

Abstract
The ends of chromosomes, called telomeres, are composed of a DNA repeat sequence and associated proteins, which prevent DNA degradation and chromosome fusion. We have previously used plasmid sequences integrated adjacent to a telomere to demonstrate that mammalian telomeres suppress gene expression, called telomere position effect (TPE). We have also shown that subtelomeric regions are highly sensitive to double-strand breaks, leading to chromosome instability, and that this instability can be prevented by the addition of a new telomere to the break, a process called chromosome healing. We have now targeted the same plasmid sequences to a site 100 kb from a telomere in a human carcinoma cell line to address the effect of telomere proximity on telomere position effect, chromosome healing, and sensitivity to double-strand breaks. The results demonstrate a substantial decrease in TPE 100 kb from the telomere, demonstrating that TPE is very limited in range. Chromosome healing was also diminished 100 kb from the telomere, consistent with our model that chromosome healing serves as a repair process for restoring lost telomeres. Conversely, the region 100 kb from the telomere was highly sensitive to double-strand breaks, demonstrating that the sensitive region is a relatively large target for ionizing radiation-induced chromosome instability.
Telomeres are composed of a six-base pair repeat sequence and associated proteins that together form a cap to protect the ends of chromosomes and prevent chromosome fusion (6). Telomeres are actively maintained by the enzyme telomerase in human germ line cells but shorten with age in most somatic cells due to the low level of expression of telomerase (12). When a telomere shortens to the point that it is recognized as a double-strand break (DSB), it serves as a signal for replicative cell senescence (13). Human cells that lose the ability to senesce continue to show telomere shortening and eventually enter crisis, which involves increased chromosome fusion, aneuploidy, and cell death (11, 15). An important step that is required for continued division of cancer cells is therefore that they possess the ability to maintain telomeres, not only to avoid senescence but also to avoid chromosome fusion brought on by crisis (11, 25).
In addition to their role in protecting the ends of chromosomes, telomeres can also inhibit the expression of nearby genes, called telomere position effect (TPE). TPE has been proposed to have a role in the cellular response to changes in telomere length (26); however, the function of TPE remains unknown. TPE has been extensively studied in Saccharomyces cerevisiae using transgenes integrated near telomeres on truncated chromosomes (1, 2, 22, 47). These studies demonstrated that TPE involves changes in chromatin conformation and is dependent upon both the distance from the telomere and telomere length (55). Subsequent studies of endogenous yeast genes, however, revealed that the influence of TPE on gene expression varies depending on the presence of insulator sequences (18, 45). TPE also occurs in mammalian cells and has been implicated in the loss of expression of genes relocated near telomeres in a variety of human syndromes (9, 16, 28, 58, 59). As in yeast, transgenes located near telomeres have been used to study TPE in the C33-A (32) and HeLa (4) human cervical carcinoma cell lines. We have also studied TPE using transgenes located adjacent to telomeres in mouse embryonic stem (ES) cells, mouse embryo fibroblasts, and transgenic mice (43). However, none of the studies of TPE in mammalian cells has addressed the distance over which TPE extends from the telomere, and so the number of genes whose expression is likely to be affected is not known.
The presence of a telomere can also influence the sensitivity of subtelomeric regions to DSBs. We previously demonstrated the sensitivity of subtelomeric regions to DSBs using selectable transgenes and a recognition site for the I-SceI endonuclease that are integrated immediately adjacent to a telomere. Unlike I-SceI-induced DSBs at most locations, which primarily result in small deletions (27, 34, 46, 50), I-SceI-induced DSBs near telomeres commonly result in large deletions, gross chromosome rearrangements (GCRs), and chromosome instability in both mouse ES cells (37) and human tumor cells (65). Therefore, depending on the size of the sensitive region, the combined targets of the subtelomeric regions on all telomeres could contribute significantly to the genomic instability caused by ionizing radiation or other agents that produce DSBs (35). This sensitivity to DSBs may result from a deficiency in DSB repair since regions near telomeres in yeast are deficient in nonhomologous end joining, resulting in an increase in GCRs (48). One possible reason for a deficiency in DSB repair near telomeres is the role of the telomere in preventing chromosome fusion. Telomeric repeat sequences in yeast have been shown to suppress the activation of cell cycle checkpoints in response to DSBs (39). Similarly, the human TRF2 protein, which is required to prevent chromosome fusion, has been demonstrated to inhibit ATM (31), whose activation is instrumental in the repair of DSBs in heterochromatin (20).
One mechanism for avoiding the consequences of DSBs near telomeres is through the addition of a new telomere to the site of a DSB, termed chromosome healing (44). Studies in yeast have shown that chromosome healing occurs through the de novo addition of telomeric repeat sequences by telomerase (14, 33, 38). Chromosome healing in S. cerevisiae is inhibited by the 5′-3′ helicase, Pif1 (52), with Pif1-deficient cells showing up to a 1,000-fold increase in chromosome healing (33, 38). The ability of Pif1 to inhibit chromosome healing has been proposed to serve as a mechanism to prevent chromosome healing from interfering with DSB repair (63). Mammalian cells that express telomerase are also capable of performing chromosome healing. We have shown that chromosome healing can also occur following spontaneous telomere loss (17, 49) or DSBs near telomeres in a human cancer cell line (65) or mouse ES cells (19, 54). We have also shown that chromosome healing can prevent the chromosome instability resulting from DSBs near telomeres (19). Because the de novo addition of telomeric repeat sequences has not been observed in mammalian cells at I-SceI-induced DSBs at interstitial sites (27, 34, 46, 50), we have proposed that chromosome healing is inhibited at most locations but serves as an important mechanism for dealing with DSBs near telomeres that would otherwise result in chromosome instability. However, an alternative possibility that has not been ruled out is that chromosome healing also occurs at interstitial sites but that the large terminal deletions that it causes at these sites results in cell death.
In the present study, we address several key questions regarding the importance of telomere proximity on TPE, chromosome healing, and sensitivity to DSBs by investigating how telomere proximity affects these processes. The first of these questions involves establishing the distance over which TPE extends from the telomere to gain insights into the numbers of genes that would be affected by changes in TPE. Second, we will investigate whether chromosome healing can occur at a site that is distant from a telomere but in which terminal deletions are known not to be lethal. This will determine for the first time whether chromosome healing is limited to regions near telomeres. Finally, we will investigate the size of the region near a telomere that is sensitive to DSBs, which will address the potential importance of the subtelomeric region as a target for ionizing radiation-induced genomic instability (35). The distance over which a telomere can exert its effects was investigated by comparing TPE, chromosome healing, and the sensitivity to DSBs at a site 100 kb from a telomere with a site immediately adjacent to the same telomere. As a control for the efficiency of generating DSBs at these sites, we have also analyzed the frequency of small deletions, the most common type of I-SceI-induced DNA rearrangement at interstitial sites in mammalian cells (27, 60). Small deletions serve as an excellent internal control for comparing the frequency of other types of rearrangements since we have previously observed a similar frequency of small deletions at telomeric and interstitial sites (65). The results provide important information on the distance over which a telomere can influence TPE, chromosome healing, and the sensitivity to DSBs.
MATERIALS AND METHODS
Plasmids.
The pNCT-tel plasmid that is integrated in clone B3 of the human bladder cell carcinoma cell line has been previously described (17). pNCT-tel contains a neomycin resistance (Neo) gene for positive selection with G418, a herpes simplex virus thymidine kinase (HSV-tk) gene for negative selection with ganciclovir, an I-SceI recognition site for introducing DSBs, and 0.8 kb of telomeric repeat sequences for seeding the formation of new telomeres (Fig. 1) .
FIG. 1.

The structure of plasmids used in this study. pNCT-tel contains a Neo gene, HSV-tk gene, and telomeric repeat sequences. pNCT-tel also contains an I-SceI recognition site, which is located between the Neo and HSV-tk genes. pNCT-tel was linearized with NotI prior to transfection. Linearization places the telomeric repeat sequences on one end to seed the formation of a new telomere following integration. pNTIL contains the Neo gene and an HSV-tk gene that contains a LoxP site in its 3′ untranslated region. pNTIL also contains an I-SceI recognition site, which is located at the 5′ end of the coding region of HSV-tk. pZTIL-tel contains a Zeo gene and a fragment of the noncoding region of the HSV-tk gene containing a LoxP site. pZTIL-tel was also linearized with NotI prior to transfection, both to facilitate targeted integration into the pNCT-tel plasmid and to place the telomeric repeat sequences on one end to seed the formation of a new telomere. pNTIL-100kb is similar to pNTIL, except that it contains a 4.5-kb fragment of the magmus gene (Target seq) for targeting to a site 100 kb from the telomere containing the pZTIL-tel plasmid. The magmus gene fragment is oriented with its distal end closest to the ampicillin gene (arrow).
The pNTIL plasmid has been previously described (65). pNTIL was derived from the pNCT plasmid (lacking telomeric repeat sequences) by insertion of a LoxP site in the 3′ untranslated region of the HSV-tk gene and moving I-SceI recognition from between the Neo and HSV-tk genes to a new site located in the 5′ coding region of the HSV-tk gene (Fig. 1). Despite being within the coding region, small deletions causing frameshifts at the I-SceI site do not inactivate the HSV-tk gene, apparently because truncated forms of HSV-tk that initiate translation from the second or third methionine are also functional (24, 29).
The pZTIL-tel plasmid that was used to target and replace the pNCT-tel plasmid in EJ-30 clone B3 contains a zeomycin resistance (Zeo) gene, a LoxP site located in a fragment of the HSV-tk gene containing the 3′untranslated region, and 0.8 kb of telomeric repeat sequences for the formation of new telomeres (Fig. 1). pZTIL-tel was derived from the pNTIL plasmid by first inserting a 1.2-kb BglII/SphI fragment containing the Zeo gene from the pCMVZeo plasmid (Invitrogen-Gibco) between the BamHI and SphI sites. This resulted in the deletion of the Neo gene, the I-SceI recognition site, and most of the HSV-tk gene but left the 5′ noncoding region of the HSV-tk gene containing the LoxP site. A 0.8-kb BamHI-BglII fragment containing the telomeric repeat sequences from pNCT-tel was then inserted into the compatible BglII site in pZTIL to create pZTIL-tel.
The pNTIL-100kb plasmid that was used to generate EJ-30 clone 10-7 was generated from the pNTIL plasmid by inserting a 4.5-kb fragment from the magmus gene (Fig. 1). The magmus gene fragment was added to allow for targeted integration into the magmus gene and was oriented with its distal end (Fig. 1, arrow) closest to the ampicillin gene so that targeted integration would result in the HSV-tk gene's being distal to the Neo gene. The magmus gene fragment was generated by PCR. PCR employed 35 cycles of 94°C for 30 s, 60°C for 30 s, and 68°C for 5 min using the primers M2-2F (5′-CAGCTCGGACTCCAACTTTC-3′) and M2-2R (5′-CTGCAGGTGAGAAGCCCTAC-3′). The 4.5-kb PCR fragment was cloned into the pCR-XL-TOPO cloning vector (Invitrogen-Gibco), excised using EagI sites in the multicloning site, and cloned into the pNTIL plasmid at an EagI-compatible NotI site between the HSV-tk and ampicillin resistance genes to generate the pNTIL-100 kb plasmid. The magmus gene fragment is oriented with its distal end closest to the ampicillin gene so that targeted integration will result in the HSV-tk gene's being distal to the Neo gene.
The pQCXIP-ISceI retroviral vector that was used for expression of the I-SceI gene was constructed as previously described (65).
Cell lines.
All of the cell lines used in this study were derived from clone B3 of the EJ-30 human bladder cell carcinoma cell line. EJ-30 is a subclone of the EJ cell line, which is also called MGH-U1 (42). All cells were grown in alpha-minimal essential medium (αMEM; University of California, San Francisco [UCSF] Cell Culture Facility) supplemented with 5% fetal calf serum (Invitrogen-Gibco), 5% newborn calf serum with iron (Invitrogen-Gibco), and 1 mM l-glutamine (Invitrogen-Gibco) and were propagated at 37°C in humidified incubators.
Cell clone B3 was derived from EJ-30 following transfection with the linearized pNCT-tel plasmid (17), which seeded the formation of a new telomere 4.29 Mb from the original telomere on the short arm of chromosome 16 (Fig. 2) . Rescue of the integrated pNCT-tel plasmid following digestion with BamHI (37) and sequencing of adjacent cellular DNA demonstrated that the plasmid integrated in the first intron at the 5′ end of the sarcalumenin gene (GenBank accession number GQ475285).
FIG. 2.
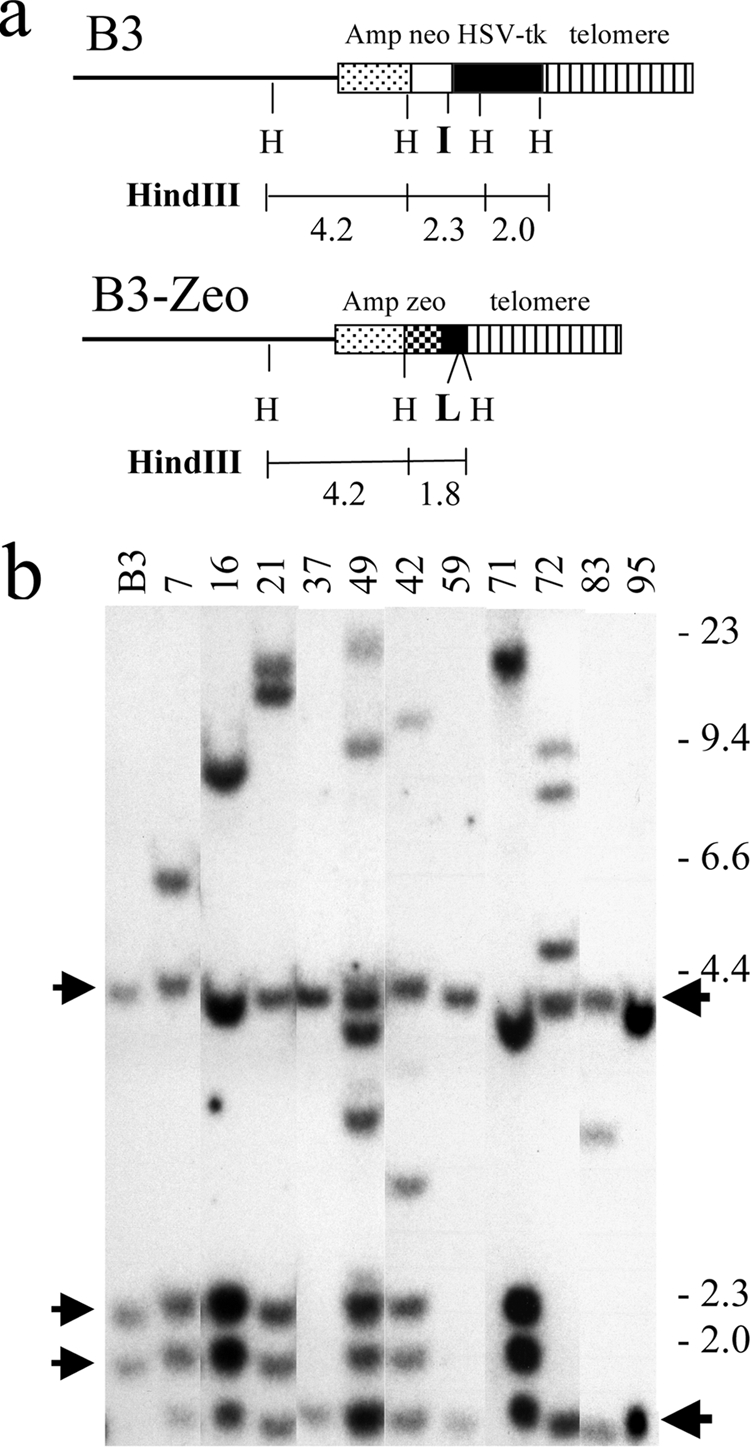
Southern blot analysis for identification of B3-Zeo clones in which the pZTIL-tel plasmid has been targeted to replace the pNCT-tel plasmid in clone B3. (a) The structure of the integrated plasmid sequences and the sizes of the predicted HindII restriction fragments are shown for the parental clone B3 and for clones containing the pZTIL-tel plasmid (B3-Zeo). (b) Southern blot analysis using the pNCTΔ plasmid of genomic DNA from clone B3 results in three HindIII restriction fragments (4.2, 2.3, and 2.0 kb) (arrows at left). B3-Zeo clones containing the targeted pZTIL-tel plasmid (clones 37, 59, and 95) contain two HindIII restriction fragments (4.2 and 1.8 kb; arrows at right). The location of the HindIII (H) restriction sites, the recognition site for the I-SceI endonuclease, and the recognition site for Cre recombinase (loxP) are shown. The molecular size markers (kb) consist of lambda bacteriophage HindIII fragments. This figure was generated by combining data from different Southern blots containing genomic DNA from B3-Zeo clones.
Cell clone B3-Zeo37 was derived from clone B3 following the replacement of the pNCT-tel plasmid with the pZTIL-tel plasmid using targeted integration (Fig. 2). Clone B3-Zeo37 therefore contains the pZTIL-tel plasmid integrated adjacent to the telomere on the short arm of chromosome 16 (Fig. 3). Upon linearization with NotI, the Amp/ori sequences in pZTIL-tel are at one end, and the telomeric repeat sequences are at the other end. The linearized plasmid was then transfected into clone B3 using Lipofectamine 2000, as recommended by the manufacturer (Invitrogen). Targeting into the pNCT-tel plasmid was facilitated by cotransfection with the pCBASce expression vector that contains the I-SceI gene (48a) to generate DSBs at the I-SceI site in the pNCT-tel plasmid. Following transfection, the cells were grown in the presence of 50 μg/ml zeomycin (Sigma) to select for cells containing the integrated pZTIL-tel plasmid, and individual colonies were picked and tested for sensitivity to G418 to identify clones that had lost the Neo gene. Southern blot analysis was then used to identify clones that had undergone targeted integration (Fig. 2b).
FIG. 3.

The structure of the integrated plasmid sequences in the various cell clones used in this study. Clone B3-Zeo37 contains a single copy of the pZTIL-tel plasmid located adjacent to the telomere on the short arm of chromosome 16. Clone 10-7 was derived by targeted integration of the pNTIL-100kb plasmid to a site 100 kb from the telomeric pZTIL-tel plasmid in clone B3-Zeo37. The targeted integration of pNTIL-100kb resulted in the duplication of the target sequence on either side of the integrated plasmid, typical of insertion-type targeting vectors (56). The 10-7Cre clones were derived from 10-7 following Cre recombinase-mediated recombination between the LoxP sites, which resulted in the repositioning of the telomere to a location immediately adjacent to the pNTIL-100kb plasmid. The location of restriction sites and the size of restriction fragments are shown for the BglII (B), HindIII (H), NcoI (N), and XbaI (X) restriction enzymes. Also shown are the locations of the I-SceI and LoxP sites (boldface I and L, respectively) recognized by the I-SceI endonuclease and Cre recombinase, respectively. Symbols are shown for the location of the telomere, plasmid vector sequences (Amp/ori), Neo gene, HSV-tk gene, Zeo gene, and cellular sequence used for targeted integration of pNTIL-100kb (Target site).
Cell clone 10-7 and its two subclones, 10-7A and 10-7B, were derived from clone B3-Zeo37 by targeting the pNTIL-100kb plasmid to a site in the magmus gene, which is located 4.39 Mb from the original end of chromosome 16p. The pNTIL-100kb integration site in clone 10-7 is therefore approximately 100 kb away from the pZTIL-tel plasmid at the newly seeded telomere on this chromosome (Fig. 3). pNTIL-100kb was cut at a unique AvrII site found near the center of the 4.5-kb magmus gene fragment prior to transfection to facilitate integration into the target sequence. Following transfection using Lipofectamine 2000 (Invitrogen-Gibco), the cultures were grown in medium containing 400 μg/ml G418 to select for B3-Zeo37 cells containing the integrated pNTIL-100kb plasmid. Individual colonies were then selected, and their genomic DNA was analyzed by Southern blot analysis for targeted integration into the magmus gene (Fig. 3).
The 10-7Cre cell clones were generated from clone 10-7 following transient transfection with the pBS185 plasmid, which contains the Cre gene under the control of the major immediate early promoter and enhancer for the human cytomegalovirus (51). The 10-7Cre cell clones contain the pNTIL-100kb plasmid located immediately adjacent to a telomere following Cre-mediated recombination between the LoxP sites in the pNTIL-100kb plasmid and the pZTIL-tel plasmid (Fig. 3).
Virus preparation and selection for virus-infected cells.
Packaging of the pQCXIP control and pQCXIP-I-SceI retroviral vectors and infection of cell cultures was performed as previously described (65). The selection for infected cells was achieved through addition of growth medium containing 2 μg/ml puromycin (Sigma). Following infection, the cells were cultured for 10 days in medium containing puromycin, with medium changes every 2 days, to allow for expression of I-SceI endonuclease and the generation of DSBs. After 10 days, the cells were trypsinized, pooled together, and replated into T-75 tissue culture flasks. The pooled cells were then either used for preparation of genomic DNA for analysis of small deletions by PCR or replated as single cells in medium with ganciclovir for analysis of large deletions or GCRs. Replating was necessary for analysis of large deletions or GCRs because the initial puromycin-resistant (puror) colonies contain a mixture of rearrangements as a result of the relatively slow rate of cutting of the I-SceI endonuclease in the infected cells. The analysis of the consequences of I-SceI-induced DSBs was performed 10 days after virus infection because our preliminary studies using PCR and ganciclovir selection demonstrated that the percentage of cells with small deletions and ganciclovir resistance (Ganr) remained constant 6 to 10 days after infection (data not shown).
Southern blot analysis and plasmid rescue.
Individual Ganr subclones were selected by ring cloning, and Southern blot analysis of genomic DNA from the individual Ganr subclones was performed following digestion with HindIII as previously described (19). Plasmid rescue for analysis of DNA rearrangements in the 10-7 subclones 10-7A and 10-7B was performed following digestion of genomic DNA with AvrII using standard laboratory protocols (19, 37). Briefly, the genomic DNA was digested with the restriction enzyme, and the DNA was circularized by ligation at a dilute concentration of 1 μg/ml overnight at 16°C, reconcentrated to 50 μl using sequential Ultra-4/Microcon 30,000 molecular-weight-cutoff (MWCO) spin filters (Millipore), and then used to transform STBL2 bacteria (Invitrogen). The selection for cells containing the plasmid was performed using ampicillin, and the rescued plasmids were mapped with restriction enzymes and sequenced using various primers specific for the plasmid DNA.
Analysis of small deletions.
The presence of small deletions was analyzed by digesting PCR products spanning the I-SceI site in the integrated pNTIL-100kb plasmid with I-SceI endonuclease. Long PCR was performed on genomic DNA isolated from the pooled puror cell cultures following infection with the retroviral vectors. Long PCR was performed using an Elongase PCR kit (Invitrogen) as described by the manufacturer, using primers CMV1-F (TATATGGAGTTCCGCGTTACA) and TK1-R (GTTTGGCCAAGACGTCCAAG). PCR involved 94°C for 30 s and then 35 cycles of 94°C for 30 s, 57°C for 30 s, and 68°C for 105 s. A total of 25 μl of the PCR product was then digested with 20 units of I-SceI endonuclease at 37°C overnight, and the products were run on 1% agarose gels. After samples were stained with ethidium bromide, digital images were analyzed using free ImageJ software (http://www.versiontracker.com/dyn/moreinfo/macosx/37303) to calculate the intensity of the bands. The percentage of cells containing small deletions at the I-SceI site was determined by dividing the intensity of the uncut band by the combined intensity of the cut and uncut bands.
Quantitative PCR.
Total RNA was prepared using a Midi purification kit (Invitrogen, Carlsbad, CA). Samples were treated with a DNA-free kit (Ambion, Austin TX) to remove traces of genomic DNA contamination. Preparation of cDNA from these RNA samples was done as described previously (10) with the following modifications: reaction mixtures were incubated for 40 min at 48°C, using 7.5 mM MgCl2, 1 mM each deoxynucleoside triphosphate (dNTP), 5 μM hexamers (Random Primers, Invitrogen), and 2.5 U/μl of Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad CA). To control for genomic DNA contamination, a mock cDNA preparation of each sample was prepared in a parallel reaction without the presence of the reverse transcriptase (RT). Real-time quantitative RT-PCR (q-RT-PCR) was performed in triplicate by the UCSF Cancer Center Genome Analysis core facility using TaqMan dually labeled probes (PerkinElmer, Norwalk, CT) and specific primers using an ABI 7700 Prism real-time thermocycler (PE Biosystems, Foster City, CA) as previously described (43).
RESULTS
Development of human tumor cell clones with transgenes and an I-SceI site located 100 kb from a telomere.
The distance over which a telomere can exert its influence on TPE, chromosome healing, and sensitivity to DSBs in mammalian cells was investigated using EJ-30 clone 10-7. Clone 10-7 was derived from clone B3, which we have previously used to investigate the frequency and types of rearrangements resulting form spontaneous and I-SceI-induced DSBs near a telomere (17, 36, 49, 65). The consequences of DSBs near a telomere in clone B3 was made possible by the presence of the pNCT-tel plasmid integrated immediately adjacent to a telomere, which was accomplished by using telomeric repeat sequences on the end of the linearized plasmid to seed the formation of a new telomere 4.29 Mb from the original end of chromosome 16p (17). Cell clones derived from clone B3 have the advantage that the rearrangements resulting from DSBs occurring at this telomere have been well characterized and compared with DSBs occurring at interstitial sites (65).
The first step in generating clone 10-7 was to replace the telomeric pNCT-tel plasmid in clone B3 with the pZTIL-tel plasmid by targeted integration. The pZTIL-tel plasmid was targeted by homologous recombination to replace the pNCT-tel plasmid that functions as the telomere on the short arm of chromosome 16p (Fig. 2a). The replacement of the pNCT-tel plasmid in clone B3 with the pZTIL-tel plasmid serves two purposes. First, it removes the Neo gene, I-SceI site, and HSV-tk gene so that they can be integrated at another site 100 kb from the telomere. Second, it introduces a LoxP site near the telomere so that the telomere can subsequently be relocated to a site immediately adjacent to the plasmid sequences integrated 100 kb from the telomere. The pZTIL-tel plasmid contains the Zeo gene for positive selection, telomeric repeat sequences, and the LoxP site at the 3′ end of the HSV-tk gene (see Materials and Methods). Southern blot analysis showed that 3 of 12 Zeor/G418s clones (B3-Zeo37, B3-Zeo59, and B3-Zeo94) had the plasmid targeted to the telomere, as shown by the presence of two HindIII restriction fragments that are 1.8 and 4.2 kb in length, rather than the three original fragments in the parental B3 clone that are 2.0, 2.3, and 4.2 kb in length (Fig. 2b). One of the clones containing the telomeric pZTIL-tel plasmid, B3-Zeo37, was used for subsequent steps.
The next step in generating clone 10-7 was to target the pNTIL-100kb plasmid to a site 100 kb from the telomere in clone B3-Zeo37 that contains the pZTIL-tel plasmid (see Materials and Methods), which resulted in the generation of clone 10-7 (Fig. 3). The pNTIL-100 kb plasmid contains a Neo gene, an HSV-tk gene, an I-SceI recognition site, and a LoxP recognition site. For targeted integration, pNTIL-100kb also contains a 4.5-kb nonrepetitive sequence found in the magmus gene, which is located 100 kb proximal to the telomeric pZTIL-tel plasmid. Southern blot analysis of genomic DNA isolated from 100 G418-resistant clones of B3-Zeo37 transfected with pNTIL-100kb demonstrated that one clone, 10-7, contained the expected restriction fragments, indicating that targeted integration had occurred (Fig. 4a). Targeted integration was confirmed by rescue of the integrated pNTIL-100kb plasmid and adjacent cellular DNA and sequencing of the cellular DNA proximal to the end of the 4.5-kb magmus gene fragment used for targeting (data not shown).
FIG. 4.
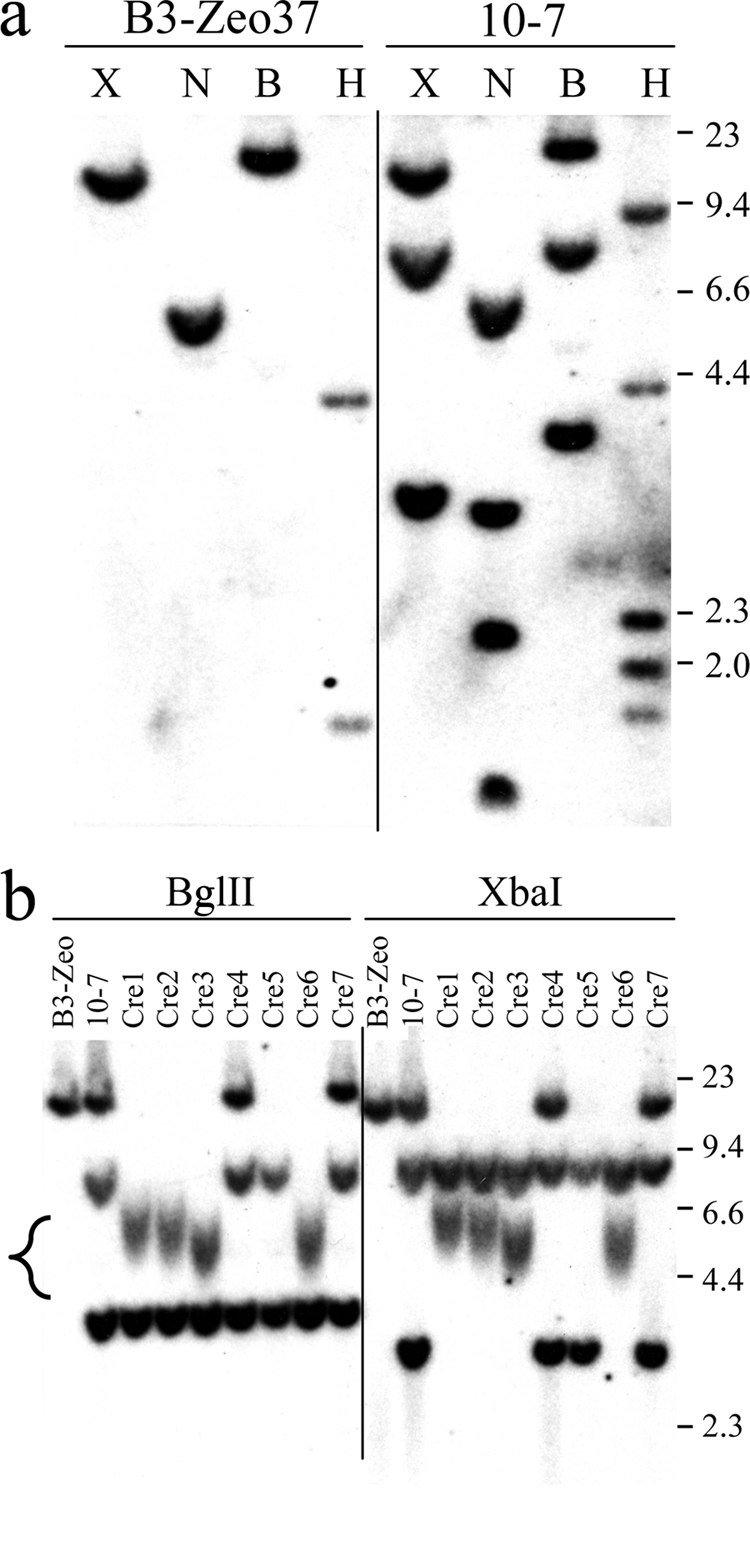
Southern blot analysis of the structure of the integrated plasmid sequences in clone 10-7 and the 10-7Cre clones. (a) Genomic DNA from the parental clone B3-Zeo37 and clone 10-h7 was digested with the BglII (B), HindIII (H), NcoI (N), and XbaI (X) restriction enzymes, and hybridization was performed using the pNCTΔ plasmid as a probe. The sizes of the restriction fragments in clone 10-7 correspond to those expected from targeted integration of the pNTIL-100kb plasmid into the site 100 kb from the telomere containing the pZTIL-tel plasmid (Fig. 3). Targeted integration in clone 10-7 was confirmed by rescue of the pNTIL-100kb plasmid and sequencing of the cellular DNA adjacent to the integration site. (b) Genomic DNA from clone B3-Zeo, clone 10-7, and seven of the 10-7Cre clones (Cre1 to Cre7) was digested with BglII or XbaI, and hybridization was performed using the pNCTΔ plasmid as a probe. The sizes of the restriction fragments in 10-7Cre1, 10-7Cre2, 10-7Cre3, and 10-7Cre6 correspond to those expected following Cre-mediated recombination between the LoxP sites in the pNTIL-100kb and pZTIL-tel plasmids (Fig. 3). Terminal restriction fragments that contain the telomeric repeat sequences are heterogeneous in length due to variability in the length of the telomere in different cells in the population (bracket, lower left). The sizes of the molecular size markers (kb) consisting of lambda bacteriophage HindIII fragments are shown.
Cell clones with the telomere relocated immediately adjacent to the pNTIL-100kb plasmid.
Cre-mediated recombination between the LoxP sites in the pNTIL-100kb and pZTIL-tel plasmids in clone 10-7 was used to generate cell clones that have the pNTIL100-kb plasmid located immediately adjacent to the telomere (Fig. 3). Following transient transfection of clone 10-7 with the pBS185 expression vector containing the gene for Cre recombinase, individual colonies were selected and analyzed for sensitivity to zeomycin to identify clones with deletions of the region between the LoxP sites that contain the Zeo gene. Southern blot analysis demonstrated that of the 72 clones that were selected at random and analyzed, 4 had undergone Cre-mediated recombination to reposition the telomere adjacent to the pNTIL-100kb plasmid (Fig. 4b). The presence of the telomere adjacent to the plasmid in these four clones, 10-7Cre1, 10-7Cre2, 10-7Cre3, and 10-7Cre6, was evident from the presence of a diffuse band containing the terminal restriction fragment, which results from the variation in the size of the telomere in different cells in the population. The average length of the telomere in these clones was estimated from the size of the band to be approximately 3.0 kb for clones 10-7Cre1 and 10-7Cre2 and 2.5 kb for clones 10-7Cre3 and 10-7Cre6, after subtracting the portion of the fragment containing plasmid DNA (3.0 kb). This telomere length is consistent with our earlier analysis of telomere length on the end of this chromosome in B3 subclones, which ranged from 3.0 to 5.0 kb (36). Proof that the pNTIL-100kb plasmid is integrated on the same chromosome 16 homolog as pZTIL-tel is provided by the high frequency of Cre-mediated recombination between the LoxP sites since recombination between LoxP sites on different chromosomes occurs at a much lower frequency (2.1 ×10−5 to 4.2 × 10−5) in cells transiently transfected with Cre recombinase (57). Additional proof that the pNTIL-100kb plasmid is integrated on the end of the same chromosome as pZTIL-tel is provided by the high frequency of loss of the pZTIL-tel plasmid following I-SceI-induced DSBs in the pNTIL-100kb plasmid (see below).
The influence of telomere proximity on TPE.
Earlier studies on human tumor cells have demonstrated that transgenes located immediately adjacent to a telomere have greatly reduced expression compared to transgenes integrated at random interstitial sites (4, 32). However, these studies did not investigate the distance over which this TPE extended from the telomere. This is a critical question because the distance that TPE extends from the telomere will determine the number of genes that will be affected by the changes in TPE brought about by telomere shortening. We therefore compared the level of expression of the Neo and HSV-tk genes in the parental 10-7 cell line with the level of expression in the four 10-7Cre clones in which the telomere had been relocated to a position adjacent to the pNTIL-100kb plasmid (Fig. 5). The level of expression of the Neo and HSV-tk genes in clone 10-7 were 1.5% and 5.0% of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression, respectively. Although relatively low compared to the highly expressed GAPDH gene, these levels of expression are comparable to the level of expression of these same transgenes at interstitial sites in mouse embryonic stem (ES) cell lines (43). As in ES cells, the HSV-tk gene in 10-7 was expressed at higher levels than the Neo gene. The level of expression of both genes was greatly reduced in all four clones in which the telomere was relocated adjacent to the pNTIL-100kb plasmid using Cre-mediated recombination (Fig. 5). The average reduction in the level of expression was 12-fold for the Neo gene and 7.8-fold for the HSV-tk gene, which is comparable to the average 10-fold lower level of expression of transgenes when they are integrated adjacent to telomeres in the HeLa human tumor cell line (4). Our results therefore demonstrate that TPE is significantly diminished at a distance of 100 kb from this telomere.
FIG. 5.

Decreased expression of transgenes following the relocation of the telomere to a site adjacent to the pNTIL-100kb plasmid. q-RT-PCR was used to determine the level of expression of the Neo gene and the HSV-tk gene in the parental clone 10-7 and four clones (10-7Cre1, 10-7Cre2, 10-7Cre3, and 10-7Cre6), that were derived from 10-7 following Cre-mediated recombination between LoxP sites in the pNTIL-100kb and pZTIL-tel plasmids. The results are given as percentage of the level of GAPDH gene expression. Error bars represent the standard deviation from three separate experiments. The levels of expression of both the Neo and HSV-tk genes were significantly reduced in all four Cre clones compared to clone 10-7 (P < 0.0001 for Neo and HSV-tk in Cre1, Cre 3, and Cre6; P < 0.0004 and P < 0.001 for Neo and HSV-tk, respectively, in Cre2).
The frequency of large deletions and GCRs at DSBs 100 kb from a telomere.
We have previously shown that DSBs occurring within the pNTIL-tel or pNCT-tel plasmids located at a telomere result in a greatly increased frequency of GCRs and large deletions compared to DSBs occurring within these plasmids integrated at random interstitial sites (65). The size of the subtelomeric region sensitive to DSBs is important because it determines the size of the target that is likely to experience DSB-induced chromosome instability as a result of exposure to ionizing radiation. Genomic instability is now considered to be an important mechanism of ionizing radiation-induced carcinogenesis (35). Our earlier results showed that the subtelomeric region on the short arm of chromosome 16 that now contains the pNTIL-100kb plasmid in clone 10-7 is highly sensitive to I-SceI-induced DSBs occurring 3 kb from the telomere (65). To determine whether this sensitivity to DSBs extends 100 kb from the telomere, we examined the frequency of large deletions and GCRs resulting from I-SceI-induced DSBs in the pNTIL-100kb plasmid in two 10-7 subclones, 10-7A and 10-7B. To introduce the DSBs, clones 10-7A and 10-7B were infected with the pQCXIP-ISceI retrovirus, and cells constitutively expressing I-SceI were selected with puromycin. After 10 days to allow for the generation of DSBs and turnover of the HSV-tk protein, the cells were plated into medium containing ganciclovir to select for cells that had lost HSV-tk gene function. The inactivation of the HSV-tk gene in the pNTIL-100kb plasmid in clone 10-7 requires a deletion of at least 76 bp at the I-SceI site to eliminate all possible start codons. The 10-7A and 10-7B subclones infected with the pQCXIP-ISceI retrovirus showed a large increase in the frequency of Ganr cells of 36% and 54%, respectively. This large increase in Ganr cells was similar to the frequency previously observed in clones with telomeric I-SceI-induced DSBs (31.5 to 48.4%) and was much higher than the frequency observed in clones with interstitial, I-SceI-induced DSBs (0.8 to 3.4%) (65). The results therefore suggested that the region 100 kb from this telomere continues to show increased sensitivity to DSBs compared to most locations in the human genome.
Southern blot analysis was performed on genomic DNA from the Ganr subclones of 10-7A and 10-7B to characterize the types of rearrangements responsible for inactivation of the HSV-tk gene (Fig. 6). The types of events that occurred were categorized by whether they resulted in complex rearrangements at the I-SceI site, large deletions at the I-SceI site, or terminal deletions (Table 1). Complex rearrangements result in the loss of one or more of the three HindIII bands containing the pNTIL-100kb plasmid (9.4, 2.3, and 2.0 kb) with or without the appearance of a new band, while large deletions result in the loss of all three of the pNTIL-100kb HindIII bands without the appearance of new bands. Terminal deletions result in the loss of the two HindIII bands containing the pZTIL-tel plasmid (4.4 and 1.8 kb), which is located 100 kb away from the I-SceI-induced DSB. Using these criteria, a variety of different types of events can be observed in the Ganr subclones of 10-7A and 10-7B, including large deletions with (lane 5) or without (lanes 1, 2, 7, and 8) terminal deletions and complex rearrangements with (lanes 4 and 6) or without terminal deletions (lane 3).
FIG. 6.
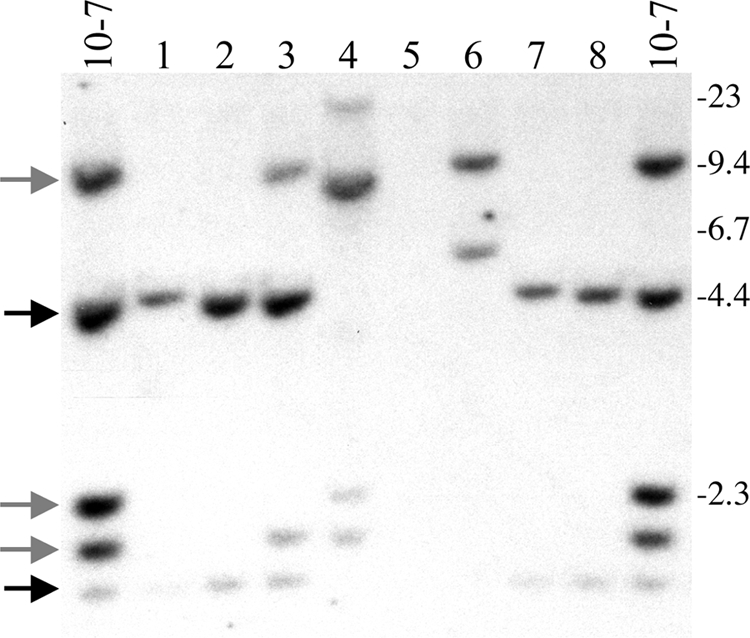
I-SceI-induced DSBs in clone 10-7A result in a high frequency of large deletions and terminal deletions. Genomic DNA from the parental clone 10-7A and eight of its Ganr subclones infected with the pQCXIP-ISceI retrovirus were analyzed by Southern blot analysis following digestion with the HindIII restriction enzyme and hybridization with the pNCTΔ plasmid as a probe. The locations of the three bands representing the pNTIL-100kb plasmid (gray arrows) and the two bands representing the pZTIL-tel plasmid (black arrows) are shown. Also shown are the sizes of the molecular size markers (kb) consisting of lambda bacteriophage HindIII fragments. Large deletions result in the loss of all three of the pNTIL-100kb-specific bands while terminal deletions result in the loss of the pZTIL-tel-specific bands.
TABLE 1.
The frequency of large deletions and terminal deletions in Ganr subclones of 10-7A and 10-7B
| Clone | No. of large deletions (%)a | No. of terminal deletions (%)b |
||
|---|---|---|---|---|
| Total | With large deletions | Without large deletions | ||
| 10-7A | 14 (35%) | 19 (47.5%) | 14 (35%) | 5 (12.5%) |
| 10-7B | 17 (43.6%) | 21 (53.8%) | 18 (46.1%) | 3 (7.7%) |
Loss of pNTIL-100kb-specific bands.
Loss of some pZTIL-tel-specific bands.
The combined results (Table 1) show that I-SceI-induced DSBs caused a high frequency of large deletions involving the complete loss of the pNTIL-100kb plasmid in both 10-7A (14 of 40, or 35%) and 10-7B (17 of 39, or 43.6%). While this was less than the frequency of large deletions that we previously observed in Ganr subclones resulting from DSBs occurring 3 kb from a telomere (>90%), it was substantially higher than the frequency of large deletions observed in Ganr subclones resulting from DSBs at interstitial sites (<10%) (65). Moreover, the I-SceI-induced DSBs also caused a high frequency of terminal deletions in both 10-7A (19 of 40, or 47.5%) and 10-7B (21 of 39, or 53.8%). Because terminal deletions involve the loss of the telomere, they would invariably result in GCRs. This conclusion is consistent with our previous analysis of rearrangements resulting from I-SceI-induced DSBs occurring 3 kb from a telomere, which demonstrated a variety of GCRs (65). Similarly, the rescue of the rearranged pNTIL-100kb plasmid and adjacent cellular DNA from one of the Ganr subclones isolated from clone 10-7 showed the presence of DNA from chromosome 7 at the recombination site, indicating that a translocation had occurred (data not shown). Therefore, although DSBs occurring 100 kb from this telomere result in fewer large deletions and terminal deletions than DSBs occurring within 3 kb, DSBs occurring 100 kb away still have a high probability of resulting in large deletions and GCRs, which rarely result from I-SceI-induced DSBs at interstitial sites (50, 60).
The frequency of small deletions at DSBs occurring 100 kb from a telomere.
Small deletions are the most common event reported for I-SceI-induced DSBs at interstitial sites (27, 60). In our earlier studies, the frequency of small deletions was similar at telomeric (15 to 24.4%) and interstitial (17.9 to 19.8%) I-SceI-induced DSBs and served as an internal control for comparing the frequency of the various types of I-SceI-induced rearrangements at different locations (65). When subclones were picked at random without ganciclovir selection, the frequency of large deletions and GCRs at interstitial DSBs (5 of 81, or 6.1%) was much less than the frequency of small deletions (17.9 to 19.8%). In contrast, the frequency of large deletions and GCRs at telomeric DSBs (50 of 101, or 49.5%) was more than double the frequency of small deletions. We therefore examined the frequency of small deletions resulting from I-SceI-induced DSBs occurring 100 kb from the telomere in clones 10-7A and 10-7B. Small deletions were analyzed by pooling puromycin-resistant cells 10 days after infection with either the pQCXIP-ISceI retrovirus or the pQCXIP retrovirus control. Long PCR was then performed on the region spanning the I-SceI site in the integrated pNTIL-100kb plasmid. The frequency of cells in the population that have small deletions at the I-SceI sites was determined by digesting the PCR product with I-SceI endonuclease to determine the fraction of genomic DNA that had lost the I-SceI site (Fig. 7). Because cells that have large deletions or rearrangements involving the pNTIL-100kb plasmid do not produce PCR products, this value must first be corrected for the frequency of Ganr cells in the population (calculated as 1 − frequency of Ganr cells). The corrected results demonstrate that the frequency of small deletions in both 10-7A and 10-7B (Table 2) was similar to that previously observed at telomeric and interstitial I-SceI-induced DSBs (65). This similarity in the number of small deletions at the different locations demonstrates a similar efficiency of cutting with the I-SceI endonuclease. These results therefore confirm that the high frequency of ganciclovir resistance, large deletions, and terminal deletions in clone 10-7 following expression of I-SceI results from an increased sensitivity to DSBs in the region 100 kb from the telomere.
FIG. 7.
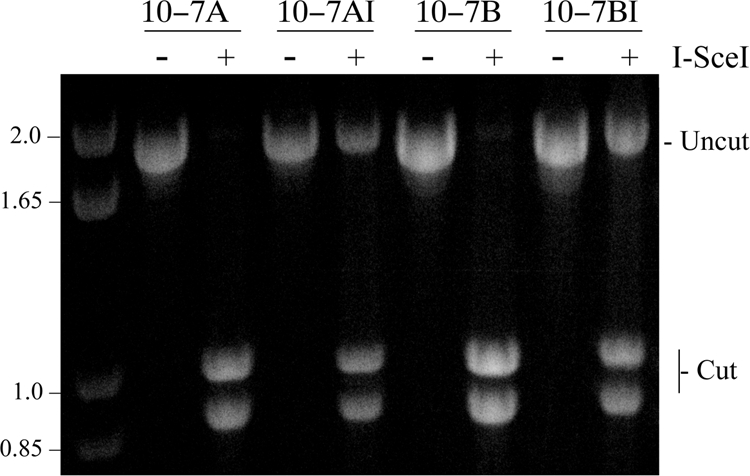
The frequency of small deletions resulting from DSBs at the I-SceI site in clones 10-7A and 10-7B. The frequency of small deletions determined by the ability of I-SceI endonuclease to cut a 1.8-kb PCR fragment spanning the I-SceI site in the integrated pNTIL-100kb plasmid. PCR was performed on genomic DNA isolated from pooled puror cultures of 10-7A and 10-7B 10 days after infection with either the pQCXIP control retrovirus (10-7A and 10-7B) or the pQCXIP-ISceI retrovirus (10-7AI and 10-7BI). The PCR product was then digested with I-SceI endonuclease, run on agarose gels, and stained with ethidium bromide, and the digital images were quantified by ImageJ analysis. The frequency of small deletions was then determined by dividing the intensity of the undigested band by the combined intensities of the digested and undigested bands.
TABLE 2.
Percentage of cells in the population that contain small deletions at the I-SceI site
| Clone | Characteristic of cell population (% of cells) |
||
|---|---|---|---|
| With uncut DNA | Ganr | With small deletionsa | |
| 10-7A | 31.2 | 36 | 20.0 |
| 10-7B | 32.5 | 54 | 15.0 |
The percentage of cells with small deletions is determined by as follows: fraction of uncut PCR product × (1 − fraction of Ganr cells).
The influence of telomere proximity on chromosome healing.
We next examined whether chromosome healing occurs at the I-SceI-induced DSB in clone 10-7. Our earlier studies demonstrated that the EJ-30 tumor cell line is capable of performing chromosome healing at I-SceI-induced DSBs that are 3 kb from a telomere (17, 65). Because chromosome healing has not been observed at interstitial I-SceI-induced DSBs (46), we have proposed that it is limited to DSBs near telomeres, where it serves as an important mechanism for preventing chromosome instability due to DSBs near telomeres (65). This model is based on our observation that chromosome healing can prevent chromosome fusion and the subsequent chromosomal instability that it causes (19). However, proof of this model requires that we rule out the possibility that chromosome healing occurs at interstitial DSBs but is not detected because the terminal deletions it causes result in cell death. To directly address whether chromosome healing occurs at sites other than telomeres, we analyzed whether chromosome healing occurs at the DSB generated by I-SceI in clones 10-7A and 10-7B. Clones 10-7A and 10-7B are ideal for this purpose since terminal deletions involving the region distal to pNTIL-100kb are not lethal, as demonstrated by our ability to generate 10-7 clones in which this region has been deleted by using Cre-mediated recombination (Fig. 4). Our previous studies using EJ-30 clones and mouse ES cells have demonstrated that chromosome healing occurs at the site of the DSB (17, 19, 54, 65). Therefore, we included G418 in the selection medium along with ganciclovir to limit the extent of degradation to ensure that a portion of the pNTIL-100kb plasmid would be retained and thereby decrease the number of subclones that would need to be analyzed. Following the selection of the Ganr/G418r subclones, Southern blot analysis was performed on genomic DNA digested with BamHI, which cuts once in the plasmid (Fig. 8a). Chromosome healing can then be detected using the pNTIL-100kb plasmid as a probe since the terminal restriction fragment will contain both a portion of the plasmid and the newly added telomere at the I-SceI site (Fig. 8a). These terminal restriction fragments are easily identifiable because they are heterogeneous in size due to the variability in the length of the telomere in different cells in the population (17, 65). Based on our analysis of telomere length in the 10-7Cre clones (Fig. 4b) and subclones derived from clone B3 (36), the newly added telomeres resulting from chromosome healing in clone 10-7 would be expected to be in the range of 2.5 to 5 kb. Chromosome healing is also detectable using PCR, with one oligonucleotide primer specific for the plasmid DNA and one specific for the telomeric repeat sequences (19, 54). Using these combined approaches, we previously found that EJ-30 clones B3 and A4 containing the I-SceI site 3 kb from a telomere had chromosome healing in 26% (9 of 35) and 28% (12 of 43) of the Ganr/G418r subclones, respectively (65).
FIG. 8.

The absence of chromosome healing in Ganr/G418r subclones of 10-7. (a) The structure of the integrated plasmid sequences and the sizes of the predicted BamHI restriction fragments are shown for the parental clone 10-7 and for subclones with chromosome healing at the I-SceI site. (b) Genomic DNA from the parental clone 10-7A and eight of its Ganr/G418r subclones was digested with the BamHI restriction enzyme, and Southern blot analysis was performed using the pNCTΔ plasmid as a probe. Terminal deletions are evident by the loss of the 6.0-kb fragment containing the pZTILtel plasmid. The absence of chromosome healing is evident by the lack of diffuse bands (Fig. 4b) that are approximately 5 to 7.5 kb in length (bracket). Instead, there are discrete bands that vary in size, typical of rearrangements resulting from GCRs. The locations of the molecular size markers (kb) that consist of lambda bacteriophage HindIII fragments are shown.
As pointed out above, approximately one-fourth of the Ganr/G418r subclones in our earlier studies demonstrated chromosome healing as a result of I-SceI-induced DSBs occurring 3 kb from this telomere. However, no chromosome healing was detected in any of the 46 Ganr/G418r subclones of 10-7A and 10-7B (Fig. 8b). Southern blot analysis of the genomic digested with BamHI demonstrated that more than half (26 of 46) had retained the 6.0-kb band representing the pZTIL-tel plasmid located at the telomere (Fig. 8b, lanes 3, 4, 6, and 14). These subclones all had deletions at the I-SceI site that inactivated the HSV-tk gene but not the more distant Neo gene that was selected for with G418. The remaining 20 Ganr/G418r subclones had all lost the 6.0-kb band containing the pZTIL-tel plasmid, demonstrating that they sustained a terminal deletion (Fig. 8b). These subclones retained the 14-kb band containing the distal portion of the pNTIL-100kb plasmid containing the ampicillin resistance gene. However, none of these subclones showed the diffuse bands demonstrating that chromosome healing had occurred. Instead, these subclones all contained new bands that varied in size (4 to 23 kb), indicating that GCRs involving the pNTIL-100kb plasmid had occurred. In several subclones, these new bands were 4 to 5 kb in length, which is the length expected in subclones in which sister chromatid fusions had occurred. Sister chromatid fusions are a common form of GCR resulting from telomere loss (36, 49, 65). The frequency of chromosome healing occurring at DSBs 100 kb from this telomere is therefore at least 10-fold lower than the frequency of chromosome healing occurring at DSBs occurring 3 kb from this telomere.
DISCUSSION
Our results demonstrate that the distance over which a telomere exerts its influence differs depending on the endpoint being analyzed. The large decrease in the level of expression of the Neo and HSV-tk transgenes (12 and 7.8-fold, respectively) upon relocation of the telomere immediately adjacent to the pNTIL-100kb plasmid clearly demonstrates a substantial reduction in TPE at a distance of 100 kb from this telomere. The reduced level of expression near the telomere is typical of the reduced expression previously reported for transgenes located near telomeres in other human tumor cells (4, 32) and mouse ES cells (43). Our results are also consistent with studies using telomeric transgenes in yeast, which demonstrate that TPE is dependent upon the distance from a telomere (55). However, the analysis of endogenous genes in yeast also shows that the influence of a telomere on expression can be discontinuous, depending on the presence of specific sequences that can serve as insulators (45). Thus, it is likely that the distance over which a telomere can influence gene expression in mammalian cells will also vary, depending on the type of sequences present in the subtelomeric DNA. Regardless, our results provide the first evidence that the influence of a telomere on gene expression is relatively limited in range, and therefore only genes relatively close to a telomere are likely to be affected by changes in TPE. Because subtelomeric regions in mammalian cells have been shown to consist of heterochromatin (7), our results suggest that the heterochromatin does not extend more than 100 kb from this telomere. It is important to note, however, that we have demonstrated that TPE in normal somatic mouse cells involves complete silencing associated with DNA methylation (43), which could increase the distance over which TPE extends into the chromosome. This complete silencing and DNA methylation have not been observed by us (unpublished observation) or others (4, 32) in telomeric transgenes in human tumor cells, even though the DNA near the ends of chromosomes in normal human cells is heavily methylated (8). The mechanisms involved in formation of TPE and the distance that TPE extends from the end of the chromosome may therefore differ between normal human cells and human cancer cells.
Although the region 100 kb from the telomere on chromosome 16p shows diminished TPE compared to the region immediately adjacent to the telomere, it continues to show increased sensitivity to DSBs. In cells constitutively expressing I-SceI, small deletions at the I-SceI site in clones 10-7A and 10-7B were present in 15 and 20% of the cells in the population, respectively. This frequency of small deletions is similar to the frequency observed at both interstitial and telomeric I-SceI sites (65). This similarity in the frequency of small deletions demonstrates that the production of DSBs by the I-SceI endonuclease is similar at the different locations. We have previously reported that small deletions are by far the most common type of event at I-SceI-induced DSBs at interstitial sites (65), consistent with the results from a number of other laboratories (27, 60). However, in clone 10-7, the combined frequency of large deletions and terminal deletions is much greater than the frequency of small deletions. In view of the fact that terminal deletions result in the loss of the telomere and therefore cause GCRs, our results demonstrate that DSBs occurring within 100 kb of a telomere are much more likely to result in chromosome instability than DSBs at interstitial sites.
The demonstration that a portion of the genome is highly sensitive to DSBs has important implications for chromosome instability resulting from oncogene-mediated replication stress and exposure to ionizing radiation. The continuous cell division resulting from oncogene expression results in stalled replication forks at regions that pose problems for DNA replication, known as fragile sites (3, 21). Stalled replication forks can lead to DSBs, and therefore oncogene-mediated replication stress can result in chromosome rearrangements. Replication forks have been demonstrated to stall near telomeres in yeast (30) and behave like fragile sites in mammalian cells (53), and therefore telomeres are likely sites of DSB formation during replication stress. Moreover, while replication stress would result in DSBs at other fragile sites, stalled replication forks near telomeres would be especially deleterious because of the sensitivity of subtelomeric regions to DSBs. Similarly, regions near telomeres would also be susceptible to chromosome instability as a result of DSBs generated by ionizing radiation. In view of the fact that there are 96 telomeres, the total length of the sensitive region is at least 9.6 ×103 kb (96 × 100 kb) and possibly longer, depending on the how far the sensitive region extends from the telomere. Because the total length of the mammalian genome is 6 × 106 kb, the frequency that random DSBs generated by ionizing radiation would occur within this region is 0.0016 (1 in 625 DSBs). Of these, approximately half experience rearrangements leading to inactivation of the HSV-tk gene, and half of these contain terminal deletions. Thus, a minimum of 1 cell in 2,500 experiencing a DSB will be likely to experience GCRs as a result of DSBs near telomeres. Moreover, many of these GCRs will involve the formation of ring chromosomes, dicentric chromosomes, or sister chromatid fusion, which result in chromosome instability (40).
The mechanism responsible for the sensitivity of subtelomeric regions to DSBs has yet to be determined. A likely explanation for this sensitivity is a deficiency in DSB repair in subtelomeric regions. Subtelomeric regions in yeast have been shown to be deficient in nonhomologous end joining (NHEJ), and as a result, I-SceI-induced DSBs near telomeres result in an increase in frequency of GCRs (48). Similarly, the sensitivity in clone 10-7 is also manifest as an increase in GCRs involving both large deletions and terminal deletions. However, these two endpoints, although related, can occur independently of one another since terminal deletions can occur without large deletions and since large deletions can occur without terminal deletions. Therefore, terminal deletions do not occur solely from extensive degradation at the I-SceI site. An increase in large deletions and terminal deletions would be consistent with a deficiency in DSB repair by either NHEJ or homologous recombination repair. Large deletions involving both strands can result from extensive resection when DSBs remain unrepaired (64). Similarly, deficient DSB repair would also lead to an increase in loss of the terminal fragment containing the telomere, which in turn would lead to GCRs and chromosome instability. However, this deficiency in DSB repair would have a minimal effect on the formation of small deletions, which have been reported to occur primarily through a mechanism involving alternative NHEJ (Alt-NHEJ) (5, 23, 62). Alt-NHEJ has also been shown to be associated with extensive degradation and chromosome rearrangements (23, 41, 61) and may therefore be responsible for the formation of large deletions and GCRs in our system.
Although the region 100 kb from the telomere is sensitive to DSBs, the frequency of chromosome healing at this location is significantly diminished relative to the frequency of chromosome healing we previously reported for DSB occurring 3 kb from this telomere (65). We have now shown that this decreased frequency of chromosome healing at interstitial sites is not due to cell lethality since we have demonstrated that clone 10-7 can survive the loss of the 100-kb terminal fragment using Cre-mediated recombination. Therefore, chromosome healing is either enhanced near telomeres or is inhibited at interstitial sites. In yeast, chromosome healing is inhibited by Pif1 helicase, which has been proposed as a mechanism for preventing de novo telomere addition from interfering with DSB repair (63). Moreover, a recent study found that Pif1 is phosphorylated in response to DSBs and that this phosphorylation is required for the inhibition of chromosome healing (37a). It is therefore likely that chromosome healing is also inhibited at most locations in mammalian chromosomes. However, unlike chromosome healing at interstitial sites, chromosome healing in subtelomeric regions would not result in the loss of substantial amounts of genetic information. The results therefore support our model that chromosome healing serves as a mechanism for stabilizing DSBs near telomeres to compensate for the deficiency of other DSB repair mechanisms in subtelomeric regions. However, our results also show that the region that is sensitive to DSBs extends farther from the telomere than the region in which chromosome healing occurs. In fact, even when DSBs occur within 3 kb of the telomere (17, 36, 49), chromosome healing is much less efficient in the EJ-30 human tumor cell line than in mouse ES cells, where chromosome healing is the most frequent event resulting from DSBs near telomeres (19). This diminished chromosome healing could contribute to the increased chromosome instability that results from a high rate of spontaneous telomere loss typically observed in human tumor cells (17, 40).
In summary, we have demonstrated that DNA methylation-independent TPE extends a relatively short distance from the telomere and therefore would affect only a small number of genes. Similarly, we have also demonstrated that chromosome healing is limited to regions near telomeres. In contrast, the sensitivity of subtelomeric regions extends at least 100 kb from a telomere, making subtelomeric regions a relatively large target for DSB-induced chromosome instability. Moreover, the fact that chromosome healing does not extend as far as the sensitivity to DSBs means that chromosome healing is not available as a mechanism to counteract the instability resulting from DSBs within subtelomeric regions. Finally, the fact that the sensitivity to DSBs extends farther than TPE suggests that the sensitivity does not result from chromatin structure or suppression of transcription but, instead, is associated with other cis-acting telomere functions.
Acknowledgments
This work was supported by National Institutes of Health grant CA12025.
Footnotes
Published ahead of print on 23 November 2009.
REFERENCES
- 1.Aparicio, O. M., B. L. Billington, and D. E. Gottschling. 1991. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell 66:1279-1287. [DOI] [PubMed] [Google Scholar]
- 2.Aparicio, O. M., and D. E. Gottschling. 1994. Overcoming telomeric silencing: a trans-activator competes to establish gene expression in a cell cycle-dependent way. Genes Dev. 8:1133-1146. [DOI] [PubMed] [Google Scholar]
- 3.Bartkova, J., Z. Horejsi, K. Koed, A. Kramer, F. Tort, K. Zieger, P. Guldberg, M. Sehested, J. M. Nesland, C. Lukas, T. Orntoft, J. Lukas, and J. Bartek. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864-870. [DOI] [PubMed] [Google Scholar]
- 4.Baur, J. A., Y. Zou, J. W. Shay, and W. E. Wright. 2001. Telomere position effect in human cells. Science 292:2075-2077. [DOI] [PubMed] [Google Scholar]
- 5.Bennardo, N., A. Cheng, N. Huang, and J. M. Stark. 2008. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4:e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackburn, E. H., C. W. Greider, and J. W. Szostak. 2006. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12:1133-1138. [DOI] [PubMed] [Google Scholar]
- 7.Blasco, M. A. 2005. Telomeres and human disease: ageing, cancer and beyond. Nat. Rev. Genet. 6:611-622. [DOI] [PubMed] [Google Scholar]
- 8.Brock, G. J., J. Charlton, and A. Bird. 1999. Densely methylated sequences that are preferentially localized at telomere-proximal regions of human chromosomes. Gene 240:269-277. [DOI] [PubMed] [Google Scholar]
- 9.Clapp, J., D. J. Bolland, and J. E. Hewitt. 2003. Genomic analysis of facioscapulohumeral muscular dystrophy. Brief Funct. Genomic. Proteomic. 2:213-223. [DOI] [PubMed] [Google Scholar]
- 10.Collins, C., J. M. Rommens, D. Kowbel, T. Godfrey, M. Tanner, S.-I. Hwang, D. Polikoff, G. Nonet, J. Cochran, K. Myambo, K. E. Jay, J. Froula, T. Cloutier, W.-L. Kou, P. Yaswen, S. Dairkee, J. Giovanola, G. B. Hutchinson, J. Isola, O.-P. Kallioniemi, M. Palazzolo, C. Martin, C. Ericsson, D. Pinkel, D. Albertson, W.-B. Li, and J. W. Gray. 1998. Positional cloning of ZNF217 and NABC1: Genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc. Natl. Acad. Sci. U. S. A. 95:8703-8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Counter, C. M., A. A. Avilion, C. E. LeFeuvre, N. G. Stewart, C. W. Greider, C. B. Harley, and S. Bacchetti. 1992. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 11:1921-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lange, T. 1995. Telomere dynamics and genome instability in human cancer, p. 265-293. In E. H. Blackburn and C. W. Greider (ed.), Telomeres. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 13.Deng, Y., S. S. Chan, and S. Chang. 2008. Telomere dysfunction and tumour suppression: the senescence connection. Nat. Rev. Cancer 8:450-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diede, S. J., and D. E. Gottschling. 1999. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerase α and δ. Cell 99:723-733. [DOI] [PubMed] [Google Scholar]
- 15.Ducray, C., J.-P. Pommier, L. Martins, F. Boussin, and L. Sabatier. 1999. Telomere dynamics, end-to-end fusions and telomerase activation during the human fibroblast immortalization process. Oncogene 18:4211-4223. [DOI] [PubMed] [Google Scholar]
- 16.Fisher, J., and M. Upadhyaya. 1997. Molecular genetics of facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 7:55-62. [DOI] [PubMed] [Google Scholar]
- 17.Fouladi, B., D. Miller, L. Sabatier, and J. P. Murnane. 2000. The relationship between spontaneous telomere loss and chromosome instability in a human tumor cell line. Neoplasia 2:540-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fourel, G., E. Revardel, C. E. Koering, and E. Gilson. 1999. Cohabitation of insulators and silencing elements in yeast subtelomeric regions. EMBO J. 18:2522-2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao, Q., G. E. Reynolds, A. Wilcox, D. Miller, P. Cheung, S. E. Artandi, and J. P. Murnane. 2008. Telomerase-dependent and -independent chromosome healing in mouse embryonic stem cells. DNA Repair 7:1233-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodarzi, A. A., A. T. Noon, D. Deckbar, Y. Ziv, Y. Shiloh, M. Lobrich, and P. A. Jeggo. 2008. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31:167-177. [DOI] [PubMed] [Google Scholar]
- 21.Gorgoulis, V. G., L. V. Vassiliou, P. Karakaidos, P. Zacharatos, A. Kotsinas, T. Liloglou, M. Venere, R. A. Ditullio, Jr., N. G. Kastrinakis, B. Levy, D. Kletsas, A. Yoneta, M. Herlyn, C. Kittas, and T. D. Halazonetis. 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907-913. [DOI] [PubMed] [Google Scholar]
- 22.Gottschling, D. E., O. M. Aparicio, B. L. Billington, and V. A. Zakian. 1990. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63:751-762. [DOI] [PubMed] [Google Scholar]
- 23.Guirouilh-Barbat, J., E. Rass, I. Plo, P. Bertrand, and B. S. Lopez. 2007. Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc. Natl. Acad. Sci. U. S. A. 104:20902-20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haarr, L., H. S. Marsden, C. M. Preston, J. R. Smiley, W. C. Summers, and W. P. Summers. 1985. Utilization of internal AUG codons for initiation of protein synthesis directed by mRNAs from normal and mutant genes encoding herpes simplex virus-specified thymidine kinase. J. Virol. 56:512-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harley, C. B. 1995. Telomeres and aging, p. 247-263. In E. H. Blackburn and C. W. Greider (ed.), Telomeres. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Harley, C. B., A. B. Futcher, and C. W. Greider. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345:458-460. [DOI] [PubMed] [Google Scholar]
- 27.Honma, M., M. Sakuraba, T. Koizumi, Y. Takashima, H. Sakamoto, and M. Hayashi. 2007. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair (Amst.) 6:781-788. [DOI] [PubMed] [Google Scholar]
- 28.Inoue, K., H. Osaka, V. C. Thurston, J. T. Clarke, A. Yoneyama, L. Rosenbarker, T. D. Bird, M. E. Hodes, L. G. Shaffer, and J. R. Lupski. 2002. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am. J. Hum. Genet. 71:838-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irmiere, A. F., M. M. Manos, J. G. Jacobson, J. S. Gibbs, and D. M. Coen. 1989. Effect of an amber mutation in the herpes simplex virus thymidine kinase gene on polypeptide synthesis and stability. Virology 168:210-220. [DOI] [PubMed] [Google Scholar]
- 30.Ivessa, A. S., J.-Q. Zhou, V. P. Schulz, E. K. Monson, and V. A. Zakian. 2002. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 16:1383-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlseder, J., K. Hoke, O. K. Mirzoeva, C. Bakkenist, M. B. Kastan, J. H. Petrini, and T. de Lange. 2004. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2:E240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koering, C. E., A. Pollice, M. P. Zibella, S. Bauwens, A. Puisieux, M. Brunori, C. Brun, L. Martins, L. Sabatier, J. F. Pulitzer, and E. Gilson. 2002. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 3:1055-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kramer, K. M., and J. E. Haber. 1993. New telomeres in yeast are initiated with a highly selected subset of TG1-3 repeats. Genes Dev. 7:2345-2356. [DOI] [PubMed] [Google Scholar]
- 34.Lin, Y., and A. S. Waldman. 2001. Capture of DNA sequences at double-strand breaks in mammalian cells. Genetics 158:1665-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Little, J. B. 2003. Genomic instability and radiation. J. Radiol. Prot. 23:173-181. [DOI] [PubMed] [Google Scholar]
- 36.Lo, A. W. I., L. Sabatier, B. Fouladi, G. Pottier, M. Ricoul, and J. P. Murnane. 2002. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia 6:531-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo, A. W. I., C. N. Sprung, B. Fouladi, M. Pedram, L. Sabatier, M. Ricoul, G. E. Reynolds, and J. P. Murnane. 2002. Chromosome instability as a result of double-strand breaks near telomeres in mouse embryonic stem cells. Mol. Cell. Biol. 22:4836-4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37a.Makovets, S., and E. H. Blackburn. 2009. DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat. Cell Biol. 11:1383-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mangahas, J. L., M. K. Alexander, L. L. Sandell, and V. A. Zakian. 2001. Repair of chromosome ends after telomere loss in Saccharomyces. Mol. Biol. Cell 12:4078-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michelson, R. J., S. Rosenstein, and T. Weinert. 2005. A telomeric repeat sequence adjacent to a DNA double-stranded break produces an anticheckpoint. Genes Dev. 19:2546-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murnane, J. P. 2006. Telomeres and chromosome instability. DNA Repair (Amst.) 5:1082-1092. [DOI] [PubMed] [Google Scholar]
- 41.Nussenzweig, A., and M. C. Nussenzweig. 2007. A backup DNA repair pathway moves to the forefront. Cell 131:223-225. [DOI] [PubMed] [Google Scholar]
- 42.O'Toole, C. M., S. Povey, P. Hepburn, and L. M. Franks. 1983. Identity of some human bladder cancer cell lines. Nature 301:429-430. [DOI] [PubMed] [Google Scholar]
- 43.Pedram, M., C. N. Sprung, Q. Gao, A. W. I. Lo, G. Reynolds, and J. P. Murnane. 2006. Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol. Cell. Biol. 26:1865-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pennaneach, V., C. D. Putnam, and R. D. Kolodner. 2006. Chromosome healing by de novo telomere addition in Saccharomyces cerevisiae. Mol. Microbiol. 59:1357-1368. [DOI] [PubMed] [Google Scholar]
- 45.Pryde, F. E., and E. J. Louis. 1999. Limitations of silencing at native yeast telomeres. EMBO J. 18:2538-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rebuzzini, P., L. Khoriauli, C. M. Azzalin, E. Magnani, C. Mondello, and E. Giulotto. 2005. New mammalian cellular systems to study mutations introduced at the break site by non-homologous end-joining. DNA Repair (Amst.) 4:546-555. [DOI] [PubMed] [Google Scholar]
- 47.Renauld, H., O. M. Aparicio, P. D. Zierath, B. L. Billington, S. K. Chhablani, and D. E. Gottschling. 1993. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev. 7:1133-1145. [DOI] [PubMed] [Google Scholar]
- 48.Ricchetti, M., B. Dujon, and C. Fairhead. 2003. Distance from the chromosome end determines the efficiency of double-strand break repair in subtelomeres of haploid yeast. J. Mol. Biol. 328:847-862. [DOI] [PubMed] [Google Scholar]
- 48a.Richardson, C., M. E. Moynahan, and M. Jasin. 1998. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 12:3831-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabatier, L., M. Ricoul, G. Pottier, and J. P. Murnane. 2005. The loss of a single telomere can result in genomic instability involving multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 3:139-150. [DOI] [PubMed] [Google Scholar]
- 50.Sargent, R. G., M. A. Brenneman, and J. H. Wilson. 1997. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol. Cell. Biol. 17:267-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sauer, B., and N. Henderson. 1990. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 2:441-449. [PubMed] [Google Scholar]
- 52.Schulz, V. P., and V. A. Zakian. 1994. The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell 76:145-155. [DOI] [PubMed] [Google Scholar]
- 53.Sfeir, A., S. T. Kosiyatrakul, D. Hockemeyer, S. L. MacRae, J. Karlseder, C. L. Schildkraut, and T. de Lange. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sprung, C. N., G. E. Reynolds, M. Jasin, and J. P. Murnane. 1999. Chromosome healing in mouse embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 96:6781-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tham, W. H., and V. A. Zakian. 2002. Transcriptional silencing at Saccharomyces telomeres: implications for other organisms. Oncogene 21:512-521. [DOI] [PubMed] [Google Scholar]
- 56.van der Weyden, L., D. J. Adams, and A. Bradley. 2002. Tools for targeted manipulation of the mouse genome. Physiol. Genomics 11:133-164. [DOI] [PubMed] [Google Scholar]
- 57.Van Deursen, J., M. Fornerod, B. Van Rees, and G. Grosveld. 1995. Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc. Natl. Acad. Sci. U. S. A. 92:7376-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Geel, M., M. C. Dickson, A. F. Beck, D. J. Bolland, R. R. Frants, S. M. van der Maarel, P. J. de Jong, and J. E. Hewitt. 2002. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics 79:210-217. [DOI] [PubMed] [Google Scholar]
- 59.van Karnebeek, C. D., S. Quik, S. Sluijter, M. M. Hulsbeek, J. M. Hoovers, and R. C. Hennekam. 2002. Further delineation of the chromosome 14q terminal deletion syndrome. Am. J. Med. Genet. 110:65-72. [DOI] [PubMed] [Google Scholar]
- 60.Varga, T., and P. D. Aplan. 2005. Chromosomal aberrations induced by double strand DNA breaks. DNA Repair (Amst.) 4:1038-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinstock, D. M., E. Brunet, and M. Jasin. 2007. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat. Cell Biol. 9:978-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie, A., A. Kwok, and R. Scully. 2009. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16:814-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou, J.-Q., E. K. Monson, S.-C. Teng, V. P. Schultz, and V. A. Zakian. 2000. Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science 289:771-774. [DOI] [PubMed] [Google Scholar]
- 64.Zierhut, C., and J. F. Diffley. 2008. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 27:1875-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zschenker, O., A. Kulkarni, D. Miller, G. E. Reynolds, M. Granger-Locatelli, G. Pottier, L. Sabatier, and J. P. Murnane. 2009. Increased sensitivity of subtelomeric regions to DNA double-strand breaks in a human tumor cell line. DNA Repair 8:886-900. [DOI] [PMC free article] [PubMed] [Google Scholar]