

Abstract
Vesicular stomatitis virus (VSV) has been shown in laboratory studies to be effective against a variety of tumors, including malignant brain tumors. However, attenuation of VSV may be necessary to balance the potential toxicity toward normal cells, particularly when targeting brain tumors. Here we compared 10 recombinant VSV variants resulting from different attenuation strategies. Attenuations included gene shifting (VSV-p1-GFP/RFP), M protein mutation (VSV-M51), G protein cytoplasmic tail truncations (VSV-CT1/CT9), G protein deletions (VSV-dG-GFP/RFP), and combinations thereof (VSV-CT9-M51). Using in vitro viability and replication assays, the VSV variants were grouped into three categories, based on their antitumor activity and non-tumor-cell attenuation. In the first group, wild-type-based VSV-G/GFP, tumor-adapted VSV-rp30, and VSV-CT9 showed a strong antitumor profile but also retained some toxicity toward noncancer control cells. The second group, VSV-CT1, VSV-dG-GFP, and VSV-dG-RFP, had significantly diminished toxicity toward normal cells but showed little oncolytic action. The third group displayed a desired combination of diminished general toxicity and effective antitumor action; this group included VSV-M51, VSV-CT9-M51, VSV-p1-GFP, and VSV-p1-RFP. A member of the last group, VSV-p1-GFP, was then compared in vivo against wild-type-based VSV-G/GFP. Intranasal inoculation of young, postnatal day 16 mice with VSV-p1-GFP showed no adverse neurological effects, whereas VSV-G/GFP was associated with high lethality (80%). Using an intracranial tumor xenograft model, we further demonstrated that attenuated VSV-p1-GFP targets and kills human U87 glioblastoma cells after systemic application. We concluded that some, but not all, attenuated VSV mutants display a favorable oncolytic profile and merit further investigation.
In the field of oncolytic virus therapy, vesicular stomatitis virus (VSV) has emerged as a promising candidate. Preclinical studies have shown effectiveness against a variety of malignancies of the lung, colon (34), liver (33), prostate (2), breast (9), and white blood cells (15). The oncolytic capabilities of VSV against brain tumors have previously been shown by us and others, both in vitro and in vivo (8, 17, 22, 36), but viral spread and neurovirulence in the brain remain challenging factors that need to be addressed in the consideration of VSV as a tool to target brain cancer.
As an oncolytic agent, VSV offers a number of advantages. Virus binding and internalization are facilitated through ubiquitous receptor mechanisms, allowing a large variety of different cancer types to be targeted (34). This is particularly important for malignant brain tumors, which often display a histologically and genetically heterogeneous nature. As we demonstrated in earlier studies, VSV targeted five different human brain tumor cell lines (38), as well as primary glioblastoma cells derived from tissue from resective brain tumor surgery (22). Another strong point of VSV oncolysis is a very fast lytic cycle, leading to fast tumor cell killing and release of new viral progeny in as little as 3 h; as the adaptive immune system mounts a defense against VSV, its rapid oncolytic action may enhance its ability to kill a brain tumor before the immune system eliminates the virus. In addition, systemic application has been shown to be effective in experimental models for targeting a variety of peripheral tumors (2, 33), widespread metastatic tumors (9, 34), and brain tumors (17, 22).
VSV, a member of the Rhabdoviridae family, is enveloped and has a negative-strand 11.2-kb RNA genome that comprises five protein-encoding genes (N, P, M, G, and L) (19). It is a nonhuman pathogen which can cause mild disease in livestock. Infection in humans is rare and usually asymptomatic, with sporadic cases of mild flu-like symptoms. As we compare different recombinant VSVs (rVSVs) with different mutational strategies, it is helpful to appreciate the roles of the five VSV genes. VSV has a short replication cycle, which starts with attachment of the viral glycoprotein spikes (G) to an unknown but ubiquitous cell membrane receptor, though nonspecific electrostatic interactions have also been proposed to facilitate viral binding (19). Upon internalization by clathrin-dependent endocytosis, the virus-containing endosome acidifies, triggering fusion of the viral membrane with the endosomal membrane. This leads to release of the viral nucleocapsid (N) and viral RNA polymerase complex (P and L) into the cytosol. The viral polymerase initiates gene transcription at the 3′ end of the nonsegmented genome, starting with expression of the first VSV gene (N). This is followed by sequential gene transcription, creating a gradient, with upstream genes expressed more strongly than downstream genes. Newly produced VSV glycoproteins are incorporated into the cellular membrane with a large extracellular domain, a 20-amino-acid transmembrane domain, and a cytoplasmic tail consisting of 29 amino acids. Trimers of G protein accumulate in plasma membrane microdomains, several of which congregate to form viral budding sites at the membrane (19). Most cells activate antiviral defense cascades upon viral entry, transcription, and replication, which in turn are counteracted by VSV matrix protein (M). VSV M protein's multitude of functions include virus assembly by linking the nucleocapsid with the envelope membrane, induction of cytopathic effects and apoptosis, inhibition of cellular gene transcription, and blocking of host cell nucleocytoplasmic RNA transfer, which includes blocking of antiviral cellular responses (3).
A number of recombinant VSVs that show attenuated virulence have been described. First, recombinant VSVs derived from DNA plasmids in general show weakened virulence (30). Nucleotide changes that alter the amino acid composition in the M protein at position 51 result in attenuated VSV phenotypes in vitro (6) and in vivo (2, 4, 34, 39). The VSV transmembrane G protein is needed for binding and internalization; truncations in the G protein to generate a reduced number of cytoplasmic amino acids are also attenuated (13, 31). Altering the order of genes also attenuates the virus (4, 5, 10). G gene deletions block the ability to produce infectious virus (8). In addition, we previously used a protocol for adapting VSV to a particular cell type through repetitive passage under evolutionary pressure, leading to the generation of VSV-rp30, a wild-type-based VSV with an enhanced oncolytic profile (38).
In the present study, we compare the oncolytic profiles of 10 different VSV variants. This study seeks to find the best compromise for potential brain tumor oncolytic targeting, balancing the need for the virus to kill cancer cells but, on the other hand, to have a weakened ability to kill normal cells.
MATERIALS AND METHODS
Human cells.
The human glioblastoma cell line U87MG was obtained from ATCC (Manassas, VA). These cells were stably transfected with the gene coding for monomeric dsRed, allowing easy detection of red human glioblastomas transplanted into mouse brains (see below) (22). The U-118, U-373, and A-172 cell lines were kindly provided by R. Matthews (Syracuse, NY). Normal human glia cells were established from tissue derived from surgery specimens from patients undergoing epilepsy surgery. Glia cell cultures were isolated through explant cultures and tested for immunoreactivity to glial fibrillary acidic protein (GFAP). Human cell preparation and use were approved by the Yale University Human Investigation Committee. All cells were kept in a humidified atmosphere containing 5% CO2 at 37°C. U87 cells were fed with minimal essential medium (MEM) supplemented with 10% fetal bovine serum, 1% sodium pyruvate, and 1% nonessential amino acids. Normal human glia cells were propagated with MEM supplemented with 10% fetal bovine serum.
Viruses.
Green fluorescent protein (GFP)-expressing VSV-G/GFP is a recombinant variant of the Indiana strain with an extra copy of the G protein fused to a GFP reporter gene downstream of the original G gene (7, 14, 35). VSV-rp30 was generated from VSV-G/GFP through repeated passage and adaptation to glioblastoma cells, as previously reported (38). VSV-M51 has a codon deletion in the gene coding for the M protein, at amino acid position 51, reducing the viral suppression of cellular immunity against VSV (3, 6, 34). VSV-CT1 and VSV-CT9 have the cytoplasmic side of the G protein reduced to a single amino acid and to 9 amino acids, respectively (25). Reducing the length of the cytoplasmic G protein reduces virulence (31). We also included a VSV with multiple sites of attenuation, including an M51 mutation together with a G protein cytoplasmic tail truncation, creating VSV-CT9-M51 (24, 36). Shifting the order of the genes downward has also been reported to reduce virulence (4, 10). To accomplish this, the GFP or red fluorescent protein (RFP; from dsRed) reporter gene sequence was added at position 1, moving all other virus genes downward, to positions 2 to 6, creating VSV-p1-GFP or VSV-p1-RFP, respectively. Insertion of a foreign gene such as one coding for a reporter at position 1 also attenuates the virus (28). Eliminating the G gene blocks the ability of the virus to infect cells; however, by adding the G protein in trans, as we did here by generating the virus in cells that express the VSV-G protein (26), the replication-restricted viruses VSV-dG-GFP and VSV-dG-RFP will at least infect a single round of cells.
Titers for all VSV variants were determined through plaque assays on BHK cells prior to experiments.
Viral infection and cytopathic effects.
For assessing infection and the appearance of cytopathic effects, cells were seeded in 12-well dishes at a density of 100,000 cells per dish in triplicate for each condition. After 12 h, fresh medium was added to each dish, containing 104 PFU (multiplicity of infection [MOI] = 0.1) of any of the 10 VSV variants. Cultures were observed for 3 days postinfection (dpi). GFP was monitored with an Olympus IX 71 fluorescence microscope, using a 485-nm excitation filter. Photomicrographs were taken with a Spot RT digital camera (Diagnostic Instruments, Sterling Heights, MI) interfaced with an Apple Macintosh computer. Contrast and color of the photomicrographs were adjusted with Adobe Photoshop.
Cell growth and viability.
U87 and human glia control cells were plated in 96-well dishes at a density of 10,000 per well, using colorless MEM without phenol red. After 12 h, the medium was replaced with either fresh medium or medium containing 100 IU alpha interferon (IFN-α; Sigma-Aldrich) for 6 h of preincubation before the addition of 5,000 PFU of the indicated VSV variants. Viability was assessed using an MTT (Molecular Probes) assay according to the manufacturer's instructions. Optical density was read at 570 nm, using a Dynatech MR500 enzyme-linked immunosorbent assay (ELISA) plate reader (Dynatech Lab Inc, Alexandria, VA), and corrected with background control subtraction. Each condition was tested in triplicate.
Quantitative real-time PCR.
Normal human glia cells were grown in T25 flasks to confluence and infected with the respective VSV variants at an MOI of 2. After 6 h, RNA was extracted with TRIzol reagent (Invitrogen). Total RNA was reverse transcribed using a SuperScriptIII RT kit (Invitrogen) and random hexamer primers (Promega, Madison, WI). Primer selection and the PCR protocol have previously been described in detail (36, 37).
Animal procedures.
For intranasal application, young mice (p16) were mildly anesthetized with ketamine-xylazine and received 25 μl of virus solution in each nostril. The head was kept reclined and in a lateral position to enhance virus delivery to the roof of the nasal cavity. Mouse health and weight were monitored daily. Animals with either significant neurological symptoms (paralysis, lateropulsion, etc.) or a body weight drop below 75% of the starting value were euthanized according to institutional guidelines.
Four- to 6-week-old immunodeficient mice with a homozygous CB17-SCID background (CB17SC-M) (Taconic Inc.) were used for tumor xenograft experiments. A total of 1 × 105 U87 glioblastoma cells expressing a red fluorescence reporter gene were injected stereotactically bilaterally into the striatum as previously described in detail (22). At 10 days postinjection, mice received a single bolus of 100 μl phosphate-buffered saline (PBS) containing 107 PFU of VSV-p1-GFP in the tail vein. Animals were monitored with daily measurements of body weight, food and water consumption, and overall health. Two or 3 days later, animals were euthanized with a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde. All animal experiments and postoperative care were performed in accordance with institutional guidelines of the Yale University Animal Care and Use Committee.
RESULTS
VSV has shown promise as an effective agent against malignant brain tumors. However, previous studies revealed the potential for infecting normal brain cells as one of the main challenges that need to be addressed before clinical trials can be pursued. In the current study, we compared 10 recombinant VSV variants for both oncolytic capabilities and normal brain cell attenuation. The VSV variants used in this study represent a systematic comparison of attenuated rVSVs, including M protein mutation (VSV-M51), mutations leading to truncated cytoplasmic tails of the VSV G protein (VSV-CT1 and VSV-CT9), gene order shifting (VSV-p1-GFP and VSV-p1-RFP), gene deletion (VSV-dG-GFP and VSV-dG-RFP), and combinations thereof (VSV-CT9-M51). In addition to these recombinant VSV variants, we also tested VSV-rp30, a glioma-passage-adapted VSV variant. VSV-rp30 has two amino acid changes, one in the VSV P protein and another in the L protein. The VSV-rp30 phenotype displayed enhanced infectivity and oncolytic activity. The reference virus for this comparative study was VSV-G/GFP, a recombinant VSV that was generated from cDNA, using sequence fragments from wild-type VSV Indiana strains (7, 29, 35). Though closely related to wild-type VSV, VSV-G/GFP has reduced virulence (30). A schematic overview of the different VSV types, with their respective variations from the wild type, is displayed in Fig. 1.
FIG. 1.

Schematic display of wild-type VSV and 10 variants. The VSV genome consists of a single RNA strand encoding five genes. The top displays a schematic of the VSV wild-type genome and a color-matched illustration of the virus structure. The next genome shows a recombinant variant, VSV-G/GFP, in which an additional copy of the G gene is inserted, tagged to a GFP reporter gene. VSV-dG-GFP has the whole gene order shifted through insertion of the GFP reporter gene at the first position and has the complete sequence for the G protein deleted. VSV-CT9 has a truncated G protein, shortening the 27-amino-acid chain of the cytoplasmic G protein tail down to 9 amino acids; the GFP reporter gene is inserted between the G and L genes. VSV-CT1 has the G gene truncated to a single amino acid in its cytoplasmic tail. VSV-M51 has a methionine deletion at position 51 of the M protein, leading to more susceptibility of the virus to the interferon-mediated antiviral defense; the GFP reporter gene is inserted between the G and L genes. VSV-CT9-M51 combines the mutation of M51 with the CT9 mutation. VSV-p1-GFP has a wild-type-related genome that is completely shifted by the insertion of the GFP reporter gene at position 1 of the gene order, leading to decreased viral transcription of downstream genes. VSV-rp30 is based on recombinant VSV-G/GFP, with one mutation each in the P and L proteins, leading to enhanced infection and oncolysis.
Potential of VSV variants to infect and kill brain tumor cells.
Since the VSV variants used in our study display features of attenuation, we first addressed to what extent this attenuation might impair the oncolytic strength of VSV shown in previous studies. We used fluorescence microscopy to detect expression of the GFP reporter gene in infected cells, phase-contrast microscopy to assess the presence of cytopathic effects, and MTT assay for quantification of cell viability and oncolytic capacity. Previous studies by us and others have demonstrated a defective interferon response in cancer cells to be a main factor in selective VSV oncolysis. On the other hand, IFN provides protection against VSV to normal cells (23). Hence, we tested the hypothesis that IFN would enhance the selectivity of some VSVs and protect normal but not cancer cells from the virus.
In the first set of experiments, U87 human glioblastoma cells were infected at an MOI of 0.1, and signs of infection were observed over the course of 2 days. One of the main advantages of replication-competent oncolytic viruses over replication-deficient vectors is the local self-amplification of the therapeutic effect, wherein even low virus concentrations can be effective against a large volume of tumor mass through ongoing tumor-selective production of viral progeny. In experimental settings, using an MOI of <1 helps in assessing infectivity at a low dose, because viral replication is required to have a strong effect on a great number of tumor cells. Representative photomicrographs taken at 36 h postinoculation are shown in Fig. 2. Under control conditions, infection of U87 cells with VSV-rp30, VSV-M51, VSV-CT9, VSV-CT9-M51, and VSV-p1-GFP led to similar, widespread, nearly complete infection and the appearance of cytopathic effects, as with wild-type-based VSV-G/GFP, and only small differences were found between the variants. In contrast, replication-impaired VSV-dG-GFP and VSV-dG-RFP infected only a fraction of the cells in the culture dish. In the presence of IFN-α, infection and spread were slightly delayed, with little difference between VSV-G/GFP and VSV-rp30, VSV-M51, or VSV-CT9. However, the double mutant VSV-CT9-M51 and gene-shifted VSV-p1-GFP showed less cytopathic effect than VSV-G/GFP. Finally, low-dose VSV-dG-GFP and VSV-dG-RFP were strongly impaired in infecting U87 cells in the presence of IFN.
FIG. 2.

VSV variants infecting U87 human glioblastoma cells. A panel of representative photomicrographs shows phase-contrast and GFP fluorescence microscopy images of cultured U87 cells after infection with VSV variants at an MOI of 0.1, at 36 h postinfection. The left two columns show control conditions, and the right two columns show experiments performed after interferon preincubation. Interferon does not protect U87 cells from VSV infection. Cytopathic effects were observed in phase-contrast mode. Replication-deficient VSV-dG-GFP and VSV-dG-RFP show significantly less infection. VSV-CT1 contains no GFP reporter, and thus no fluorescence image is shown for it.
For quantitative assessment of cell viability, we used a colorimetric MTT assay and studied the oncolytic action of 10 VSV variants on U87 cells and on normal human glia cells. To test the hypothesis that IFN would increase the selectivity of some of the viruses for cancer cells, cells were grown in 96-well dishes in the presence or absence of IFN-α (100 U/ml). To investigate which viruses performed well at a low virus concentration, virus was applied at an MOI of 0.5, and the MTT assay was performed at the indicated time points. Thirty-six hours after inoculation of the 10 VSV variants, little effect on cell viability was seen in human glia control cells, with all viruses (except VSV-CT9 and VSV-dG-RFP) causing a <20% decrease in viability (Fig. 3A). After 72 h, a significant decrease in cell viability was noted with VSV-G/GFP, VSV-rp30, and VSV-CT9. In contrast, cultures infected with VSV-M51, VSV-CT9-M51, VSV-p1-GFP, VSV-p1-RFP, and VSV-CT1 maintained viabilities of over 80% compared to mock-infected control cells (Fig. 3B). On the other hand, complete protection from infection of any VSV variant was seen in control cells after preincubation with IFN-α (gray shaded boxes in Fig. 3A and B). Control cultures pretreated with IFN-α lacked any signs of GFP expression (data not shown), further supporting the protective role of IFN in controlling VSV infection in normal cells. In contrast, cell viability of U87 glioblastoma cells was significantly reduced, by 25 to 60%, compared to that of mock-treated control cells 36 h after infection with 6 of the 10 VSV variants, with VSV-rp30, VSV-M51, and VSV-CT9-M51 showing the most tumor cell killing (Fig. 3C). In the presence of IFN, VSV-rp30, VSV-M51, and VSV-CT9-M51 caused reductions of U87 cell viability of about 20%. Underscoring the strong oncolytic potential of the viruses, tumor cell killing was nearly complete at 72 h postinfection (hpi), despite the low initial MOI of 0.5, in all but the two replication-restricted viruses, VSV-dG-GFP and VSV-dG-RFP (Fig. 3D). In addition, even after preincubation with IFN-α, 7 of the 10 VSV variants continued to infect and kill tumor cells, with a reduction in viability of 40 to 70%. The two replication-incompetent viruses, VSV-dG-GFP and VSV-dG-RFP, showed a poor ability to kill tumor cells in the presence of IFN and also showed only a modest effect under control conditions at a low MOI of 0.5; this was due in part to the inability of the viruses to generate second rounds of infectivity. VSV-CT1, which showed strong tumor cell killing under control conditions, was not effective at killing tumor cells in the presence of IFN. There is an apparent difference in that replication-restricted VSV-dG variants suppressed viability on human glia cell control cultures but not on human U87 glioblastoma cells. Since U87 tumor cells divide rapidly, the number of initially infected cells was outgrown by the dividing culture in 36 and 72 h (for VSV-dG-GFP and VSV-dG-RFP, respectively). In contrast, the proportion of infected glia control cells remained approximately the same in the course of the 3-day experiment. Together, these initial in vitro experiments showed a number of VSV variants to be highly attenuated for control human glia cells but ineffective against U87 cells. These include the two replication-incompetent VSV-dG-GFP and VSV-dG-RFP variants and VSV-CT1. On the other hand, we found a number of VSVs to be excellent in their antitumor action, but with noticeable toxicity on human control glia cells. These included VSV-G/GFP, VSV-rp30, and VSV-CT9. Finally, a third group emerged, with little toxicity against control cells yet reasonably good tumor cell killing, comprised of VSV-M51, VSV-CT9-M51, VSV-p1-GFP, and VSV-p1-RFP.
FIG. 3.

Cell viability of human glioblastoma and normal human glia cells after infection with VSV variants. Using an MTT cell viability assay, the cytotoxic effects of 10 VSV variants were tested on human glia control cells and U87 glioblastoma cells. (A) At 36 hpi at an MOI of 0.5, little effect on cell viability was seen for all VSV variants on control cells. (B) VSV-G/GFP, VSV-rp30, and VSV-CT9 showed signs of toxicity after 72 h. The presence of IFN (shaded boxes) completely protected normal human glia cells from infection with all VSV variants. (C) Human U87 glioblastoma cells showed a pronounced loss of viability at 36 hpi with 6 of 10 VSV variants. (D) At 72 hpi, all VSV variants, except the two replication-restricted VSV-dG mutants, completely killed U87 cells. Interferon provided little protection for U87 cells at 72 hpi (shaded box).
Effect of VSV attenuation on viral replication in tumor and control cells.
Local self-amplification is one of the mainstays of oncolytic virus therapy. Viruses selectively replicating faster in tumor cells than in normal cells would be expected to have a stronger oncolytic profile. Here we used standard plaque assay techniques to determine viral replication of the eight replication-competent VSV variants on U87 cells and compared it to replication on normal human control cells. Two replication-restricted VSV-dG variants were also included in the replication assay to provide a baseline value for noninternalized parent viral particles. Cell culture supernatants were collected at 1, 2, and 3 days postinfection and frozen until further analysis. Experiments were again performed in the presence and absence of IFN-α. Of the 10 VSV variants tested, VSV-rp30, VSV-M51, VSV-CT9-M51, VSV-CT9, and VSV-CT1 all had similar growth curves on normal human glia cells to that of wild-type-based VSV-G/GFP, in contrast to VSV-p1-GFP and VSV-p1-RFP, which showed reduced replication, by ∼100-fold, and VSV-dG-GFP and VSV-dG-RFP, which, as expected, showed no replication (Fig. 4A). In the absence of plaque formation for replication-restricted variant titers, VSV-dG variants were assessed by the number of individual infected cells expressing either the red or green fluorescence reporter gene. For all replication-competent VSV variants, viral replication was greatly reduced by IFN-α pretreatment.
FIG. 4.

Replication of VSV variants on U87 cells and normal human glia cells. Virus replication was compared for each variant between brain tumor cells and normal human glia cells, using a plaque assay. Monolayers of each culture were infected at an MOI of 1 with the respective VSV variant, and supernatants were collected at the indicated time points. Virus replication was attenuated on human glia cells compared to that on U87 cells, by about 2 log. Also, VSV-p1-GFP and VSV-p1-RFP showed decreased viral replication. Differences between the other variants were marginal. (A) Note the complete block of viral replication after IFN preincubation. (B) In contrast, viral replication in U87 brain tumor cells was higher for each variant and productive even in the presence of IFN. Graphs for replication-restricted VSV-dG variants display the baseline for the original inoculum.
On U87 cells, viral replication was significantly higher (∼100-fold) than that on control cells for all but the two replication-deficient viruses, VSV-dG-GFP and VSV-dG-RFP. As on normal human glia cells, little difference was seen between VSV-rp30, VSV-M51, VSV-CT9-M51, VSV-CT9, VSV-CT1, and wild-type-based VSV-G/GFP (Fig. 4B). Calculating the maximum titer difference at 2 dpi for viruses under non-IFN control conditions between normal human glia cells and U87 cells resulted in the following ratios. These ratios are relevant and serve as an important index of the relative levels of VSV replication in normal and cancer cells. A large ratio suggests a virus that shows substantially greater replication in cancer cells than in control cells. The ratios were as follows: VSV-G/GFP, 1:100; VSV-rp30, 1:121; VSV-M51, 1:287; VSV-CT9-M51, 1:341; VSV-CT9, 1:237; VSV-CT1, 1:74; VSV-p1-GFP, 1:386; and VSV-p1-RFP, 1:602. In contrast to the case with control human glia cells, interferon pretreatment did not prevent viral replication in U87 cells, with viral titers reaching similar values to those in non-IFN-treated controls by 3 dpi (Fig. 4B).
Infection and growth suppression of additional human glioma cultures.
Glioblastoma tumors are characterized by heterogenous histology and mutation profiles. To test whether the effects of attenuated VSV mutants on U87 glioma infection and oncolysis can be generalized to other human glioblastoma cell lines, we analyzed infections of three human cell lines by the four most effective antitumor VSV variants, VSV-rp30, VSV-M51, VSV-CT9-M51, and VSV-p1-GFP. U118, U373, and A-172 cells were plated in 24-well dishes, infected at an MOI of 2, and analyzed 24 h later. Cell counting revealed cell growth suppression compared to noninfected controls for all VSV variants tested in all tumors (Fig. 5A). As in U87 cells, VSV-rp30 displayed the strongest suppression of tumor growth and cell lysis of up to 80% in U118 cells and 50% in both U373 and A172 cells. By 48 h, all cells were dead (data not shown). As seen with U87 cells, the other tested VSV variants displayed increasingly attenuated tumor suppression, in the order of VSV-M51, VSV-CT9-M51, and VSV-p1-GFP. Using GFP fluorescence-reported infection, we studied the infectivity of these VSV variants. VSV-rp30-infected cultures displayed the highest number of infected cells compared to VSV-p1-GFP, which showed the fewest cells infected (Fig. 5B). Together, these data mirror the trend that was seen with U87 glioblastoma cells. VSV-rp30 was found to be highly effective at targeting and killing glioblastoma cells, with the tested alternative VSV variants displaying an attenuated yet still effective antitumor profile.
FIG. 5.
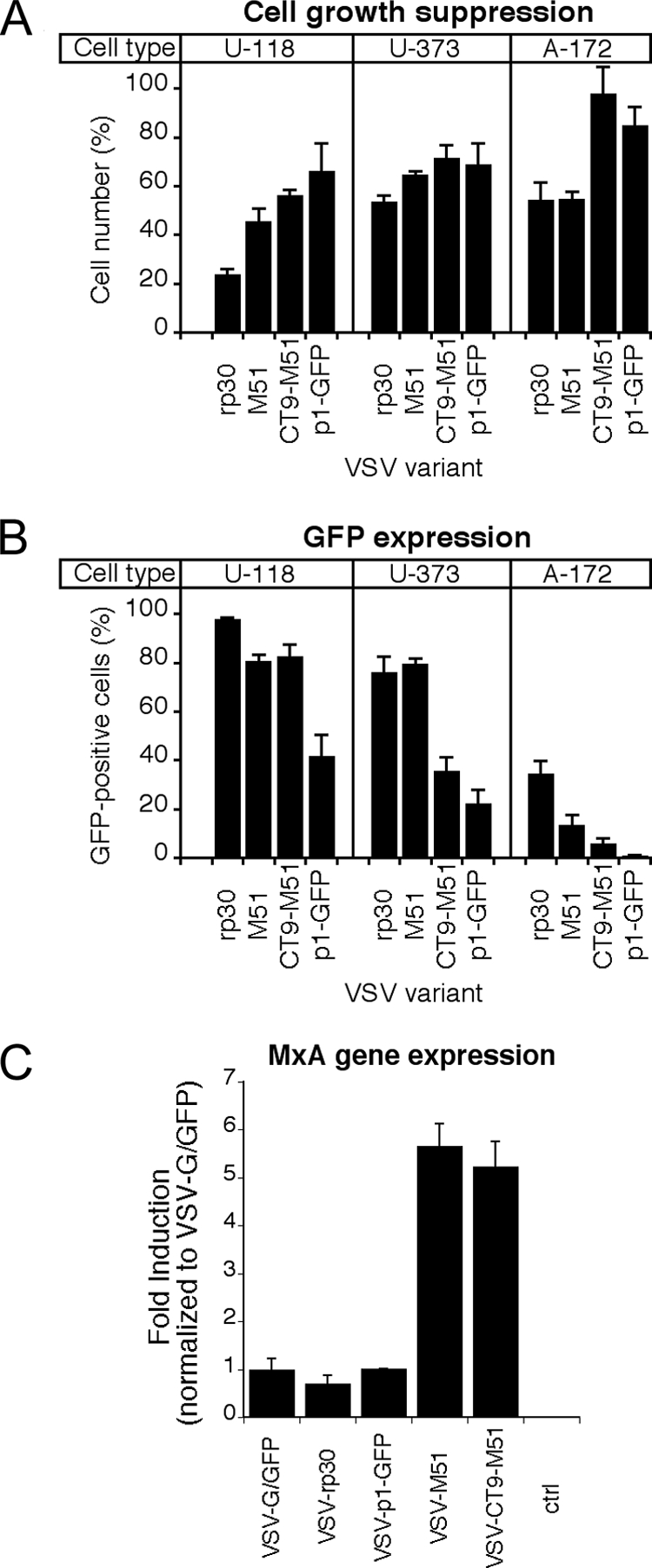
Infection and growth suppression of alternate human glioma cultures and MxA gene induction of VSV variants on control cells. Tumor cell infectivity and growth suppression were tested for human glioblastoma cell types U118, U373, and A-172. VSV variants VSV-rp30, VSV-M51, VSV-CT9-M51, and VSV-p1-GFP were applied at an MOI of 2 and analyzed at 24 hpi. (A) As on U87 cells, VSV-rp30 had the strongest growth-suppressing effect, and VSV-CT9-M51 and VSV-p1-GFP suppressed tumor growth to a lesser extent. (B) GFP fluorescence revealed a similar picture, with VSV-rp30 having the highest rates of infection compared to the attenuated VSV variants. Bars show mean values for five microscopic fields. Error bars indicate standard errors of the means. (C) Using quantitative real-time PCR, the expression of the interferon-induced antiviral gene MxA was compared after infection of cells with VSV-G/GFP, VSV-rp30, VSV-p1-GFP, VSV-M51, and VSV-CT9-M51. M51 mutation-containing mutants induced about 5 to 6 times more MxA than did VSV variants with wild-type M protein. Results are means for triplicate cultures. Error bars indicate standard errors of the means.
Differential induction of interferon downstream gene MxA.
The innate cellular immune response plays a crucial role in controlling VSV infection in normal cells. MxA is a potent downstream gene of the activated interferon path. We previously found significant differences in expression profiles of MxA after VSV-rp30 infection between five glioblastoma cell lines and a panel of three normal human glia cell cultures (37). To address differences in the expressional response to different VSV variants, we tested the induction of MxA. A representative selection of different VSV mutants was used to infect triplicate cultures of normal human control glia cells at an MOI of 2. After 6 h, RNA was extracted and reverse transcribed. Quantitative real-time PCR revealed a five- to sixfold higher induction of MxA gene expression in cultures infected with VSV-M51 or VSV-CT9-M51 than in those infected with VSV-G/GFP, VSV-rp30, and VSV-1p-GFP (Fig. 5C), confirming the previously described ability of M51 mutants to increase the cellular interferon response due to the inability to block cellular gene expression (34).
Reduced neurovirulence of intranasally applied VSV-p1-GFP.
VSV may display neurovirulence in developing mice upon intranasal application (18, 35). Based on our initial sets of in vitro experiments, we sought to test an attenuated virus that still retained a good antitumor profile, VSV-p1-GFP, on young mice and then compare it with wild-type-based VSV-G/GFP. Sixteen-day-old mice were given 250,000 PFU of either VSV-p1-GFP or VSV-G/GFP in each nostril, and mice were observed for neurological symptoms and weighed on a daily basis. Figure 6A shows complete survival of 16-day-old mice (n = 10) after VSV-p1-GFP application, compared to 80% lethality in VSV-G/GFP-treated mice (n = 10). The corresponding body weight graph (Fig. 6B) displays a steady increase in weight in VSV-p1-GFP-treated mice and a significant drop in body weight in VSV-G/GFP-treated mice; the decrease in body weight was apparent after 5 dpi.
FIG. 6.
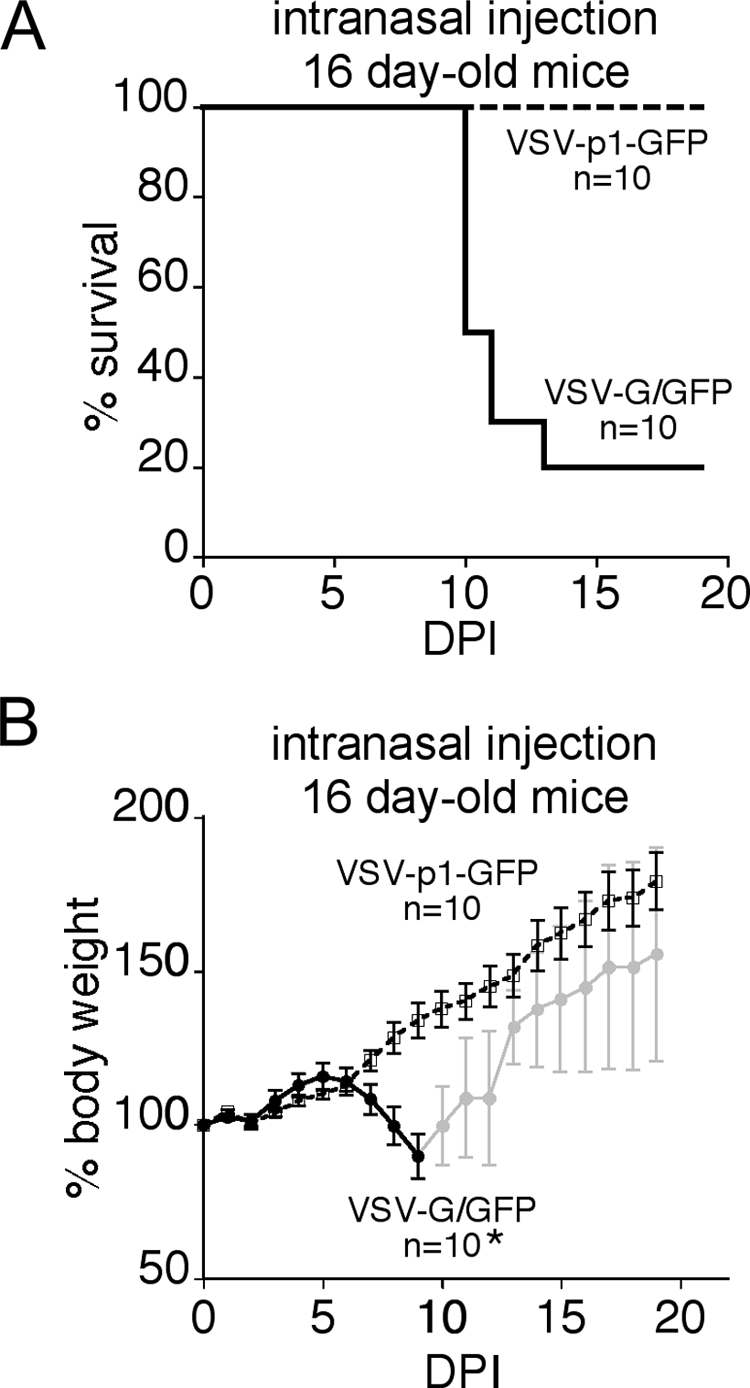
Intranasal application of VSV-G/GFP and VSV-p1-GFP. VSV has been shown to cause neurotoxicity in young mice when administered through an olfactory entry route. A total of 500,000 PFU of either wild-type-related VSV-G/GFP or attenuated VSV-p1-GFP was inoculated intranasally into young mice. The mice lost body weight after VSV-G/GFP inoculation (B), and 8 of 10 mice ultimately succumbed (A). In contrast, littermates that received VSV-p1-GFP gained weight steadily, and no mortality was seen. n = 10, the number of mice initially infected with the virus.
Intravenous application of VSV-p1-GFP targets intracranial brain tumor xenografts.
We and others previously presented studies using VSV-rp30 (22) and VSV-M51 (17) to systemically target intracranial brain tumor xenografts after intravenous virus injection. Based on our initial in vitro experiments and the display of neuroattenuation after intranasal application of VSV-p1-GFP, we sought to test the capability of this attenuated VSV variant to find and infect intracranial U87 xenografts after a single intravenous application in a proof-of-principle setup. As in our previous study, we used U87 cells that were stably transfected with monomeric RFP for tumor transplantation, allowing easy tracing and distinction from surrounding normal brain parenchyma. Human glioblastoma cells were injected bilaterally into the striatum of SCID mice. Ten days later, mice were given a single intravenous injection of 100 μl sterile PBS containing 5 × 106 PFU of VSV-p1-GFP. Two mice each were sacrificed at 2 dpi and 3 dpi for histological analysis of virus infection of the tumor xenografts. All four animals bore sizeable tumors. All tumors were selectively infected with VSV-p1-GFP (Fig. 7). A smaller tumor was completely infected at 3 dpi (Fig. 7A). Figure 7B displays a section of a larger tumor with partial infection (at 2 dpi). Finally, we observed VSV-p1-GFP infection not only in the tumor bulk but also in small tumor islands dispersed around the main tumor (Fig. 7C).
FIG. 7.

Targeting of human brain tumor transplants after intravenous injection of VSV-p1-GFP. Human glioblastoma cells expressing red fluorescent protein were stereotactically injected into the brains of SCID mice and given 10 days to form sizeable tumors. After a single injection of 107 PFU of VSV-p1-GFP into the tail vein, the virus targeted the tumors, as indicated by GFP fluorescence. (A) Complete spread through the tumor mass at 3 dpi. (B) Initiating focus of viral infection at an early time point (2 dpi). (C) Remote tumor cell clusters were also successfully targeted by VSV-p1-GFP without infecting the surrounding, nontumor brain parenchyma.
The ability of VSV-p1-GFP to infect smaller tumor islands is important, as one of the chief clinical problems associated with glioblastoma is its tendency to migrate into normal brain tissue and thereby spread the cancer. Importantly, at the two time points analyzed, GFP expression was seen nearly exclusively in red fluorescent U87 cells, whereas the surrounding brain parenchyma was left largely uninfected. In a previous study (22), we confirmed the activation of apoptosis in tumor cells infected with VSV-rp30, using the same in vivo xenotransplant model. We did observe similar morphological changes to those described before. The tumors analyzed at 3 dpi showed cellular disintegration and blebbing of infected cells, which are typical of virally mediated oncolysis (Fig. 7A and C).
DISCUSSION
There is currently no cure for glioblastoma in the brain, and patients diagnosed with this type of cancer generally die within a year (21). Oncolytic viruses that can infect and destroy malignant glioblastomas have emerged as a potential approach to combating this cancer (1). A potential complication in using oncolytic viruses to attack cancer is the problem of infection of normal cells. This is particularly problematic in the brain, where neurons do not replicate, and virally mediated neuronal loss could lead to unwanted dysfunction. We therefore generated and compared a series of VSVs to determine which had reduced general virulence against normal brain cells but still retained the ability to infect and destroy glioblastoma cancer cells. An ideal oncolytic virus would show high levels of infection and replication in cancer cells but low levels in noncancer control cells. Of the 10 VSVs examined, the group 3 types showed an optimal phenotype, including VSV-M51, VSV-CT9-M51, VSV-p1-GFP, and VSV-p1-RFP. The other VSVs tested either showed a limited ability to destroy tumor cells (group 2 [VSV-dG-GFP, VSV-dG-RFP, and VSV-CT1]) or did not show sufficiently attenuated virulence against normal cells (group 1 [VSV-G/GFP, VSV-rp30, and VSV-CT9]).
Multiple mechanisms of attenuation in top candidate oncolytic VSVs.
Here we focus on the recombinant VSVs that showed the greatest promise for selective infection of glioblastomas. Each of these viruses had multiple sites of molecular attenuation. The virus generated from a DNA plasmid (14) is substantively attenuated for virulence compared with wild-type VSV (29). Second, each virus (except VSV-CT1) included a gene coding for a fluorescent reporter, either GFP or RFP. Adding this transgene to the viral genome helped in identifying infected cells but, importantly, also served to attenuate the resultant virus. This was particularly effective when the reporter gene was added at the first position, resulting in greater expression of the reporter gene than when it was placed in a secondary position and also causing a reduction in the expression of all five of the viral structural genes (4, 5, 28, 36). VSV-CT9-M51, with a shortened cytoplasmic tail of the G protein and an M51 codon-deleted M gene, was further attenuated by a GFP reporter and by DNA derivation. The CT9 mutant by itself showed attenuated virulence, but interestingly, the combination of the CT9 mutation together with the M51 mutation gave a virus that behaved in a fashion roughly similar to that of virus with the M51 mutation alone.
Of significant interest, two viruses that emerged as top candidates were first-position reporter gene viruses, VSV-p1-GFP and VSV-p1-RFP, that both showed promise in retaining oncolytic capacity combined with reduced infection of normal cells. The two fluorescent reporters are different in more than just color. The RFP (dsRed) we used here combines to form a red tetramer, and this tetramer may have slightly greater toxicity than GFP (16). This may serve to reduce replication and budding of progeny VSV-p1-RFP and to increase the toxicity of the virus when a cancer cell is infected. Previous studies have shown that VSV M51 mutants are attenuated in normal cells but still infect many cancer cells. M51 mutants have been used to target brain cancer (17). We show here that 1p-VSV mutants are similarly attenuated, show substantially reduced neurotoxicity after intranasal inoculation, and are able to target glioblastoma in the brain after peripheral intravenous inoculation.
We used a semiquantitative measure of relative viral replication in control versus glioblastoma cells, i.e., the ratio of replication. The largest ratios were indicative of the most ideal viral candidates, namely, viruses that replicated more efficiently in cancer cells than in noncancer cells. The largest ratios were 1:386 and 1:602, for VSV-p1-GFP and VSV-p1-RFP, respectively. These contrasted with those of poor oncolytic performers, such as VSV-CT1, which had a ratio of 1:74 and was relatively ineffective at killing glioblastoma cells.
In a proof-of-principle experiment, we transplanted human glioblastomas into the brains of mice. After peripheral tail vein inoculation with VSV-p1-GFP, we found that all tumors were selectively infected with the virus, yet the surrounding brain appeared largely uninfected. We have previously shown that peripheral inoculation with VSV does not target noncancer mouse or human control cells transplanted into the brain and does not target local brain injury at the same 10-day interval as that between cancer cell implantation and virus inoculation (22).
Interferon selectively protects normal cells from rVSV.
A primary mechanism of protection of normal cells against RNA viruses such as VSV is the activation of innate IFN pathways (23). Several studies have suggested that many cancer cells have defective IFN response pathways (34). Together, these findings suggest that IFN may selectively enhance the survival of normal cells over tumor cells in the presence of VSV. We found that IFN was effective at protecting normal cells from all VSV variants tested. However, in the presence of interferon, the oncolytic action against brain tumor cells was impaired with the strongly attenuated variants, VSV-CT1, VSV-dG-GFP, and VSV-dG-RFP. Three rVSVs (VSV-rp30, -CT9, and -G/GFP) showed more toxicity and greater replication on normal cells than did the other rVSVs. IFN completely reduced infection and replication in normal cells by all VSV variants. IFN has already been approved for use in the human CNS for treatment of multiple sclerosis (12), indicating that it has a reasonably strong safety margin within the brain. Thus, treatment of human brain tumors with recombinant VSVs may derive further benefit from coapplication of IFN in the brain to enhance the selectivity of the virus for the tumor, particularly with those viruses (VSV-G/GFP, VSV-rp30, and VSV-CT9) where infection of noncancer cells may be a problem. Although IFN may reduce infection by VSV, it did not greatly alter the ratio of infections in normal versus tumor cells for the top VSV candidates.
Mutation or deletion of the amino acid at position 51 of the M protein impairs the virus's capability to shut down host cell gene expression while remaining functional for virus assembly (6). Effective mutations at position 51 of the matrix protein, by amino acid substitution (arginine for methionine) (6) or methionine deletion (24, 34), prevent the normal ability of VSV to block nuclear pores and thereby block cellular mRNA transport through the nuclear membrane. Without inhibition of gene expression, cells infected by VSV-M51 mutants mount a significantly greater interferon response, hence creating a stronger antiviral defense. This makes normal cells more resistant to VSV infection. However, tumor cells, which are often deficient in their interferon pathways (34, 37), largely remain susceptible to VSV oncolysis, even with M51 attenuation. VSV-M51 lacks some of VSV's inherent oncolytic potency in vivo, in part due to an effective activation of the systemic immune response to virally infected cells that can reduce the time interval during which VSV can act to infect tumors (39). In addition, whereas VSV and, probably, first-position mutants induce apoptosis through the caspase-independent mitochondrial pathway, VSV-M51 may induce apoptosis through a caspase-dependent pathway (11), which may have consequences for antitumor targeting. Thus, although the M51 and first-position mutants both performed well, their mechanisms of attenuation are different. Another strategy that may facilitate targeting of cancer cells is the inclusion of an interferon gene directly in the VSV genome (20); this has not been tested on brain cancers.
VSV G protein mutations.
The 29-amino-acid cytoplasmic tail of VSV G mediates sorting and anchoring of the viral core to the cellular membrane before initialization in the budding process. The cytoplasmic portion of the G protein can be truncated, leading to an attenuated phenotype (31). Although the CT1 mutant may show an attenuated phenotype in vivo (13, 25), low titers of this virus were not effective at killing glioblastoma cells in our study. The VSV-CT9 mutant, with a G protein cytoplasmic domain truncated down to 9 amino acids, was only mildly impaired in viral budding but showed a greater degree of infection of glioblastomas than did the VSV-CT1 mutant. However, neither of these two VSV G-protein-truncated viruses was as effective as those viruses in the lead group.
The extreme version of VSV-G modification is the complete deletion of the glycoprotein gene, as in VSV-dG-GFP and VSV-dG-RFP. In the absence of VSV glycoprotein, viral budding is severely impaired, with viral particle yields around 30 times lower than those with the G protein present (32). Though virus progeny can still be produced and leave the cell (31, 36), the absence of G protein spikes leaves the viral particle incapable of binding to any new cell, thereby terminating the viral infectious cycle. This virus is effective at killing the cells it infects, but as its progeny are not infective, it would primarily be useful as a direct tumor toxin (8). While increasing its safety profile in the brain, it would ultimately eliminate only those cancer cells directly infected upon direct inoculation into the tumor. However, VSV-dG-GFP may still generate an immune response and merits further consideration relating to whether it might be effective in stimulating an antitumor immune response after selective infection of tumors. VSV can enhance destruction of tumors both by direct oncolytic actions and by recruiting the immune system to attack tumor cells (27).
In conclusion, our data suggest that in addition to the mutated M51 VSV variants that have previously been shown to show promise for targeting cancer cells with reduced infection of normal cells (17), VSVs with a reporter gene in the first position, VSV-p1-GFP and VSV-p1-RFP, show a similarly attenuated infection of normal cells but retain the ability to target and infect brain cancer both in vitro and in vivo.
Acknowledgments
Support for this work was provided by NIH grant CA124737.
Footnotes
Published ahead of print on 11 November 2009.
REFERENCES
- 1.Aghi, M., and S. Rabkin. 2005. Viral vectors as therapeutic agents for glioblastoma. Curr. Opin. Mol. Ther. 7:419-430. [PubMed] [Google Scholar]
- 2.Ahmed, M., S. D. Cramer, and D. S. Lyles. 2004. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 330:34-49. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed, M., and D. S. Lyles. 1997. Identification of a consensus mutation in M protein of vesicular stomatitis virus from persistently infected cells that affects inhibition of host-directed gene expression. Virology 237:378-388. [DOI] [PubMed] [Google Scholar]
- 4.Clarke, D. K., F. Nasar, M. Lee, J. E. Johnson, K. Wright, P. Calderon, M. Guo, R. Natuk, D. Cooper, R. M. Hendry, and S. A. Udem. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J. Virol. 81:2056-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper, D., K. J. Wright, P. C. Calderon, M. Guo, F. Nasar, J. E. Johnson, J. W. Coleman, M. Lee, C. Kotash, I. Yurgelonis, R. J. Natuk, R. M. Hendry, S. A. Udem, and D. K. Clarke. 2008. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and G gene truncation reduces neurovirulence and enhances immunogenicity in mice. J. Virol. 82:207-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulon, P., V. Deutsch, F. Lafay, C. Martinet-Edelist, F. Wyers, R. C. Herman, and A. Flamand. 1990. Genetic evidence for multiple functions of the matrix protein of vesicular stomatitis virus. J. Gen. Virol. 71:991-996. [DOI] [PubMed] [Google Scholar]
- 7.Dalton, K. P., and J. K. Rose. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414-421. [DOI] [PubMed] [Google Scholar]
- 8.Duntsch, C. D., Q. Zhou, H. R. Jayakar, J. D. Weimar, J. H. Robertson, L. M. Pfeffer, L. Wang, Z. Xiang, and M. A. Whitt. 2004. Recombinant vesicular stomatitis virus vectors as oncolytic agents in the treatment of high-grade gliomas in an organotypic brain tissue slice-glioma coculture model. J. Neurosurg. 100:1049-1059. [DOI] [PubMed] [Google Scholar]
- 9.Ebert, O., S. Harbaran, K. Shinozaki, and S. L. Woo. 2005. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 12:350-358. [DOI] [PubMed] [Google Scholar]
- 10.Flanagan, E. B., J. M. Zamparo, L. A. Ball, L. L. Rodriguez, and G. W. Wertz. 2001. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J. Virol. 75:6107-6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaddy, D. F., and D. S. Lyles. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodin, D. S. 2005. Treatment of multiple sclerosis with human beta interferon. Int. MS J. 12:96-108. [PubMed] [Google Scholar]
- 13.Johnson, J. E., F. Nasar, J. W. Coleman, R. E. Price, A. Javadian, K. Draper, M. Lee, P. A. Reilly, D. K. Clarke, R. M. Hendry, and S. A. Udem. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawson, N. D., E. A. Stillman, M. A. Whitt, and J. K. Rose. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. USA 92:4477-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lichty, B. D., D. F. Stojdl, R. A. Taylor, L. Miller, I. Frenkel, H. Atkins, and J. C. Bell. 2004. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum. Gene Ther. 15:821-831. [DOI] [PubMed] [Google Scholar]
- 16.Long, J. Z., C. S. Lackan, and A. K. Hadjantonakis. 2005. Genetic and spectrally distinct in vivo imaging: embryonic stem cells and mice with widespread expression of a monomeric red fluorescent protein. BMC Biotechnol. 5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lun, X., D. L. Senger, T. Alain, A. Oprea, K. Parato, D. Stojdl, B. Lichty, A. Power, R. N. Johnston, M. Hamilton, I. Parney, J. C. Bell, and P. A. Forsyth. 2006. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J. Natl. Cancer Inst. 98:1546-1557. [DOI] [PubMed] [Google Scholar]
- 18.Lundh, B., A. Löve, K. Kristensson, and E. Norrby. 1988. Non-lethal infection of aminergic reticular core neurons: age-dependent spread of ts mutant vesicular stomatitis virus from the nose. J. Neuropathol. Exp. Neurol. 47:497-506. [DOI] [PubMed] [Google Scholar]
- 19.Lyles, D. S., and C. E. Rupprecht. 2007. Rhabdoviridae, p. 1363-1408. In D. M. Knipe et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 20.Obuchi, M., M. Fernandez, and G. N. Barber. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 77:8843-8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohgaki, H., and P. Kleihues. 2005. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 64:479-489. [DOI] [PubMed] [Google Scholar]
- 22.Özduman, K., G. Wollmann, J. Piepmeier, and A. N. van den Pol. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry, A. K., G. Chen, D. Zheng, H. Tang, and G. Cheng. 2005. The host type I interferon response to viral and bacterial infections. Cell Res. 5:407-422. [DOI] [PubMed] [Google Scholar]
- 24.Publicover, J., E. Ramsburg, M. Robek, and J. K. Rose. 2006. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J. Virol. 80:7028-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Publicover, J., E. Ramsburg, and J. K. Rose. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 78:9317-9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Publicover, J., E. Ramsburg, and J. K. Rose. 2005. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J. Virol. 79:13231-13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiao, J., H. Wang, T. Kottke, R. M. Diaz, C. Willmon, A. Hudacek, J. Thompson, K. Parato, J. Bell, J. Naik, J. Chester, P. Selby, K. Harrington, A. Melcher, and R. G. Vile. 2008. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 15:604-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsburg, E., J. Publicover, L. Buonocore, A. Poholek, M. Robek, A. Palin, and J. K. Rose. 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J. Virol. 79:15043-15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts, A., E. Kretzschmar, A. S. Perkins, J. Forman, R. Price, L. Buonocore, Y. Kawaoka, and J. K. Rose. 1998. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J. Virol. 72:4704-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose, N. F., P. A. Marx, A. Luckay, D. F. Nixon, W. F. Moretto, S. M. Donahoe, D. Montefiori, A. Roberts, L. Buonocore, and J. K. Rose. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539-549. [DOI] [PubMed] [Google Scholar]
- 31.Schnell, M. J., L. Buonocore, E. Boritz, H. P. Ghosh, R. Chernish, and J. K. Rose. 1998. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 17:1289-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnell, M. J., J. E. Johnson, L. Buonocore, and J. K. Rose. 1997. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell 90:849-857. [DOI] [PubMed] [Google Scholar]
- 33.Shinozaki, K., O. Ebert, and S. L. Woo. 2005. Eradication of advanced hepatocellular carcinoma in rats via repeated hepatic arterial infusions of recombinant VSV. Hepatology 41:196-203. [DOI] [PubMed] [Google Scholar]
- 34.Stojdl, D. F., B. D. Lichty, B. R. tenOever, J. M. Paterson, A. T. Power, S. Knowles, R. Marius, J. Reynard, L. Poliquin, H. Atkins, E. G. Brown, R. K. Durbin, J. E. Durbin, J. Hiscott, and J. C. Bell. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263-275. [DOI] [PubMed] [Google Scholar]
- 35.van den Pol, A. N., K. P. Dalton, and J. K. Rose. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 76:1309-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Pol, A. N., K. Ozduman, G. Wollmann, W. S. Ho, I. Simon, Y. Yao, J. K. Rose, and P. Ghosh. 2009. Viral strategies for studying the brain, including a replication-restricted self-amplifying delta-G vesicular stomatitis virus that rapidly expresses transgenes in brain and can generate a multicolor Golgi-like expression. J. Comp. Neurol. 516:456-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wollmann, G., M. D. Robek, and A. N. van den Pol. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J. Virol. 81:1479-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wollmann, G., P. Tattersall, and A. N. van den Pol. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005-6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu, L., T. G. Huang, M. Meseck, J. Altomonte, O. Ebert, K. Shinozaki, A. García-Sastre, J. Fallon, J. Mandeli, and S. L. Woo. 2008. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 19:635-647. [DOI] [PMC free article] [PubMed] [Google Scholar]