC; ALP forward, 5′-ATGGTAACGGGCCTGGCTACA; ALP reverse, 5′-AGTTCTGCTCATGGACGCCGT; Runx2 forward, 5′-TGGCCGGGAATGATGAGAACTA; Runx2 reverse, 5′-GGACCGTCCACTGTCACTTTAA; Msx1 forward, 5′-TGCCACTCGGTGTCAAAGTGG; Msx1 reverse, 5′-CCGACTGAGAAATGGCCGAGA.
TAZ nuclear translocation.
C3H10T1/2 cells were cotransfected with pEGFP-TAZ or pEGFP-N-TAZ and pcDNA3-ephrin B1 and treated with soluble clustered EphB2-Fc protein (2 μg/ml) or Fc 24 h after transfection. The transfected cells were examined under an Olympus fluorescence microscope at 4 h after treatment. The green fluorescence images were captured by using the computer-driven camera with MagnaFire software (v2.1; Olympus America, Melville, NY). For examination of endogenous TAZ nuclear trafficking of BMS cells, the cells were treated with soluble clustered EphB2-Fc or Fc for 4 h. The cytoplasmic and nuclear proteins were extracted and analyzed by Western blotting with specific antibodies to mouse TAZ, β-tubulin, and histone H3 as described previously (56).
Immunoprecipitation and Western blotting.
Primary BMS cells were cultured until 70% confluence, treated with clustered EphB2-Fc protein at 37°C for 5 min, and cross-linked with 5 mM dithiobis (succinimidyl) propionate (Pierce) for 30 min at room temperature. The cells were lysed with lysis buffer (50 mM HEPES [pH 7.5], 100 mM NaCl, 10 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail, and 1× phosphatase inhibitor cocktail). Cell lysate (300 μg of total protein) was first precleared by using protein A/G-Sepharose and then incubated with 4 μg of TAZ-specific monoclonal antibody or control IgG for 1 h at 4°C with gentle shaking, followed by the addition of protein A/G-Sepharose and an additional overnight incubation at 4°C. After centrifugation, the protein A/G-Sepharose beads were washed five times with cold lysis buffer and then boiled with SDS-PAGE sample buffer to dissociate the proteins. The immunoprecipitated proteins were separated by SDS-PAGE under reducing conditions for Western blotting with polyclonal antibodies against NHERF1 and ephrin B1.
Statistical analyses.
Data were analyzed by analysis of variance (ANOVA) or Student's t test.
RESULTS
Characterization of conditional ephrin B1 KO mice.
Conditional KO mice with disruption of ephrin B1 in type 1α2 collagen producing cells were generated as described in Fig. 1A. To confirm loss of ephrin B1 expression in the Cre+ mice, we extracted the total cellular proteins from primary calvarial osteoblasts from 2-week-old mice for immunoblotting analyses with antibodies to ephrin B1 and Cre. As expected, the Cre recombinase was expressed in type Iα2 collagen producing osteoblasts isolated from Cre+, efnb1lox/lox mice, but not in Cre−, efnb1lox/lox mice. Accordingly, the expression of the ephrin B1 protein was absent in osteoblasts from Cre+ mice but was detected in the cells from Cre− control mice (Fig. 1B).
Ephrin B1 deficiency in osteoblasts resulted in skull defects and reductions in bone size and peak bone BMD caused by reduced bone formation and mineralization.
Upon breeding efnb1lox/y hemizygous males carrying the Col1α2-Cre allele with efnb1lox/lox homozygous females, the ephrin B1 conditional KO mice were not retrieved at the expected Mendelian ratio, while the ratio of WT male to WT female offspring is almost 1:1 at 3 weeks of age. We, therefore, compared the litter sizes for pups generated from Cre+ versus Cre− males that were bred with efnb1lox/lox homozygous females. We found that the average litter sizes were approximately 5 and 9 live pups for Cre+, efnb1lox/y and Cre−, efnb1lox/y males, respectively. Among all live pups (n = 64) from the cross of efnb1 loxp homozygous mice with Cre+ loxp hemizygous mice, 60% of pups were males and 40% were females. Among them, 15% of the females and 20% of the males were Cre+. However, the ephrin B1-deficient fetuses were recovered at the expected Mendelian ratio at E19.5. Of these, more than 40% of the Cre+ females that were efnb1 loxp homozygous and 20% of Cre+ males that were efnb1 loxp hemizygous had severe defects of the skull and exencephaly (Fig. 1C). The size of the skull was reduced and was not large enough to accommodate the growing brain. Skeletal preparations revealed that the basal aspect of the skull and the jaw were intact. However, the frontal, parietal, and interparietal bones were missing in both KO males and females with skull defects. The mandible and maxilla were smaller. The overall size of the skull of the ephrin B1 gene KO mice was about 35% smaller than that of the WT littermates. As a result of calvarial bone defects, the basal elements of the skull became visible when viewed from above (Fig. 1D). Besides the calvarial defects, the conditional KO mice were much smaller than the WT and bone mineralization was reduced. Alizarin red staining of conditional KO embryos also revealed delayed mineralization in both skull bones formed by the intramembranous route and long bones formed by the endochondral bone formation route (Fig. 1E).
Though nearly 30% of ephrin B1 conditional KO male and female mice died before or shortly after birth, we obtained an adequate number of male and female mutants for phenotypic characterization. The body weights of both male and females were significantly reduced by 29 and 17% at 3 and 8 weeks of age, respectively (71% ± 13% and 83% ± 9% of the control littermates; n = 14; P < 0.05). As shown in Fig. 1F and G, the body length of ephrin B1 mutants was reduced by 15% compared to the corresponding littermates at 3 weeks of age. Femur length was reduced by 14% and 13% in KO males and females, respectively, at 8 weeks of age (Fig. 2A and B). To determine if the targeted disruption of the ephrin B1 gene in osteoblasts affects bone mass, we performed DEXA measurements. We found that total body BMC was reduced by 30% and 23% at 3 and 8 weeks of age, respectively (Fig. 2C). Total body areal BMD was reduced by 10% at both time points, while total body bone area was reduced by 25% and 15% at 3 and 8 weeks of age, respectively. To further characterize the skeletal phenotypes of ephrin B1 conditional KO mice, we collected the femurs from 8-week-old mice and carried out pQCT and μ-CT analyses. Our pQCT analyses demonstrated that the periosteal circumference and endosteal circumference of the femurs at middiaphysis were reduced by 12% and 16%, respectively, in ephrin B1 conditional KO mice compared to the control littermates (Fig. 2D). Consistent with the pQCT data, μ-CT analyses confirmed that both bone volume and tissue volume were reduced by 25% at the middiaphysis of the femurs isolated from 8-week-old KO mice compared to the littermate controls (Fig. 2E to G). The percentages of bone volume to tissue volume were not different at this site of the femur. However, trabecular bone volume adjusted for tissue volume was significantly decreased at the metaphysis of the femur (Fig. 3A to D). Three-dimensional μ-CT analyses found that trabecular numbers were reduced 50% in the distal femurs isolated from KO females. The ratio of bone volume to tissue volume was reduced by 30% compared to WT controls.
FIG. 2.

Ephrin B1 deficiency in osteoblasts caused reduced bone size and peak bone BMD. (A) Femurs of WT and KO mice at 8 weeks of age. (B) Femur lengths of WT and KO mice at 8 weeks of age (n = 14). (C) Percentages of total body BMC, BMD, and bone area of KO mice compared with the gender-matched WT littermates at 3 and 8 weeks of age, as measured by DEXA (n = 14). SE, standard error. (D) μ-CT images of cross sections, showing the periosteal circumferences (PC) and endosteal circumferences (EC), of the femurs at middiaphysis from 8-week-old female mice. (E to G) Cortical bone volumes (BV), total tissue volumes (TV), and BV/TV ratios of the femurs isolated from 8-week-old female mice measured by μ-CT. Asterisks indicate a significant difference in KO mice compared with WT mice (n = 6, P < 0.05).
FIG. 3.

Reduced trabecular bone volume, bone formation rate (BFR), and mineral apposition rate (MAR) in ephrin B1 KO mice. (A) Three-dimensional μ-CT images of distal femurs isolated from 8-week-old females. (B) Two-dimensional μ-CT images of trabecular bone of distal femurs. (C) Trabecular numbers (Tb. N) measured by μ-CT at distal femurs of ephrin B1 KO and WT females (n = 6). (D) Trabecular bone volume/total tissue volume (Tb. BV/TV) (n = 6). (E) Images of calcein double labeling of the femurs from 3-week-old WT and KO mice. (F) BFR (n = 8). (G) MAR (n = 8). Asterisks indicate a significant difference in KO mice compared with WT mice (P < 0.05).
To identify the target cell types and cellular processes that contribute to reduced bone formation and decreased peak bone mass in the ephrin B1 conditional KO mice, we performed histomorphometric studies at the middiaphysis of the femurs from 3-week-old KO and control mice. Figure 3E shows a decreased width of newly formed bone between two calcein labels in the KO mice compared to control mice. Because of reduced bone size in the KO mice, we adjusted bone formation rate (BFR) for bone surface (BS). The BFR/BS ratio was reduced by 50% at the femur midshaft in the KO mice (Fig. 3F). In order to determine if the reduced bone formation rate is caused by reduced activity of osteoblasts, we measured mineral apposition rate (MAR) and found a 43% reduction in MAR in the KO mice compared to the WT littermates (Fig. 3G).
We next determined if loss of ephrin B1 in type 1α2 collagen-producing cells influences bone resorption. Figure 4 shows the data from tartrate-resistant acid phosphatase (TRAP) staining for periosteal and endosteal surfaces examined at the middiaphysis of the femurs of 3-week-old mice. The percentages of TRAP-labeled surface at both periosteum and endosteum were not affected in the conditional KO mice compared to the littermate controls (Fig. 4A and B). In addition, the serum level of C telopeptide, a bone resorption marker, was not changed (Fig. 4C). The lack of difference in bone resorption between conditional KO and WT mice was not surprising because conditional disruption of ephrin B1 in collagen 1-producing cells did not influence expression of ephrin B1 in osteoclast precursors derived from mouse spleen as shown by Western blot analyses (Fig. 4D).
FIG. 4.
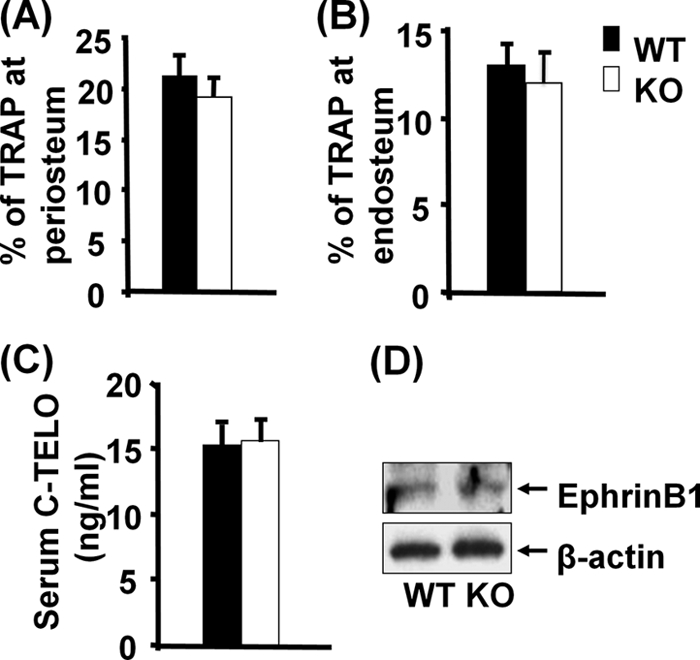
Lack of ephrin B1 expression in osteoblastic lineage cells does not affect bone resorption. (A and B) TRAP staining for periosteal and endosteal bones examined at the middiaphysis of femurs of 3-week-old mice (n = 8). (C) Serum levels of C telopeptide (C-TELO) of 3-week-old mice (n = 12). (D) Expression of ephrin B1 by Western blotting using cell lysates from osteoclast precursors from 3-week-old KO and WT mice.
Interaction of ephrin B1 and EphB2 expressed in bone cells mediates osteoblast differentiation.
Our finding that conditional disruption of ephrin B1 produces a dramatic change in skeletal phenotypes predicts that ephrin B1 produced locally in bone is important. Accordingly, we found that undifferentiated BMS cells express a large amount of ephrin B1 protein (Fig. 5A). However, only a trace amount of ephrin B2 protein was expressed in BMS cells when the signals of ephrin B1 and B2 were normalized with expression levels of β-actin (Fig. 5B). Because ephrin B1 has been shown to preferentially bind to EphB2 and EphB3 receptors and weakly interacts with EphB1 and EphB4, we examined the expression of these receptors in bone cells by reverse transcription (RT) real-time PCR (29). We found that EphB2 mRNA, as measured by RT real-time PCR, was expressed at a level 20 times higher than that of EphB3 and EphB4 in BMS cells, while EphB1 was undetectable (data shown). Because mice with disruption of the EphB3 receptor showed behavior phenotypes only related to the central nervous system (39), we decided to focus on the EphB2 and EphB4 receptors in this study. We therefore determined the relative expression of these two receptors at the protein level. Figures 5C and D show that EphB2 protein, as determined by Western blot analyses, was highly expressed while expression of EphB4 was undetectable in BMS cells. Because both ephrin B1 and its multiple receptors are expressed in BMS cells, we next examined the role of osteoblast produced ephrin B1 by using BMS cells derived from ephrin B1 conditional KO and WT mice. We found that the amount of mineralized nodule was reduced by 66% in BMS cells derived from KO mice compared to control mice after 24 days of culture in mineralization medium (Fig. 5E). To confirm the relative contribution of ephrin B-mediated reverse signaling to osteoblast differentiation, we seeded BMS cells derived from 3-week-old WT and ephrin B1 conditional KO mice at low density to minimize cell-cell contact, and treated them with control Fc and EphB2-Fc in a differentiation medium for 6 days to activate reverse signaling. We found that treatment of EphB2-Fc increased ALP activity by 118% compared to that of cells treated with control Fc. In contrast, treatment of BMS cells derived from ephrin B1 conditional KO mice with EphB2-Fc failed to stimulate ALP activity (Fig. 5G).
FIG. 5.

Interaction of ephrin B1 and EphB2 by cell-cell contact mediates osteoblast differentiation. (A) Ephrin B1 expression in BMS cells (BMS) measured by Western blotting. +C, positive control of cellular lysate from C3H10T1/2 cells overexpressing ephrin B1. (B) Trace amount of ephrin B2 expression in BMS cells. +C, positive control of cellular lysate from C3H10T1/2 cells overexpressing ephrin B2. (C) EphB2 expression in BMS cells. +C, positive control of cellular lysate from C3H10T1/2 cells overexpressing EphB2. (D) EphB4 was not detectable in BMS cells. +C, positive control of recombinant EphB4 protein (5 and 10 ng). (E) Images of nodule formation from BMS cells of ephrin B1 KO and WT mice, stained by alizarin red. (F) Quantitative mineralization area measured by the OsteoMeasure system. (G) Activation of ephrin B1 stimulates osteoblast differentiation. BMS cells from WT and ephrin B1 conditional KO mice were stimulated with 2 μg/ml of EphB2-Fc in a differentiation medium for 6 days, followed by ALP staining and ALP activity assay. ALP activity values are means ± standard deviations (SD) and are expressed as percentages of the Fc control. The asterisk indicates a significant difference versus the Fc control (n = 6, P < 0.01).
Ephrin B1 reverse signaling regulates osterix expression.
To further determine the cause for reduced osteoblastic function in the conditional ephrin B1 KO mice, we measured expression levels of transcription factors that are critical in osteoblast differentiation. Total RNA was extracted from the marrow-free long bones of 3-week-old KO mice and corresponding littermates and used for RT real-time PCR with primers specific for ALP, osterix, Runx2, Msx1, ephrin B1, EphB2, and PPIA. As expected, the expression of ephrin B1 was reduced by 85% in the bones of conditional KO mice compared to the WT controls. There was no change of EphB2 receptor expression between the bones isolated from WT and KO mice. The expression of ALP was decreased by 80% in the bones of conditional KO mice compared to corresponding controls (Fig. 6A). There was a significant 75% reduction in the expression of osterix in the femurs of the KO mice compared to the WT littermates. However, the expression of Runx2 and Msx1 was not significantly altered. To further examine whether ephrin B1-mediated reverse signaling regulates osterix expression, we treated BMS cells from WT and conditional KO mice with the EphB2-Fc soluble receptor. We predicted that the soluble receptor, upon binding to ephrin B1 ligand, would activate ephrin B1 reverse signaling in WT but not KO cells. We also predicted that the EphB2-Fc soluble receptor would inhibit forward signaling by competing with the endogenous EphB receptor to bind to the ephrin B1 ligand in WT cells. As expected, we found that treatment of WT BMS cells with 2 μg/ml EphB2-Fc for 72 h in a mineralization medium increased osterix expression by approximately 4-fold as determined by Western blotting using an osterix-specific antibody. As expected, osterix protein levels were considerably lower in the ephrin B1 KO cells than in WT control cells. Furthermore, addition of the EphB2-Fc soluble receptor did not induce osterix expression in the ephrin B1-deficient cells (Fig. 6B and C). There were no changes in TAZ, NHERF1, or Runx2 expression in the BMS cells derived from KO mice compared to the cells from WT control mice. Furthermore, treatment of BMS cells derived from WT and KO mice with EphB2-Fc did not stimulate the expression of TAZ, NHERF1, or Runx2.
FIG. 6.

Ephrin B1 reverse signaling regulates osterix expression. (A) Expression of ALP, osterix, Runx2, MSX1, ephrin B1, and EphB2 in bones from 3-week-old mice, measured by RT real-time-PCR. Values are means ± SD from 6 replicates. Asterisks indicate a significant difference in KO mice compared with WT mice (P < 0.01). (B) Activation of ephrin B1 reverse signaling stimulates osterix expression in BMS cells. BMS cells from WT and KO mice were treated with clustered EphB2-Fc or control Fc in a differentiation medium. After 72 h of treatment, cellular protein was extracted for Western blotting. (C) The intensity of Western blot signals of osterix expression was quantitated by ImageQuant, normalized to the signals of β-actin, and expressed as fold change over the intensity of the WT control.
Activation of ephrin B1 recruits PTPN13, NHERF1, and TAZ to phosphorylated ephrin B1 and stimulates TAZ nuclear translocation.
In terms of the mechanism by which ephrin B1 regulates osterix expression, it is known that the cytoplasmic tail of ephrin B1 contains a PDZ binding motif that can interact with PDZ domain-containing proteins such as PTPN13 and NHERF1 and -2 (34). Since NHERF1 and -2 contain two conserved PDZ domains that can potentially bind to both the membrane proteins and the transcriptional coactivator TAZ, we predicted that activation of ephrin B1 can induce formation of a protein complex containing PTPN13, NHERF1 and -2, and TAZ. To test this prediction, we first examined whether BMS cells express NHERF1 and/or NHEFR2 protein by Western blotting. We found that NHERF1 was expressed in the BMS cells at very high levels but that NHERF2 could not be detected at the protein level using a commercial polyclonal antibody that detects only high levels of NHERF2 (data not shown). We then examined the interaction of ephrin B1, TAZ, NHERF1, and PTPN13 in BMS cells. The BMS cells were stimulated with clustered EphB2-Fc for 5 min, cross-linked with dithiobis (succinimidyl) propionate, and lysed for immunoprecipitation with anti-TAZ and anti-PTPN13, respectively. Because we used a strategy that involved a membrane-permeable and reversible cross-linker to stabilize the protein complex, we were able to immunoprecipitate both ephrin B1 and NHERF1 with an antibody specific to TAZ and ephrin B1 with antibody specific to PTPN13 (Fig. 7A).
FIG. 7.

Activation of ephrin B1 recruits PTPN13, NHERF1, and TAZ to phosphorylated ephrin B1 and stimulates TAZ nuclear translocation. (A) Interaction of ephrin B1 with TAZ, NHERF1, and PTPN13 in BMS cells by immunoprecipitation (IP). IB, immunoblotting. (B) Dynamic protein complex formation of ephrin B1, NHERF1, and PTPN13 in C3H10T1/2 cells overexpressing ephrin B1 by immunoprecipitation. (C) Time-dependent ephrin B phosphorylation and TAZ dephosphorylation in C3H10T1/2 cells overexpressing ephrin B1 and TAZ by Western blotting. (D) Nuclear trafficking of TAZ. C3H10T1/2 cells were cotransfected with pEGFP-N-TAZ and pcDNA-ephrinB1 (1 and 2), pEGFP-TAZ alone (3 and 4), or pEGFP-TAZ and pcDNA-ephrinB1 (5 and 6). Twenty-four hours later, cells were treated with clustered control Fc (1, 2, and 3) or EphB2-Fc (4, 5, and 6) for 4 h, followed by examination under a fluorescence microscope. Arrows indicate nucleus-localized TAZ. (E) Endogenous TAZ nuclear translocation in BMS cells by Western blotting. BMS cells from WT and ephrin B1 KO mice were treated with clustered EphB2-Fc for 4 h.
Because PTPN13 can dephosphorylate tyrosine residues of the clustered ephrin B1, we predicted that the formation of ephrin B1, NHERF1, and other PDZ domain-containing protein phosphatases (PP) such as PTPN13 and PP2C can produce a scaffold membrane-associated protein cluster and that the protein complex can dephosphorylate ephrin B1 and TAZ (25, 40). To test the prediction, we performed coimmunoprecipitation again with the antibody specific to NHERF1 to pull down the NHERF1 protein complex. The coimmunoprecipitated proteins were blotted with antibodies against ephrin B1 and PTPN13, respectively. Figure 7B shows stronger interactions among ephrin B1, NHERF1 and PTPN13 30 min after addition of EphB2-Fc compared to prior EphB2-Fc treatment. As expected, the scaffold protein clustering was transient and returned to baseline by 120 min after EphB2-Fc stimulation. Activation of ephrin B1 reverse signaling with soluble clustered EphB2-Fc receptors also led to a time-dependent increase in the phosphorylation of ephrin B1 (7.3-, 15.4-, and 9.6-fold at 5, 30, and 120 min, respectively) and the dephosphorylation of TAZ (1.5-, 2.2-, and 3.3-fold at 5, 30, and 120 min, respectively) in C3H10T1/2 cells overexpressing ephrin B1 and TAZ (Fig. 7C).
To test if ephrin B1 reverse signaling-mediated TAZ dephosphorylation is concomitant with TAZ trafficking from the cytoplasmic compartment to the nucleus, we transfected C3H10T1/2 cells that do not express endogenous ephrin B1 with pEGFP-TAZ alone, pEGFP-N-TAZ and pcDNA-ephrinB1, or pEGFP-TAZ and pcDNA-ephrinB1. The cells were then stimulated with clustered EphB2-Fc or control Fc for 4 h for examination of TAZ and N-TAZ nuclear translocation. As expected, cotransfection of C3H10T1/2 cells with N-TAZ and ephrin B1 resulted in the cytoplasmic distribution of N-TAZ whether or not cells were stimulated by EphB2-Fc because N-TAZ-GFP fusion protein lacks C terminus that can be modified to interact with NHERF1 (Fig. 7D1 and D4). The same cellular localization of TAZ was observed in the cells transfected with TAZ alone without overexpression of ephrin B1 (Fig. 7D2 and D5) because C3H10T1/2 cells produce no detectable levels of ephrin B1 protein under the culture conditions used (data not shown). Cotransfection with both intact TAZ and ephrin B1 also resulted in cytoplasmic localization of TAZ without EphB2-Fc stimulation (Fig. 7D3). However, EphB2-Fc treatment led to nuclear translocation of TAZ in cells cotransfected with TAZ and ephrin B1 (Fig. 7D6). To rule out the possibility that the observed changes in TAZ localization in response to activation of ephrin B1 signaling are an artifact of overexpression of ephrin B1 and TAZ, we also examined endogenous TAZ nuclear translocation induced by ephrin B1 reverse signaling. Primary BMS cells isolated from WT and KO mice were cultured and treated with soluble clustered EphB2-Fc or Fc for 4 h. The cytoplasmic and nuclear proteins were extracted and analyzed by Western blotting with specific antibodies to mouse TAZ, β-tubulin, and histone H3. Consistent with the data above, we found that TAZ was predominantly localized in the cytoplasmic compartment in the absence of EphB2-Fc stimulation. However, activation of ephrin B1 reverse signaling by EphB2-Fc increased accumulation of TAZ in the nuclei of BMS cells, as measured by Western immunoblotting (Fig. 7E). The nuclear level of TAZ was elevated 3-fold after 4 h of treatment with clustered EphB2-Fc compared to the Fc control. In contrast, treatment of BMS cells derived from ephrin B1 KO mice with EphB2-Fc failed to induce TAZ nuclear translocation.
Knockdown of TAZ and NHERF1 expression decreases ephrin B1 reverse signaling-mediated osterix expression.
To evaluate the role of TAZ in ephrin B1 reverse signaling, we knocked down TAZ expression with a lentivirus expressing shRNA and then examined osterix expression in response to clustered EphB2-Fc stimulation in BMS cells. We found that TAZ expression was reduced by nearly 80% at the protein level and 78% at the mRNA level in the cells infected with mouse TAZ shRNA compared to the cells transduced with GFP control shRNA (Fig. 8A & B). As expected, the expression of osterix mRNA was increased by 8-fold upon activation of ephrin B1 reverse signaling by EphB2-Fc in BMS cells expressing GFP control shRNA. However, EphB2-Fc-induced osterix induction was reduced by 75% in the BMS cells expressing TAZ shRNA. Similarly, knockdown of NHERF1 expression significantly reduced basal level of osterix expression by 90% (data not shown) and abolished ephrin B1-mediated induction of osterix expression (Fig. 8C).
FIG. 8.

Knockdown of TAZ and NHERF1 expression decreases ephrin B1 reverse signaling-mediated osterix expression. BMS cells were infected with lentivirus shRNA and treated with clustered EphB2 for 3 days. (A) TAZ and NHERF1 expression analyzed by Western blotting. (B) TAZ and NHERF1 expression quantified by RT real-time PCR. (C) Osterix expression quantified by RT real-time PCR. Asterisks indicate a significant difference in KO mice compared with WT mice (n = 3, P < 0.01).
DISCUSSION
Ephrin B1 has been shown to be expressed in various types of cells and to play key roles in the growth and development of multiple tissues (1, 14, 26, 31, 50, 57). Although total loss of ephrin B1 function or reverse signaling in every cell type results in perinatal lethality and defects in skeletal patterning in mice, little is known of the role of ephrin B1 produced locally by bone cells (13, 14). In this study, we used transgenic mice expressing an improved Cre recombinase under the control of the type I collagen promoter to disrupt ephrin B1 in type 1α2 collagen-producing cells (17, 21) and examined the consequences of conditional disruption of ephrin B1 in bone cells for the skeletal phenotypes in vivo. With this targeting strategy, we showed that disruption of ephrin B1 in osteoblasts was almost complete and that calvarial defects in the conditional KO embryos were much more severe than those in PGK-Cre- or Meox2-Cre-mediated conditional KO mice reported previously (13, 14). Furthermore, we found that exencephaly occurred in both mutant hemizygous males and homozygous females, contrary to a previous study that reported that skull defects were restricted in ephrin B1 heterozygous KO females (9). The heterozygous conditional KO females with one efnb1 allele in type 1α2 collagen producing cells appeared normal, with no obvious bone phenotypes although they were mosaic because of random X chromosome inactivation. We did not observe phenotypes of polydactyly, cleft palate, or tooth defects in the ephrin B1 homozygous/hemizygous conditional KO mice or ephrin B1 heterozygous females. We believe that there are two main reasons for the observed differences between the magnitude of the skeletal phenotype of our conditional ephrin B1 KO mice and those reported earlier. First, the promoters used to drive Cre expression were different in the three studies, which targeted different cell types and/or different stages of embryonic development. In our study, we used the collagen1α2 promoter to express Cre recombinase in the conditional KO mice. Cre-mediated recombination could be detected in osteoblasts at the site of intramembranous bone formation at E13.5. Thus, the ephrin B1 gene is deleted at later stages of embryonic development in our conditional KO mice, which apparently contributes to reduction in embryonic lethality but severe skull defects and reduced bone size and bone density. Second, the differences in genetic background could also in part contribute to the observed differences in the severity of the skeletal phenotype in the three studies.
Recent studies have demonstrated that the 3.6 rat collagen 1 promoter is active in osteoclasts in a mouse transgenic model (5). In our experiments, we found that expression levels of ephrin B1 in osteoclast precursors isolated from the spleens of the KO mice were comparable to those levels in WT littermates. In addition, our histomorphometric and serum C telopeptide analyses demonstrated that bone resorption was not affected in the ephrin B1 conditional KO mice. Based on our data, we conclude that the reduced bone size, bone density, and trabecular numbers of the conditional KO mice were due to impaired osteoblast-mediated bone formation but not increased osteoclast-mediated bone resorption.
All three ephrin B ligands have the same structure: a single transmembrane domain and a well-conserved cytoplasmic domain that includes 33 amino acids with nearly 100% identity (4, 12). Among ephrin B proteins, B1 and B2 have been shown to be expressed in osteoblasts while ephrin B3 is not expressed in bone cells (13, 58). The C-terminal structural similarity between ephrin B1 and B2 raises the question of whether a functional compensation exists between these two family members. In our studies, we found that only a trace amount of ephrin B2 protein, compared to ephrin B1, was expressed in BMS cells derived from WT mice. Furthermore, loss of ephrin B1 did not cause upregulation of ephrin B2 in BMS cells (data not shown). Accordingly, ephrin B1-deficient BMS cells exhibit impaired formation of mineralized nodules in vitro. Our in vitro data together with in vivo data showing severe bone phenotypes in ephrin B1 conditional KO mice strongly support the prediction that the amount of ephrin B2 protein in osteoblasts cannot compensate for the effect of the loss of ephrin B1 on the skeletal phenotype in the ephrin B1 conditional KO mice.
It should be noted that BMS cells express both ephrin B1 and its cognate EphB2 receptor. Thus, both forward and reverse signals are feasible upon cell to cell contact. In this regard, Zhao et al. suggested that ephrin B2 produced by osteoclasts might interact with the EphB4 receptor in osteoblasts to induce forward signaling and osteoblast differentiation based on the findings that exogenous addition of clustered ephrin B2-Fc stimulated ALP activity of calvarial osteoblasts and overexpression of EphB4 in transgenic mice increased bone formation (58). While these studies provided evidence that EphB4-mediated forward signaling might be involved in osteoblast differentiation, the relative contributions of forward and reverse signaling in mediating ephrin B1 effects on skull development and bone formation need to be further evaluated in vivo by appropriate genetic rescue experiments. In our studies, we used low-density cell cultures to minimize reverse signaling via the interaction between endogenously produced EphB2 receptor from one cell and the ephrin B1 ligand of a neighboring cell. We found that exogenous addition of soluble EphB2-Fc receptor capable of binding to ephrin B1 caused an increase in osterix expression and ALP activity in BMS cells expressing ephrin B1. Knockout of EphB2 or EphB3 produced only mild phenotypes of the digestive or nervous system, while KO of both EphB2 and EphB3 receptors capable of activating ephrin B1 reverse signaling resulted in embryonic lethality (15, 24, 26, 39, 52, 58). These data, supported by previous genetic studies that a single amino acid mutation in the PDZ domain of ephrin B1 in mice has been shown to cause skeletal defects (14, 23), in aggregate indicate that ephrin B1-mediated reverse signaling is critically important in regulating osteoblast differentiation. Besides the activation of ephrin B1 reverse signaling, the role and molecular pathways of EphB2 receptor in bone cells are unknown. In mouse small intestine and colon, EphB2 signaling directs stem cell migration, promotes cell cycle reentry of progenitor cells, and stimulates cell proliferation (26). Whether EphB2 can also modulate mesenchymal stem cell adhesion, migration, proliferation, and/or differentiation in bone in vivo remains to be established. Therefore, future studies involving generation of conditional knock-in mice in which the WT copy of ephrin B1 is replaced by truncated or mutated ephrin B1 incapable of binding PDZ domain proteins and/or WT copy of EphB2 receptor is replaced by intracellular domains-truncated EphB2 that can bind to ephrin B1 but does not initiate forward signaling are necessary to convincingly demonstrate the relative contribution of ephrin B1 reverse and forward signaling in regulating bone formation.
In terms of the molecular pathway by which ephrin B1-mediated reverse signaling could regulate osteoblasts, investigations using endothelial cells and Xenopus oocytes have shown that ephrin B1 interaction with its receptors can induce a rapid coclustering of ephrin B1 and Src family kinases, leading to rapid phosphorylation of the tyrosine sites at the C terminus (7, 40) and subsequent recruitment of PDZ adaptor proteins (10). However, it is currently unknown what downstream signaling molecules bind to the phosphorylated tails of ephrin B1 in bone cells. In our study, we have demonstrated that ephrin B1 interacts with PTPN13 and TAZ via NHEFR1 in BMS cells. The activation of ephrin B1 reverse signaling via exogenous addition of soluble EphB2-Fc receptor resulted in a significant increase in TAZ dephosphorylation and nuclear translocation in BMS cells as well as in C3H10T1/2 cells that overexpress GFP-TAZ and ephrin B1. We have also shown that disruption of TAZ expression using specific lentivirus shRNA resulted in significant blockade of ephrin B1 reverse signaling-mediated increases in osterix in BMS cells. Our data provide, for the first time, experimental evidence that TAZ and NHERF1 are important downstream players in ephrin B1 reverse signaling.
While our in vitro studies and published data on the skeletal phenotypes of mice lacking TAZ and NHERF1 are consistent with our proposed model that an interaction between ephrin B1, NHERF1, and TAZ is involved in regulating osteoblast differentiation, it remains to be determined whether additional pathways are involved in mediating ephrin B1 effects on bone. In this regard, recent studies have shown that TAZ can also promote tumor cell migration, cell proliferation, epithelial-mesenchymal transition, and E3 ligase-mediated protein degradation (8, 32, 48). Thus, TAZ may have other functions in bone besides regulating expression of genes related to the osteoblast differentiation pathway. In addition, NHERF1 functions to control renal phosphate transport (45). Therefore, the relative contributions of the NHERF1 pathway in bone cells, via regulating ephrin B1 signaling, versus kidney cells, via regulation of phosphate transport, to the skeletal phenotype in NHERF1 KO mice remains to be determined. Furthermore, the reduced severity of skeletal phenotype in NHERF1 or TAZ KO mice compared to that in ephrin B1 conditional KO mice suggests potential involvement of other signaling pathways besides TAZ-mediated signaling in mediating ephrin B1 effects on bone formation in vivo.
In conclusion, we have demonstrated that targeted deletion of ephrin B1 in type 1α2 collagen producing cells results in severe calvarial defects, decreased bone size, BMD, and trabecular bone. The impairment of osteoblast differentiation and bone formation in the growing mice with conditional disruption of ephrin B1 appears to be due to reduced expression of the osterix gene. Activation of ephrin B1 reverse signaling upon interaction with its receptors stimulates TAZ dephosphorylation, nuclear trafficking, transactivation, and TAZ target gene transcription. Therefore, our findings provide a novel molecular pathway by which ephrin B1 regulates bone formation.
Acknowledgments
This work was supported by Assistance Award no. DAMD17-03-2-0021, provided by the U.S. Army Medical Research Acquisition Activity, Fort Detrick, MD, and a VA Merit Review Award provided by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. All work was performed at facilities provided by the Jerry L. Pettis Memorial VA Medical Center in Loma Linda, CA.
The information contained in this publication does not necessarily reflect the position or the policy of the U.S. Government, and no official endorsement should be inferred.
We thank Catrina M. Alarcon and Anil Kapoor for their technical assistance. We are also grateful to Philippe Soriano at the Mount Sinai School of Medicine, Koichi Matsuo at Keio University, Cai Bin Cui at the University of North Carolina, Bert Vogelstein at Howard Hughes Medical Institute at Johns Hopkins, and Michael B. Yaffe and Mary Q. Stewart at the Massachusetts Institute of Technology for providing reagents.
Footnotes
Published ahead of print on 7 December 2009.
REFERENCES
- 1.Adams, R. H., G. A. Wilkinson, C. Weiss, F. Diella, N. W. Gale, U. Deutsch, W. Risau, and R. Klein. 1999. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 13:295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beamer, W. G., L. R. Donahue, C. J. Rosen, and D. J. Baylink. 1996. Genetic variability in adult bone density among inbred strains of mice. Bone 18:397-403. [DOI] [PubMed] [Google Scholar]
- 3.Beck, L., A. C. Karaplis, N. Amizuka, A. S. Hewson, H. Ozawa, and H. S. Tenenhouse. 1998. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. U. S. A. 95:5372-5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckmann, M. P., D. P. Cerretti, P. Baum, T. Vanden Bos, L. James, T. Farrah, C. Kozlosky, T. Hollingsworth, H. Shilling, E. Maraskovsky, et al. 1994. Molecular characterization of a family of ligands for eph-related tyrosine kinase receptors. EMBO J. 13:3757-3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boban, I., C. Jacquin, K. Prior, T. Barisic-Dujmovic, P. Maye, S. H. Clark, and H. L. Aguila. 2006. The 3.6 kb DNA fragment from the rat Col1a1 gene promoter drives the expression of genes in both osteoblast and osteoclast lineage cells. Bone 39:1302-1312. [DOI] [PubMed] [Google Scholar]
- 6.Bong, Y. S., H. S. Lee, L. Carim-Todd, K. Mood, T. G. Nishanian, L. Tessarollo, and I. O. Daar. 2007. ephrinB1 signals from the cell surface to the nucleus by recruitment of STAT3. Proc. Natl. Acad. Sci. U. S. A. 104:17305-17310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bong, Y. S., Y. H. Park, H. S. Lee, K. Mood, A. Ishimura, and I. O. Daar. 2004. Tyr-298 in ephrinB1 is critical for an interaction with the Grb4 adaptor protein. Biochem. J. 377:499-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan, S. W., C. J. Lim, K. Guo, C. P. Ng, I. Lee, W. Hunziker, Q. Zeng, and W. Hong. 2008. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 68:2592-2598. [DOI] [PubMed] [Google Scholar]
- 9.Compagni, A., M. Logan, R. Klein, and R. H. Adams. 2003. Control of skeletal patterning by ephrinB1-EphB interactions. Dev. Cell 5:217-230. [DOI] [PubMed] [Google Scholar]
- 10.Cowan, C. A., and M. Henkemeyer. 2001. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature 413:174-179. [DOI] [PubMed] [Google Scholar]
- 11.Cui, C. B., L. F. Cooper, X. Yang, G. Karsenty, and I. Aukhil. 2003. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell. Biol. 23:1004-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis, S., N. W. Gale, T. H. Aldrich, P. C. Maisonpierre, V. Lhotak, T. Pawson, M. Goldfarb, and G. D. Yancopoulos. 1994. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 266:816-819. [DOI] [PubMed] [Google Scholar]
- 13.Davy, A., J. Aubin, and P. Soriano. 2004. Ephrin-B1 forward and reverse signaling are required during mouse development. Genes Dev. 18:572-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davy, A., J. O. Bush, and P. Soriano. 2006. Inhibition of gap junction communication at ectopic Eph/ephrin boundaries underlies craniofrontonasal syndrome. PLoS Biol. 4:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dottori, M., L. Hartley, M. Galea, G. Paxinos, M. Polizzotto, T. Kilpatrick, P. F. Bartlett, M. Murphy, F. Kontgen, and A. W. Boyd. 1998. EphA4 (Sek1) receptor tyrosine kinase is required for the development of the corticospinal tract. Proc. Natl. Acad. Sci. U. S. A. 95:13248-13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dravis, C., N. Yokoyama, M. J. Chumley, C. A. Cowan, R. E. Silvany, J. Shay, L. A. Baker, and M. Henkemeyer. 2004. Bidirectional signaling mediated by ephrin-B2 and EphB2 controls urorectal development. Dev. Biol. 271:272-290. [DOI] [PubMed] [Google Scholar]
- 17.Florin, L., H. Alter, H. J. Grone, A. Szabowski, G. Schutz, and P. Angel. 2004. Cre recombinase-mediated gene targeting of mesenchymal cells. Genesis 38:139-144. [DOI] [PubMed] [Google Scholar]
- 18.Gerety, S. S., H. U. Wang, Z. F. Chen, and D. J. Anderson. 1999. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 4:403-414. [DOI] [PubMed] [Google Scholar]
- 19.Glaser, D. L., and F. S. Kaplan. 1997. Osteoporosis. Definition and clinical presentation. Spine 22:12S-16S. [DOI] [PubMed] [Google Scholar]
- 20.Govoni, K. E., J. E. Wergedal, R. Chadwick, A. Srivastava, and S. Mohan. 2008. Pre-pubertal OVX increases IGF-I expression and bone accretion in C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab. 295:E1172-E1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Govoni, K. E., J. E. Wergedal, L. Florin, P. Angel, D. J. Baylink, and S. Mohan. 2007. Conditional deletion of IGF-I in collagen type 1{alpha}2 (Col1{alpha}2) expressing cells results in postnatal lethality and a dramatic reduction in bone accretion. Endocrinology 148:5706-5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gubbay, J., J. Collignon, P. Koopman, B. Capel, A. Economou, A. Munsterberg, N. Vivian, P. Goodfellow, and R. Lovell-Badge. 1990. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346:245-250. [DOI] [PubMed] [Google Scholar]
- 23.Hafner, C., S. Meyer, I. Hagen, B. Becker, A. Roesch, M. Landthaler, and T. Vogt. 2005. Ephrin-B reverse signaling induces expression of wound healing associated genes in IEC-6 intestinal epithelial cells. World J. Gastroenterol. 11:4511-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henkemeyer, M., D. Orioli, J. T. Henderson, T. M. Saxton, J. Roder, T. Pawson, and R. Klein. 1996. Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell 86:35-46. [DOI] [PubMed] [Google Scholar]
- 25.Hirai, A., M. Tada, K. Furuuchi, S. Ishikawa, K. Makiyama, J. Hamada, F. Okada, I. Kobayashi, H. Fukuda, and T. Moriuchi. 2004. Expression of AIE-75 PDZ-domain protein induces G2/M cell cycle arrest in human colorectal adenocarcinoma SW480 cells. Cancer Lett. 211:209-218. [DOI] [PubMed] [Google Scholar]
- 26.Holmberg, J., M. Genander, M. M. Halford, C. Anneren, M. Sondell, M. J. Chumley, R. E. Silvany, M. Henkemeyer, and J. Frisen. 2006. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell 125:1151-1163. [DOI] [PubMed] [Google Scholar]
- 27.Hong, J. H., E. S. Hwang, M. T. McManus, A. Amsterdam, Y. Tian, R. Kalmukova, E. Mueller, T. Benjamin, B. M. Spiegelman, P. A. Sharp, N. Hopkins, and M. B. Yaffe. 2005. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309:1074-1078. [DOI] [PubMed] [Google Scholar]
- 28.Hong, J. H., and M. B. Yaffe. 2006. TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell Cycle 5:176-179. [DOI] [PubMed] [Google Scholar]
- 29.Jensen, P. L. 2000. Eph receptors and ephrins. Stem Cells. 18:63-64. [DOI] [PubMed] [Google Scholar]
- 30.Kanai, F., P. A. Marignani, D. Sarbassova, R. Yagi, R. A. Hall, M. Donowitz, A. Hisaminato, T. Fujiwara, Y. Ito, L. C. Cantley, and M. B. Yaffe. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19:6778-6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, H. S., T. G. Nishanian, K. Mood, Y. S. Bong, and I. O. Daar. 2008. EphrinB1 controls cell-cell junctions through the Par polarity complex. Nat. Cell Biol. 10:979-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei, Q. Y., H. Zhang, B. Zhao, Z. Y. Zha, F. Bai, X. H. Pei, S. Zhao, Y. Xiong, and K. L. Guan. 2008. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the Hippo pathway. Mol. Cell. Biol. 28:2426-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin, D., G. D. Gish, Z. Songyang, and T. Pawson. 1999. The carboxyl terminus of B class ephrins constitutes a PDZ domain binding motif. J. Biol. Chem. 274:3726-3733. [DOI] [PubMed] [Google Scholar]
- 34.Lu, Q., E. E. Sun, R. S. Klein, and J. G. Flanagan. 2001. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell 105:69-79. [DOI] [PubMed] [Google Scholar]
- 35.Makinen, T., R. H. Adams, J. Bailey, Q. Lu, A. Ziemiecki, K. Alitalo, R. Klein, and G. A. Wilkinson. 2005. PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev. 19:397-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marquardt, T., R. Shirasaki, S. Ghosh, S. E. Andrews, N. Carter, T. Hunter, and S. L. Pfaff. 2005. Coexpressed EphA receptors and ephrin-A ligands mediate opposing actions on growth cone navigation from distinct membrane domains. Cell 121:127-139. [DOI] [PubMed] [Google Scholar]
- 37.McLeod, M. J. 1980. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology 22:299-301. [DOI] [PubMed] [Google Scholar]
- 38.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063-1066. [DOI] [PubMed] [Google Scholar]
- 39.Orioli, D., M. Henkemeyer, G. Lemke, R. Klein, and T. Pawson. 1996. Sek4 and Nuk receptors cooperate in guidance of commissural axons and in palate formation. EMBO J. 15:6035-6049. [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer, A., M. Zimmer, K. S. Erdmann, V. Eulenburg, A. Porthin, R. Heumann, U. Deutsch, and R. Klein. 2002. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol. Cell 9:725-737. [DOI] [PubMed] [Google Scholar]
- 41.Parfitt, A. M., M. K. Drezner, F. H. Glorieux, J. A. Kanis, H. Malluche, P. J. Meunier, S. M. Ott, and R. R. Recker. 1987. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner Res. 2:595-610. [DOI] [PubMed] [Google Scholar]
- 42.Qin, X., J. E. Wergedal, M. Rehage, K. Tran, J. Newton, P. Lam, D. J. Baylink, and S. Mohan. 2006. Pregnancy-associated plasma protein-A increases osteoblast proliferation in vitro and bone formation in vivo. Endocrinology 147:5653-5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubinson, D. A., C. P. Dillon, A. V. Kwiatkowski, C. Sievers, L. Yang, J. Kopinja, D. L. Rooney, M. Zhang, M. M. Ihrig, M. T. McManus, F. B. Gertler, M. L. Scott, and L. Van Parijs. 2003. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 33:401-406. [DOI] [PubMed] [Google Scholar]
- 44.Schroeder, T. M., A. K. Nair, R. Staggs, A. F. Lamblin, and J. J. Westendorf. 2007. Gene profile analysis of osteoblast genes differentially regulated by histone deacetylase inhibitors. BMC Genomics 8:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shenolikar, S., J. W. Voltz, C. M. Minkoff, J. B. Wade, and E. J. Weinman. 2002. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc. Natl. Acad. Sci. U. S. A. 99:11470-11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Songyang, Z., A. S. Fanning, C. Fu, J. Xu, S. M. Marfatia, A. H. Chishti, A. Crompton, A. C. Chan, J. M. Anderson, and L. C. Cantley. 1997. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73-77. [DOI] [PubMed] [Google Scholar]
- 47.Srivastava, A. K., S. Bhattacharyya, G. Castillo, N. Miyakoshi, S. Mohan, and D. J. Baylink. 2000. Development and evaluation of C-telopeptide enzyme-linked immunoassay for measurement of bone resorption in mouse serum. Bone 27:529-533. [DOI] [PubMed] [Google Scholar]
- 48.Tian, Y., R. Kolb, J. H. Hong, J. Carroll, D. Li, J. You, R. Bronson, M. B. Yaffe, J. Zhou, and T. Benjamin. 2007. TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol. Cell. Biol. 27:6383-6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torres, R., B. L. Firestein, H. Dong, J. Staudinger, E. N. Olson, R. L. Huganir, D. S. Bredt, N. W. Gale, and G. D. Yancopoulos. 1998. PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron 21:1453-1463. [DOI] [PubMed] [Google Scholar]
- 50.Twigg, S. R., R. Kan, C. Babbs, E. G. Bochukova, S. P. Robertson, S. A. Wall, G. M. Morriss-Kay, and A. O. Wilkie. 2004. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. U. S. A. 101:8652-8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Twigg, S. R., K. Matsumoto, A. M. Kidd, A. Goriely, I. B. Taylor, R. B. Fisher, A. J. Hoogeboom, I. M. Mathijssen, M. T. Lourenco, J. E. Morton, E. Sweeney, L. C. Wilson, H. G. Brunner, J. B. Mulliken, S. A. Wall, and A. O. Wilkie. 2006. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. Am. J. Hum. Genet. 78:999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams, S. E., F. Mann, L. Erskine, T. Sakurai, S. Wei, D. J. Rossi, N. W. Gale, C. E. Holt, C. A. Mason, and M. Henkemeyer. 2003. Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron 39:919-935. [DOI] [PubMed] [Google Scholar]
- 53.Wong, G. L., and D. V. Cohn. 1975. Target cells in bone for parathormone and calcitonin are different: enrichment for each cell type by sequential digestion of mouse calvaria and selective adhesion to polymeric surfaces. Proc. Natl. Acad. Sci. U. S. A. 72:3167-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xing, W., D. Baylink, C. Kesavan, Y. Hu, S. Kapoor, R. B. Chadwick, and S. Mohan. 2005. Global gene expression analysis in the bones reveals involvement of several novel genes and pathways in mediating an anabolic response of mechanical loading in mice. J. Cell. Biochem. 96:1049-1060. [DOI] [PubMed] [Google Scholar]
- 55.Xing, W., D. Baylink, C. Kesavan, and S. Mohan. 2004. HSV-1 amplicon-mediated transfer of 128-kb BMP-2 genomic locus stimulates osteoblast differentiation in vitro. Biochem. Biophys. Res. Commun. 319:781-786. [DOI] [PubMed] [Google Scholar]
- 56.Xing, W., A. Singgih, A. Kapoor, C. M. Alarcon, D. J. Baylink, and S. Mohan. 2007. Nuclear factor-E2-related factor-1 mediates ascorbic acid induction of osterix expression via interaction with antioxidant-responsive element in bone cells. J. Biol. Chem. 282:22052-22061. [DOI] [PubMed] [Google Scholar]
- 57.Yu, G., H. Luo, Y. Wu, and J. Wu. 2004. EphrinB1 is essential in T-cell-T-cell cooperation during T-cell activation. J. Biol. Chem. 279:55531-55539. [DOI] [PubMed] [Google Scholar]
- 58.Zhao, C., N. Irie, Y. Takada, K. Shimoda, T. Miyamoto, T. Nishiwaki, T. Suda, and K. Matsuo. 2006. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 4:111-121. [DOI] [PubMed] [Google Scholar]