

Abstract
In infected cells, virus components must be organized at the right place and time to ensure assembly of infectious virions. From a different perspective, assembly must be prevented until all components are available. Hypothetically, this can be achieved by allosterically controlling assembly. Consistent with this hypothesis, here we show that the structure of the hepatitis B virus (HBV) core protein dimer, which can spontaneously self-assemble, is incompatible with capsid assembly. Systematic differences between core protein dimer and capsid conformations demonstrate linkage between the intradimer interface and interdimer contact surface. These structures also provide explanations for the capsid-dimer selectivity of some antibodies and the activities of assembly effectors. Solution studies suggest that the assembly-inactive state is more accurately an ensemble of conformations. Simulations show that allostery supports controlled assembly and results in capsids that are resistant to dissociation. We propose that allostery, as demonstrated in HBV, is common to most self-assembling viruses.
Viruses are incredibly efficient molecular machines. It is not surprising that they have certain commonalities in their life cycles. For example, most of them self-assemble to specifically encapsidate the right components in a crowded and complex intracellular environment. They interact with host cells to enter the host and to traffic therein. Even under less than ideal circumstances, they persist while between hosts. It is likely that the physical basis for these common behaviors is a few common mechanisms. The critical hypothesis addressed in this paper is whether virus capsid assembly is subject to allosteric regulation. Allostery would explain how capsid proteins avoid packaging the wrong nucleic acid to unproductively assemble into extremely stable dead-end complexes. Conversely, allostery provides a mechanism for selectively packaging the correct content. Therefore, allostery may be generalizable for any self-assembling virus.
Hepatitis B virus (HBV) is one of the most widespread infectious public health problems (43). HBV has infected 2 billion people, 360 million chronically, and it leads to approximately 600,000 deaths each year (44). The virus itself, among the smallest human pathogens, has an unusual life cycle that is critically dependent on accurate assembly. The immature core of this DNA virus is composed of an icosahedral capsid containing the RNA pregenome (the RNA template for the viral genome), reverse transcriptase, and some host proteins (21). The core assembles in the cytoplasm, after which the linear single-stranded RNA pregenome is reverse transcribed within the core to generate the circular, partially double-stranded DNA genome of the infectious particle (21, 35, 38).
The predominant form of the HBV capsid is constructed from 120 copies of the homodimeric core protein (Cp; also known as the HBV core antigen [HBcAg]) arranged with icosahedral symmetry (see Fig. 1). The local quaternary arrangement in a capsid is a lattice of triangles, i.e., trimers of dimers (15). Cp itself is a 183-residue protein comprising a 149-residue assembly domain and a 34-residue RNA-binding C terminus. Cp dimers are held together by a four-helix bundle (amino acids 49 to 109), with two helices contributed by each half dimer. This bundle forms the spikes that punctuate the virus surface (5, 48). The dimer interface also incorporates a pair of conserved cysteines that can form a stabilizing, but not required, disulfide (34, 52). The interdimer contact is a helix-turn-extended structure (amino acids 111 to 143) that forms overlapping “shingles” around icosahedral fivefold and quasi-sixfold vertices. The RNA-binding domain (amino acids 150 to 183), which appears to be disordered based on image reconstructions (57), also has roles in signaling intracellular transport of the virus (49). In heterologous expression systems, the RNA-binding domain tenaciously binds and encapsidates random host RNA (2).
FIG. 1.

The crystallographic asymmetric unit is a trimer of dimers arranged as a broken triangle or one turn of a helix. (a) Cp149-Y132A dimers. Icosahedral HBV capsids can be considered to be arrays of triangular trimers of dimers. A threefold symmetric trimer is highlighted using yellow and green surface shading. Such triangles are the fundamental quaternary structure in HBV capsids; a trimer of dimers has been implicated in nucleating capsid assembly (58). (b and c) Asymmetric unit of Cp149-Y132A as a ribbon diagram (b) and a surface-shaded diagram (c). Dimers are labeled AB, CD, and EF (inset). The mutated residue, Y132A, is identified by the pink spheres at the ends of each dimer. The B-C and D-E interdimer contacts resemble those found in capsid. The F subunit passes under the A subunit and does not make an equivalent contact. The broken triangle of the Cp149-Y132A asymmetric unit suggests that interprotein contacts between mutant dimers are not strong enough to force a planar quaternary structure, likely the basis for their inability to nucleate capsid assembly.
Self-assembly has been demonstrated previously using the 149-residue assembly domain (Cp149) of the homodimeric core protein (2, 11, 12, 40, 41, 46). Based on kinetic analyses of in vitro assembly reactions, the formation of a trimer of Cp dimers has been identified as the nucleating event of assembly (58). Evidence indicates that Cp undergoes a conformational change(s) associated with assembly. It has been suggested that the surprisingly weak interdimer association energy in capsids is due to the energetic expense of a conformational change related to assembly (12). Further support for conformational change comes from observations of ions, peptides, and small molecules that bind to Cp. Zinc ions are bound cooperatively by Cp dimers and stimulate assembly kinetics, promoting kinetic traps (41). Similarly, a cooperative change in dimer structure is induced by a capsid-binding peptide, as observed in nuclear magnetic resonance (NMR) studies (20). Cooperative effects strongly imply concerted structural changes concomitant with binding. Heteroaryldihydropyrimidine (HAP) molecules, a family of assembly effectors, activate assembly both kinetically and thermodynamically (40, 42); assembly at an inappropriate time or place has great potential as an antiviral strategy. HAPs overfill a cavity at the interdimer contact, explaining their thermodynamic effect and their ability to misdirect assembly (5) but not their kinetic effect, which has been proposed to be the result of an assembly-activating conformational change. However, any change must be relatively subtle, as circular dichroism (39, 45) and NMR (20) spectroscopy data indicate little change in secondary structure between dimer and capsid states. Immunological results are consistent with the findings of in vitro studies: though the tips of the intradimer spikes are the major antigenic region in both capsids and free dimer, some antibodies are specific for one form or the other (37).
These results have led to the hypothesis that HBV assembly is subject to allosteric regulation (2, 11, 12, 40, 41, 46). That is, the structure of free Cp dimer is not compatible with assembly and therefore a structural transition activates the assembly reaction. This mechanism would minimize inappropriate assembly. Furthermore, it implies that some molecule(s), an assembly effector, induces the transition. Though they show less fidelity for forming regular structures, similar allosteric mechanisms have also been implicated in retrovirus assembly (16, 17, 26, 29, 31). However, the structural basis of allostery in regulating virus assembly has not been demonstrated previously.
To test the allostery hypothesis, we have determined the structure of dimeric Cp out of the context of the capsid. To do so, we developed an assembly-incompetent form of dimeric Cp, comprising Cp149 with the mutation Y132A (Cp149-Y132A) (7). Y132 is almost completely buried in the capsid, accounting for nearly 10% of the total buried surface area (48), but should be completely exposed in a free dimer. Thus, unless dimeric Cp149 is completely refolded compared to the capsid form, it is unlikely that the mutation Y132A will affect dimer structure. Cp149-Y132A has been purified as a dimer and has circular dichroism, fluorescence, and chromatographic properties that are essentially identical to those of the wild-type protein. By itself, Cp149-Y132A is assembly incompetent (28), even under conditions in which the wild-type assembly domain assembles nearly quantitatively (7). However, Cp149-Y132A will coassemble with wild-type protein to yield extremely fragile capsids (7). Here, we examine the structure of free dimer and examine the implications of that structure for virus self-assembly.
MATERIALS AND METHODS
Protein purification.
Cp149-Y132A expression and purification were performed as described previously (7), except for the final ion exchange step. Partly purified Cp149-Y132A in buffer QA (50 mM Tris-HCl, pH 9.0, 80 mM NaCl, 2 mM dithiothreitol [DTT]) was adsorbed to a HiTrap Q column (GE Health Sciences) and eluted in a multistep gradient with buffer QB (50 mM Tris-HCl, pH 9.0, 2 M NaCl, 2 mM DTT). The sample was initially loaded onto a 5-ml column and washed with 5 volumes of buffer QA. The wash was followed by a 0 to 5% buffer QB gradient in 2 column volumes, a 5 to 10% QB gradient in 10 column volumes (wherein Cp149-Y132A elutes), a 10 to 30% QB gradient in 10 column volumes, a 30 to 100% QB gradient in 2 volumes, and then 5 column volumes of 100% QB to clear the column. Fractions were pooled based on SDS-PAGE and absorbance, concentrated to at least 8 mg/ml, and then dialyzed into 10 mM Tris-HCl (pH 9.0)-2 mM DTT. Cp149-C61S was purified as described previously for wild-type Cp149 (59).
Crystallization and structure determination.
Crystals of Cp149-Y132A were grown from a 10-mg/ml solution of Cp in 50 mM Tris-HCl (pH 9.0) in an equal volume of precipitating solution containing 100 mM sodium cacodylate (pH 6.5), 9% polyethylene glycol 1500, and 0.1 M calcium acetate. Small rhombohedral crystals were transferred into synthetic mother liquor with 20% glycerol immediately prior to freezing at liquid nitrogen temperatures. Crystallographic data were collected on a Rigaku 3HR X-ray source equipped with an R-Axis IV++ detector at a crystal-to-detector distance of 150 mm. Diffraction data were indexed, integrated, and scaled with the HKL-2000 program (36). Crystals belonged to space group P31 with cell dimensions as follows: a = b = 104.16 Å, c = 86.91 Å, α = β = 90°, γ = 120°. We also collected data on similar crystals but at lower resolution, with no significant structural differences.
The structure was determined by molecular replacement. Initial phases were obtained using the program PHASER (32) with atomic coordinates of one AB dimer of HBV core protein from a capsid (Protein Data Bank [PDB] identification no. 1QGT) (48). The 1QGT structure was chosen as the highest-resolution HBV structure available. The initial structure determination was performed with a 2.9-Å data set. The first stages of structure refinement were performed using REFMAC 5.0 (33, 47) implemented in the CCP4 suite (13). About 5% of the reflections were used for the test set. An initial examination of the asymmetric unit showed that subunits were not arranged with point symmetry or in a continuous helix, and thus we did not use molecular averaging to improve phases. The resultant model obtained from the structure solution was subjected to rigid body refinement using individual proteins first and then protein fragments, which led to an R-factor of 42.2% (Rfree = 46.0%). The improved model was subjected to restrained refinement with REFMAC, followed by manual rebuilding with COOT (18), which brought the R-factor to 24.7% (Rfree = 30.9%). After several iterations of rebuilding with COOT and restrained refinement with REFMAC, further refinement and energy minimization were performed with CNS 1.2 (9). This model was then applied to a newly acquired 2.25-Å data set, and a similar process of molecular replacement from the PHASER to CNS steps was carried out. In the refined model, improved with iterative rounds of model building, there remained areas of weak density, the spike tips for the A, B, C, and D subunits (but not the E and F subunits) and the C terminus of the A subunit, that were iteratively rebuilt by making use of omit maps to eliminate model bias. Finally, 213 water molecules were added, which lead to a final R-factor of 21.7% (Rfree = 26.4%). Density was unambiguous for the main chain from residue 1 to at least residue 141 for all six polypeptides (see Fig. S1 and S2 in the supplemental material). The Ramachandran plot for the final model was calculated using the program PROCHECK (30) and shows that only 1.1% of φψ angles were in disallowed regions, 89.1% of the residues were in the most favored region, and the remaining residues were in the additionally allowed regions. Residues in the disallowed regions were from regions of poorly defined density also characterized by high B-factors: the A, C, and D spike tips and the C-terminal extended region of the A chain. Statistics for the data set used in this structure determination and the final refined structure are detailed in Table 1.
TABLE 1.
Data and refinement statistics
| Statistics | Value(s)a |
|---|---|
| Data statistics | |
| Space group | P31 |
| Unit cell dimensions | a = b = 104.16 Å, c = 86.91 Å; α = β = 90°, γ = 120° |
| Resolution range (Å) | 25.32-2.25 (2.30-2.25) |
| Completeness of data (%) | 99.96 (99.9) |
| Redundancy of data | 4.5 (4.4) |
| I/σ | 41.8 (5.34) |
| Rmerge | 0.076 (0.572) |
| Refinement statistics | |
| Resolution (Å) | 2.25 |
| No. of unique reflections | 47,650 (3,506) |
| R (%) | 21.7 (27.8) |
| Rfree (%) | 26.4 (32.5) |
| Asymmetric unit | Trimer of dimers |
| No. of nonhydrogen protein atoms | 6,729 |
| No. of water molecules | 213 |
| Mean B-factor | |
| Protein | 48.3 |
| Water | 48.8 |
| Ramachandran plot—residues in: | |
| Most favored regions (%) | 89.1 |
| Additionally allowed regions (%) | 8.7 |
| Generously allowed regions (%) | 1.1 |
| Disallowed region (%) | 1.1 |
| Nonideality of: | |
| Bond angles (°) | 1.926 |
| Bond lengths (Å) | 0.020 |
Values in parentheses are for the highest-resolution shell.
Assembly studies.
Coassembly reactions were carried out at either 23 or 37°C in 50 mM HEPES, pH 7.5, in the presence or absence of 5% β-mercaptoethanol (βME). Assembly was initiated by the addition of 2× NaCl to a final concentration of 0.5 M. Long-term kinetics of assembly were observed using size exclusion chromatography performed with a 21-ml Superose 6 column equilibrated in a solution of 50 mM HEPES, pH 7.5, and 1.0 M NaCl. The column was mounted on a Shimadzu high-performance liquid chromatography system equipped with a temperature-controlled autoinjector in order to facilitate the long time course. Peaks at 280 nm were quantified after manual baseline correction using the supplied LCSolutions software (Shimadzu).
Kinetic modeling.
Mathematical models are based on a master-equation approach that treats assembly and disassembly as a completely reversible cascade of second-order reactions. The underlying math has been extensively reviewed (19). The equations used in this study are for an icosahedral structure assembled from 30 tetravalent “dimers” (19). The dimers are designed to resemble the HBV subunit topology. For computational simplicity, only single forward and backward paths were allowed. Similar models have been used previously to qualitatively fit the assembly of HBV (41, 58) and cowpea chlorotic mottle virus (25, 56).
RESULTS
The crystallographic asymmetric unit suggests aborted self-assembly.
Cp149-Y132A crystallizes under conditions that are conducive to assembly of the wild-type Cp149 (see Materials and Methods); under these same conditions, the mutant does not assemble into capsid. Crystals diffracted to 2.25 Å, allowing in-house collection of a high-quality data set and a reliable molecular replacement solution of the structure based on a dimer from capsid (Table 1). The crystallographic asymmetric unit is a trimer of Cp149-Y132A dimers (Fig. 1b and c) that is suggestive of an abortive assembly intermediate or nucleation complex (58). The trimer itself resembles the quaternary arrangement of dimers in capsid (Fig. 1). The crystallographically unique dimers, named AB, CD, and EF, are at first glance structurally similar to dimers from capsid (Fig. 1 and 2). However, unlike a trimer that might be extracted from a capsid, the crystallographic asymmetric unit is not a closed triangle. Instead, the asymmetric unit is a one-turn right-handed helix.
FIG. 2.

Overlays of dimers show asymmetry and divergence from capsid structure viewed tangent to the virus surface. (a) Cp149-Y132A dimers are asymmetric and structurally unique. The monomers fall into two structural classes based on the conformation of the long helices at the intradimer interface. Even so, obvious differences at the tip of the spike (top center of the figure) and at the turn at the end of the interdimer contacts are apparent. (b) Overlay of the EF Cp149-Y132A dimer (with the E and F subunits colored dark blue and marine blue, as in panel a) and dimer from capsid (AB from the PDB no. 1QGT structure [48], in yellow) to allow comparison of free and dimer structures. There are substantial differences between the two conformations at the periphery, even though central cores, or chasses, are identical. Unlike Cp149-Y132A, the dimer from capsid has well-maintained twofold symmetry (see the supplemental material). (c) For quantitative analysis, as well for all overlays in this figure, chassis α carbons (corresponding to residues 1 to 10, 26 to 62, and 95 to 110) of A, D, and E subunits of Cp149-Y132A were aligned with equivalent residues from a capsid A subunit; distances between equivalent α carbons throughout the dimers are reported. Though not specifically constrained, both half dimers show the same pattern of similarity and difference. (d) A side-by-side comparison of the E (left) and F (right) halves of the four-helix bundle spike shows the asymmetry of the Cp149-Y132A dimers. This view shows the complementary surfaces that are buried at the dimer interface. The segments from A54 to W71 have approximately the same alignments to facilitate comparison. Note that residues 79 and 80 are part of the random coil turn in chain F.
The trimer of dimers in the asymmetric unit has two capsid-like interdimer contacts, the B-C and D-E junctions (Fig. 1). There is no F-A contact. The two junctions cover the same respective surfaces as in capsid, although the Y132A mutation allows closer interaction than that observed in capsids (5, 48). The lack of closure of the trimer results largely from changes in dimer geometry (detailed in the following paragraphs) that are propagated by quaternary structure. Clearly, this broken triangle with two contacts, rather than a closed triangle with three contacts, is stable enough to persist as the crystallographic asymmetric unit. This suggests that closure of the triangle, as seen in capsids, represents a compromise between the stability of dimer geometry and energy obtained by forming one more interdimer contact. This compromise can be described explicitly, by comparing the energies of triangle and asymmetric unit formation:
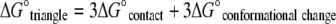 |
(1) |
The weaker contact interaction of Cp149-Y132A results in an asymmetric unit where the association energy, neglecting crystal packing contacts, is
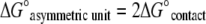 |
(2) |
For the Cp149-Y132A mutant, we observe that ΔG°asymmetric unit is <ΔG°triangle, that is, the broken triangle is favored over the closed triangle, which leads to the conclusion that ΔG°conformational change is positive.
Conformations of free dimers are distinct from dimers in capsid.
Cp149-Y132A has distinct structural differences from subunits extracted from a capsid that are not directly related to the mutation itself. The comparison of the Cp149-Y132A and capsid structures is most readily viewed from the perspective of dimers. First consider the capsid structure: the four classes of monomers from a T = 4 capsid, A, B, C, and D, are nearly identical to one another; the AB and CD dimers show well-preserved twofold symmetry (5, 48). This similarity results in a root mean square deviation (RMSD) between dimers of about 1 Å, in either orientation.
In contrast, the Cp149-Y132A AB, CD, and EF dimers are asymmetric and show obvious differences from one another in (i) the upper half of the four-helix bundle that forms the spikes at the dimer interface and (ii) the orientation of the C-terminal interdimer contact domain (Fig. 2a and d). The spike tips have two different conformations in every dimer, an obvious difference from the capsid form. In the B, C, and F half dimers, the spike tip is capsid-like. In A, D, and E half dimers, residues 79 to 81 are unraveled from a helical conformation but are relatively upright (Fig. 2d; see also Fig. S1 and S2 in the supplemental material). Thus, all spike tips show a large displacement compared to capsid (Fig. 2c). There is no obvious reason that the different conformations are not in sequence (e.g., alternating capsid-like/unraveled spike tips). The loops at the tips of the spikes also vary in their crystallographic order, with the clearest density for the EF spike tip. The structural change at the intradimer interface cannot be directly related to the Y132A mutation, which is at the periphery of the subunit (the far left and right of Fig. 2a and b), at an interdimer contact (Fig. 1), not at the intradimer interface.
When Y132A dimers are aligned together and against dimer from capsid, the overall RMSD for α carbons between any pair is about 2 Å (Table 2), but this average misses the systematic structural changes. Visual examination of the overlays reveals an underlying constant substructure, or chassis, that is attached by hinges to three subdomains (Fig. 2c). Using this chassis as the basis for alignment reveals systematic structural changes. When the capsid and Y132A dimer forms are aligned based on the chassis of one half dimer, the chassis of the other half dimer remains in register (Fig. 2c). The RMSD between α carbons of the chassis domains from the three Y132A dimers and a capsid dimer is about 0.6 to 0.8 Å (Table 2; see also Table S1 in the supplemental material), which is close to the uncertainty of atomic position predicted for a structure at this resolution and level of refinement by Luzzatti and Sigma A analyses implemented in the REFMAC refinement program (33). Thus, the chassis subdomain is discontinuous but structurally well conserved from free dimer to capsid. In our structure, there is a disulfide at the dimer interface between dimer-related C61 residues, also observed in capsid. Because this disulfide, which forms an intradimer cross-link, is not likely to be present in authentic cytoplasmic assembly, we examined its effect in solution studies described later in this paper.
TABLE 2.
Comparison of Cp149-Y132A dimers with an AB dimer from capsid
| Region | RMSD (Å) between PDB no. 1QCT dimer and Cp149-Y132A dimer: |
||
|---|---|---|---|
| AB | DC | FE | |
| Overall | 1.80 | 2.30 | 2.82 |
| Chassisa | 0.68 | 0.79 | 0.82 |
The chassis domain, residues 1 to 10, 26 to 62, and 95 to 110, is discontinuous with respect to the primary structure. For this comparison, the chasses of the Y132A mutant A, D, and E subunits were aligned with a capsid A subunit; the chassis for the other half dimer was not constrained.
In dimers aligned based on the chasses, there are three obvious subdomains that have moved relative to their positions in the capsid conformation (Fig. 2b and c and Fig. 3). We refer to these subdomains as the fulcrum helix (residues 10 to 25), the spike tip (residues 63 to 94), and the C-terminal interdimer contact (residue 111 to the C terminus). As noted previously, the spike tips in A, D, and E subunits are not capsid-like. However, the spike tips from B, C, and F, the fulcrums, and the contact subdomains appear to move as rigid bodies based on their structures and positions in the capsid form. The fulcrum is constrained by both the spike and the contact domain and can mechanically link these two domains. The intersubunit contact domain adopts a range of positions in the Y132A dimers. Because the contact domains have not changed their tertiary structure appreciably, they are able to form capsid-like contacts (Fig. 1), but the higher-order structural changes prevent propagation of assembly. Because subdomain movement is hinged to the chassis, the greatest displacement from the capsid conformation is at the subdomain periphery, a Cα displacement by as much as 9 Å at the spike tip and intersubunit contact subdomains (Fig. 2c).
FIG. 3.
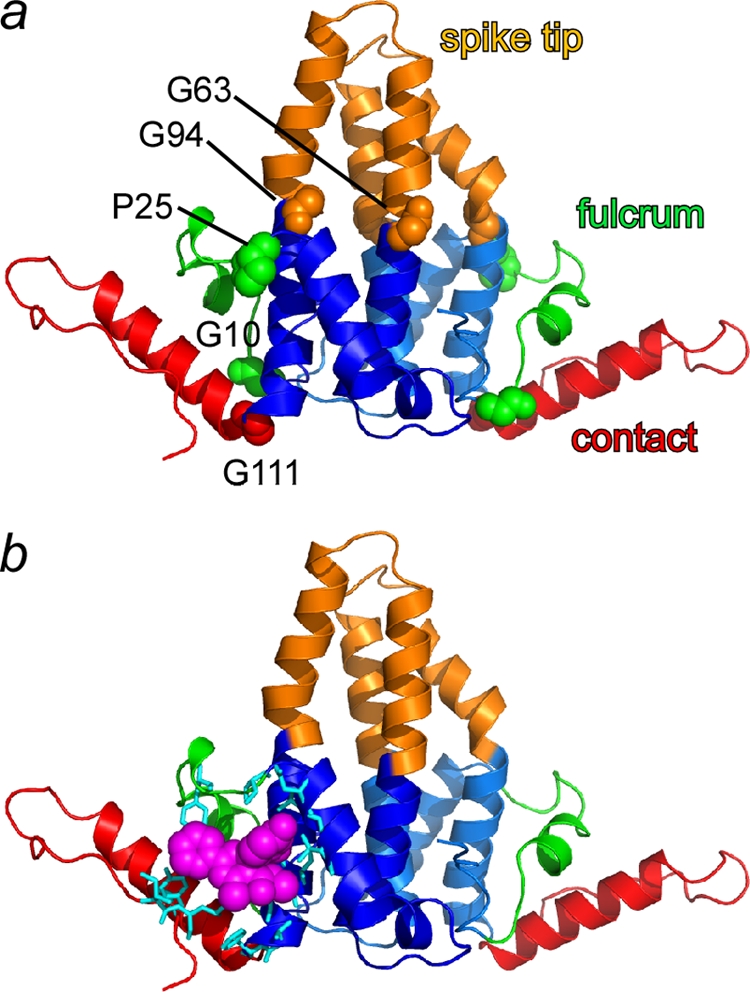
Free dimers have subdomains connected to a chassis by flexible hinges. (a) Based on molecular displacement data (Fig. 2c), subdomains have been color coded and labeled. Domain junctions are flexible hinges (shown in a space-filling representation): Gly10 and Pro25 delineate the fulcrum domain (green), Gly63 and Gly94 delineate the spike tip (copper), and Gly111 is the C-terminal interdimer contact domain (red). The structurally invariant chassis is shown in blue. (b) An assembly effector HAP molecule (magenta) and adjacent residues (cyan), modeled on the basis of a capsid-HAP cocrystal structure (5), noncovalently links the flexible contact and fulcrum domains with the chassis. This stabilization may explain HAP's assembly-accelerating activity.
Movement of the subdomains relative to the chassis is facilitated by amino acid hinges: glycines at positions 10, 63, 94, and 111 and a proline at position 25 (Fig. 3). Though glycine is uncommon in α helices and is considered to be helix breaking, glycines at positions 63 and 94 are conserved in HBV core protein sequences (22), resulting in a hinge that cuts right through a four-helix bundle. Mutational analysis supports a biological role for a hinge in the spike: residue 97 (Phe in this structure, Ile in other HBV strains) in the chassis domain forms a large part of the base through which the spike tip interacts with the chassis; mutation of this residue to Leu is known to result in phenotypic changes in cell culture (50, 51) that correlate with enhanced in vitro assembly (11). The changed positions of these domains suggest that dimers are highly dynamic. However, subdomains do not show evidence of any concerted molecular motion of subdomains within the crystal (as would be detected in TLS refinement [33]). We thus hypothesize that the movement of these domains results in a conformational switch between assembly-active and -inactive states (described in Discussion).
Implications of dimer structure for antigenicity.
The Cp149-Y132A structure provides a clear explanation for the differential antigenicities of capsid and dimer. In both capsid and dimer contexts, one of the major antigenic sites of the core protein is the tip of the spike (37). Cryo-electron microscopy structures of capsid-Fab complexes show that the binding sites for many such antibodies include the loops at the tips of both half dimers (1, 14). In the free dimer structure, the spike tip is grossly distorted compared to the capsid form. The spike tip subdomain of one half dimer (B, C, or F) is very similar to the equivalent region in a capsid dimer, allowing unambiguous alignment of the tip from that half dimer (Fig. 4). However, in that alignment, the loop from the other subunit of the free dimer (A, D, or E) adopts a noncapsid conformation which, combined with the quaternary changes, results in a unique dimer epitope. In this alignment, the α carbons of this second loop are displaced by up to 10 Å from the equivalent residues in the dimer from capsid. The flexibility of the free dimer (this structure) and dimer in the context of capsid (4) suggests that it is possible for dimer to transiently remodel into other alternative conformations, resulting in a broad range of selectivity/specificity (1).
FIG. 4.
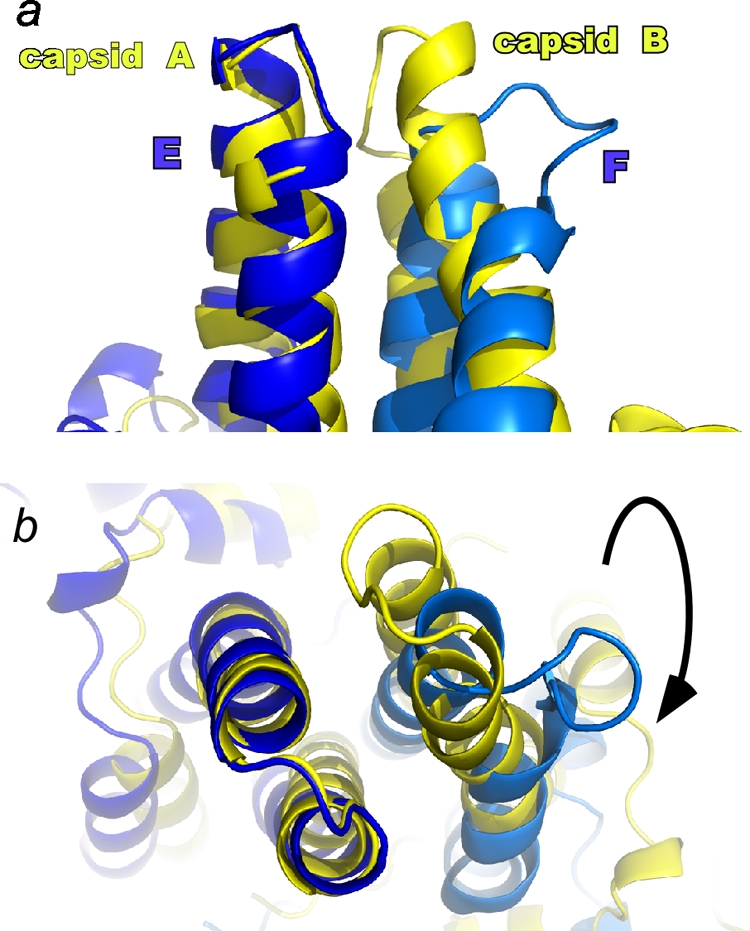
The major Cp epitopes, the spike tips at the dimer interfaces, are radically different in free and capsid forms. In many capsid-antibody complexes, the epitope includes both half dimers (24). The structural difference is made clear in this overlay of Y132A mutant and capsid dimers (colored as in Fig. 2), where alignment is based on α carbons of the structurally similar components of the spike tips (e.g., residues 70 to 85 of the Cp149-Y132A E subunit and the corresponding residues of a capsid A subunit). In the side view (a) and top view (b), the displacement of the α carbons of the F subunit spike tip is as much as 10 Å.
Role of the chassis intradimer disulfide in self-assembly.
This crystal structure has an intermolecular disulfide (C61-C61) at the dimer interface within the chassis domain (Fig. 5a). Crystal structures of capsid, even with a C61A mutation (5), indicate that the conformation of capsid is conducive to disulfide formation. However, a disulfide would not be expected for free dimer in the reducing environment within a cell. Thus, we ask whether the structure of the oxidized chassis domain is an accurate representation of the reduced form. It is clear that in the wild-type protein, both reduced and oxidized forms are assembly competent (unpublished data). We tested the structural and dynamic constraints that the disulfide may impose on the free dimer by examining coassembly reactions with reduced and oxidized forms of Cp149-Y132A.
FIG. 5.

Chassis domain allostery is affected by the presence or absence of an intradimer disulfide. (a) Cysteine 61 is a conserved residue at the dimer interface in the chassis domain (domains are colored as in Fig. 3). It readily oxidizes to a C61-C61 disulfide, which may constrain chassis dynamics and conformation. (b) The presence or absence of the chassis domain disulfide in the Y132A mutant affects coassembly with Cp149-C61S at 23°C. The amounts of capsid and dimer were evaluated by size exclusion chromatography at a specified time after assembly was induced. The capsid assembly rate correlates directly to the nucleation rate (19). The assembly-competent C61S mutant was used for these comparisons, as it assembles the same way in oxidizing and reducing environments (indicated by open and closed squares, respectively). Oxidized 20 μM Cp149-Y132A (oxY132A [open triangles]) and reduced 20 μM Cp149-Y132A (redY132A [closed triangles]) were coassembled with 5 μM Cp149-C61S (C61S). While partially oxidized Cp149-Y132A was able to participate in normal assembly, reduced Cp149-Y132A poisons the reaction, slowing the nucleation rate and decreasing the extent of assembly.
Previously, we observed that Cp149-Y132A can coassemble with assembly-competent Cp (7). To separate the effects of the disulfide in the Y132A mutant from a disulfide in the assembly-competent Cp149, coassembly reactions were carried out using a Cp149 mutant in which cysteine 61 has been mutated to a serine; this mutant displays a normal extent and normal kinetics of assembly (unpublished data). Cp149-Y132A was either allowed to partially air oxidize or reduced by the addition of 5% βME. The oxidation state was verified by nonreducing SDS-PAGE; oxidized protein was cross-linked by the intradimer disulfide and ran at twice the weight of the monomer (see Fig. S3 in the supplemental material).
The most striking result was observed in assembly reactions run at 23°C. As a control, we show here that Cp149-C61S assembles normally in the presence or absence of βME (Fig. 5b). When 10 μM oxidized Cp149-Y132A (yielding an oxidized/reduced protein ratio of approximately 1:1) was included in coassembly reactions with 5 μM Cp149-C61S, results indicate that the oxidized mutant participated in assembly but did not appreciably change the rate of nucleating capsid formation. We observed that the yield of capsid increased, indicating coassembly. The rate of capsid formation at later times, which is proportional to the nucleation rate (19), did not change compared to the rate of the control reaction, suggesting that either the Y132A mutant does not participate in nucleation or its effect on the nucleation rate is exactly balanced by its concentration (7). However, when reduced Cp149-Y132 was used in coassembly reactions, there was a dramatic decrease in the amount of capsid produced over a 24-h period. In the first hour after the initiation of assembly, the oxidized-form and control reactions were >50% complete, while no assembly was detected with reduced Cp149-Y132A (Fig. 5b). When this reaction was repeated at 37°C, the coassembly reaction, while still slowed, had a much greater yield of capsid; it appeared to be approaching the same final extent of assembly as the reaction with Cp149-C61S alone (see Fig. S4 in the supplemental material). This suggests that reduced Cp149-Y132A actually inhibits nucleation. If the mutant simply did not participate in assembly, neither the rate of capsid formation nor the yield of capsid would have changed.
DISCUSSION
The structural differences between the free dimer and dimer from capsid strongly suggest a correlated movement of subdomains, consistent with the allostery hypothesis. Movement of the spike tips from the dimer to capsid conformations shifts the positions of adjacent fulcrum helices, which in turn shifts the intersubunit contact domain. Conversely, removing structural constraints at the intersubunit contact domains, such as by release of a dimer from a capsid, will shift the fulcrum helices and allow the spike tip to relax. These movements could be further amplified by changes in the chassis subdomain related to disulfide reduction. This correlated movement is consistent with the propagated change induced by a capsid-binding peptide examined in detail previously by NMR (20) and with the cooperative binding of zinc ions to free dimer observed previously using intrinsic protein fluorescence (41). The absence of major refolding of the protein is in agreement with published circular dichroism and NMR data (20, 39).
Results from the NMR study of unassembled HBV dimer, equivalent to Cp149 (20), bear comparison to our structure. To maintain the dimeric state, the NMR sample was examined at pH 9.5, which prevented assembly but also raises barriers to a more complete analysis. As we did with our crystal structure, the researchers in the NMR study observed essentially the same secondary structure as in the capsid state. Unlike our near-neutral-pH asymmetric dimer, their pH 9.5 structure appears to be symmetrical based on the overlap of transverse relaxation optimized spectroscopy (TROSY) cross-peaks. In the NMR paper, the authors also observed that a peptide, which binds at the spike tip (3), bound with a stoichiometry of one molecule per Cp dimer and induced conformational change and asymmetry, indicated by shifts, splitting, or the disappearance of peaks corresponding to about 30% of the protein. Most of the residues involved were within the upper half of the spike or close to hinges in the spike and fulcrum subdomains, areas predicted from our structure to have the greatest opportunities for correlated conformational response to outside stimulus.
The differences between the free dimer structure we present in this report and the symmetrical dimer from capsid are not artifacts of the Y132A mutation or of packing contacts. The mutation is located in the contact subdomain, a helix-turn-extended structure distant from the spike or fulcrum helix. Though the contact subdomains are in different positions with respect to the chassis, all six crystallographically independent examples (A through F) presented in this paper are essentially the same as in the capsid structure. The Y132A mutation does not alter local tertiary structure. Similarly, crystal packing contacts cannot be blamed for the gross structural differences between free and capsid conformations of dimer. The three Y132A dimers have similar asymmetric conformations (Fig. 2a). The asymmetry is most obvious for the helices that form the spike at the dimer interface (Fig. 2a and d). Though the asymmetry patterns are approximately the same, the packing environments of the three dimers are crystallographically independent of one another. Thus, the asymmetric dimers with mobile subdomains, presented here, are an accurate representation of at least some of the conformations available to free dimer.
The Cp149-Y132A free dimer structure and the apparent allosteric nature of the protein shed light on the basis of the antiviral mechanism of HAPs. HAPs accelerate assembly, strengthen protein-protein interactions, and lead to assembly of aberrant particles (40). In a capsid-HAP complex crystal structure, HAP fills a pocket at the interface between two dimers to improve packing at the cost of slightly distorting the geometry of the contact (5, 6). In free dimer, the HAP-binding site is framed by residues from the static chassis and two mobile subdomains: the fulcrum and the interdimer contact (Fig. 3). Chassis residues that contribute to the HAP site are mainly from the base of the helix bundle (residues 29 to 34 and 101 to 110). The mobile components of the HAP site are the fulcrum residues 23 to 25, including hinge Pro25 and contact subdomain residues 114 to 118 and 138 to 140. This model for the HAP site suggests that HAPs noncovalently cross-link subdomains in the assembly-active conformation, jump-starting assembly nucleation. Thus, HAPs are able to circumvent a major barrier to initiating assembly.
We tested coassembly of reduced and oxidized Cp149-Y132A with a wild type-like mutant and observed effects related to the disulfide within the chassis region. Oxidized Cp149-Y132A coassembled with Cp149-C61S. Pure reduced Cp149-Y132A inhibited capsid formation, acting as a sort of competitive inhibitor of assembly. The presence of the disulfide and the presumed restriction of chassis dynamics increase the association of Cp149-Y132A. Conversely, the reduced form of the protein seems to poison the assembly. The lack of the disulfide may favor the inactive state, allow unconstrained motion of the chassis, or allow a different conformation of the chassis domain from that observed in the structure. This indication that assembly is affected by the dynamics of free dimer (within the chassis) or structural change within the dimer suggests a direction for further structural and analytical studies. Thus, for reduced dimer, the likely cellular form of Cp, allosteric activation of assembly is far more critical for assembly.
Allostery provides a rationale for in vivo behavior. It also substantively alters quantitative models of assembly and disassembly. Simulations of assembly-disassembly reactions suggest that conformational change is imposed on HBV by induced fit. As in previous studies (27, 39, 54, 58), we simulated assembly as a bulk reaction where each capsid starts with a nucleation event followed by a cascade of second-order reactions adding one dimer at a time. We consider three scenarios for incorporating allostery: (i) sudden activation of all subunits, (ii) an equilibrium where only a small fraction of a subunit is in the assembly-active state, and (iii) induced fit, where activated subunits are required for nucleation but inactive subunits can add to a growing polymer. The first two alternatives simply do not work. If dimer conformational changes were induced all at one time, the high association energy would trump nucleation, resulting in a kinetic trap with many partially assembled fragments; such kinetic traps have been observed when assembly was “overnucleated” by Zn2+ (41). If assembly were based on equilibrium between active and inactive Cp, the low concentration of reactant combined with the strong association energy necessary to drive assembly at low concentrations would create a reaction starved for intermediates that stalled far short of equilibrium—as illustrated in kinetic simulations (19) and line tension arguments (8).
In contrast, induced fit provides a structural and energetic basis for a nucleation step to generate the initial template for assembly. In this scenario, the kinetic basis for nucleation is the low probability of collision between assembly-active molecules. Elongation based on induced fit, a process also termed autostery (10), allows the common assembly-inactive state to participate in assembly. Such collisions are likely and fast (diffusion limited). Induced fit indicates that the assembly-inactive subunit in this complex undergoes a very fast intramolecular structural transition to the assembly-active state, which results in a more stable complex. Because the induced-fit effect does not affect the collision rate, simulations show a typical, efficient, nucleated assembly reaction (Fig. 6a) (19, 53, 58). Induced fit is consistent with the weak net association energy observed experimentally (12): the sum of the strong association energy of the assembly-active state and the energetic cost of the conformational change.
FIG. 6.

Structural differences support allostery and an induced-fit model of assembly that causes dramatic hysteretic stabilization of capsids. (a) In assembly, the limiting step is the kinetically controlled association. The subsequent allosteric change cannot be discerned. Thus, assembly kinetics are the same for simple association and induced fit. In disassembly, the limiting step is the breaking of the strong interactions between the complex (the capsid or intermediate) and the assembly-active Cp. Assuming that the on rates for active and inactive states are the same, the difference in the association constant will be reflected in a proportionately lower off rate. (b) Simulations of disassembly for induced-fit and nonallosteric reactions where both have net ΔGcontact values of −3.1 kcal/mol. Dissociation in the induced-fit simulation over a 160,000-s period was not detectable in this graphic representation. Simulations were calculated for a 30-subunit capsid using master equations (19, 55).
Though induced fit shows no discernible effect on assembly kinetics, there is a dramatic effect in kinetic simulations of capsid disassembly (Fig. 6b). The difference between the association energy of the activated state and net association energy contributes to the hysteresis in response to dissociation, where hysteresis is markedly enhanced stability of capsids in resistance to dissociation compared to the stability expected based on association energy and kinetics (23, 39). In the disassembly of induced-fit capsids, the release of subunits requires breaking of the strong interactions of the assembly-active state. The overall effect is that the model (and any other virus that incorporates an induced-fit mechanism) will demonstrate remarkable stability and persistence while between hosts (Fig. 6b). Such capsids can persist as metastable structures that are primed for dissociation in response to an appropriate stimulus such as pH or a receptor.
The hinged movement of the subdomains and the structural differences between the three Cp149-Y132A dimers in the crystallographic asymmetric unit and a dimer from capsid strongly suggest that the actual protein is dynamic. The relatively unconstrained reduced free dimer is likely to have greater dynamic freedom to search for the biologically relevant binding partners that may trigger the allosteric change that initiates assembly. We propose that allosteric regulation of assembly and induced fit, with their biological, kinetic, and energetic implications, are a common mechanism of ensuring the specificity and fidelity of capsid assembly in viruses. The allosteric effector that controls HBV assembly in vivo is probably related to the complex of viral RNA and reverse transcriptase. The allostery hypothesis suggests that most self-assembling viruses will have natural allosteric effectors that ought to be identified. The HAP molecules are artificial allosteric effectors of HBV assembly. Functionally similar effectors are likely to have antiviral effects on any self-assembling virus that incorporates an allosteric mechanism of regulation.
Supplementary Material
Acknowledgments
We acknowledge help from present and former members of the Zlotnick lab, including Reeha Dhasan, Jennifer M. Johnson, and Matthew R. Fultz. Data collection took place at the Indiana University Macromolecular Crystallography Lab with trials at Argonne National Lab's Advanced Photon Source beamline 14-BMC and the Berkeley Advanced Light Source beamline 4.2.2 (associated with the Molecular Biology Consortium).
Footnotes
Published ahead of print on 25 November 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Belnap, D. M., N. R. Watts, J. F. Conway, N. Cheng, S. J. Stahl, P. T. Wingfield, and A. C. Steven. 2003. Diversity of core antigen epitopes of hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 100:10884-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birnbaum, F., and M. Nassal. 1990. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol. 64:3319-3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottcher, B., N. Tsuji, H. Takahashi, M. R. Dyson, S. Zhao, R. A. Crowther, and K. Murray. 1998. Peptides that block hepatitis B virus assembly: analysis by cryomicroscopy, mutagenesis and transfection. EMBO J. 17:6839-6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottcher, B., M. Vogel, M. Ploss, and M. Nassal. 2006. High plasticity of the hepatitis B virus capsid revealed by conformational stress. J. Mol. Biol. 356:812-822. [DOI] [PubMed] [Google Scholar]
- 5.Bourne, C., M. G. Finn, and A. Zlotnick. 2006. Global structural changes in hepatitis B capsids induced by the assembly effector HAP1. J. Virol. 80:11055-11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourne, C., S. Lee, B. Venkataiah, A. Lee, B. Korba, M. G. Finn, and A. Zlotnick. 2008. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 82:10262-10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourne, C. R., S. P. Katen, M. R. Fulz, C. Packianathan, and A. Zlotnick. 2009. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry 48:1736-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruinsma, R. F., W. M. Gelbart, D. Reguera, J. Rudnick, and R. Zandi. 2003. Viral self-assembly as a thermodynamic process. Phys. Rev. Lett. 90:248101. [DOI] [PubMed] [Google Scholar]
- 9.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. Delano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, N. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905-921. [DOI] [PubMed] [Google Scholar]
- 10.Caspar, D. L. 1980. Movement and self-control in protein assemblies. Quasi-equivalence revisited. Biophys. J. 32:103-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceres, P., S. J. Stray, and A. Zlotnick. 2004. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J. Virol. 78:9538-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceres, P., and A. Zlotnick. 2002. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 41:11525-11531. [DOI] [PubMed] [Google Scholar]
- 13.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760-763. [DOI] [PubMed] [Google Scholar]
- 14.Conway, J. F., N. Cheng, A. Zlotnick, S. J. Stahl, P. T. Wingfield, D. M. Belnap, U. Kanngiesser, M. Noah, and A. C. Steven. 1998. Hepatitis B virus capsid: localization of the putative immunodominant loop (residues 78 to 83) on the capsid surface, and implications for the distinction between c and e-antigens. J. Mol. Biol. 279:1111-1121. [DOI] [PubMed] [Google Scholar]
- 15.Crowther, R. A., N. A. Kiselev, B. Bottcher, J. A. Berriman, G. P. Borisova, V. Ose, and P. Pumpens. 1994. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 77:943-950. [DOI] [PubMed] [Google Scholar]
- 16.Datta, S. A., J. E. Curtis, W. Ratcliff, P. K. Clark, R. M. Crist, J. Lebowitz, S. Krueger, and A. Rein. 2007. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 365:812-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datta, S. A., Z. Zhao, P. K. Clark, S. Tarasov, J. N. Alexandratos, S. J. Campbell, M. Kvaratskhelia, J. Lebowitz, and A. Rein. 2007. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J. Mol. Biol. 365:799-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 19.Endres, D., and A. Zlotnick. 2002. Model-based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys. J. 83:1217-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freund, S. M. V., C. M. Johnson, A. M. Jaulent, and N. Ferguson. 2008. Moving towards high-resolution descriptions of the molecular interactions and structural rearrangements of the human hepatitis B core protein. J. Mol. Biol. 384:1301-1313. [DOI] [PubMed] [Google Scholar]
- 21.Ganem, D., and A. M. Prince. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118-1129. [DOI] [PubMed] [Google Scholar]
- 22.Gunther, S., L. Fischer, I. Pult, M. Sterneck, and H. Will. 1999. Naturally occurring variants of hepatitis B virus. Adv. Virus Res. 52:25-137. [DOI] [PubMed] [Google Scholar]
- 23.Hagan, M. F., and D. Chandler. 2006. Dynamic pathways for viral capsid assembly. Biophys. J. 91:42-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris, A., D. M. Belnap, N. R. Watts, J. F. Conway, N. Cheng, S. J. Stahl, J. G. Vethanayagam, P. T. Wingfield, and A. C. Steven. 2006. Epitope diversity of hepatitis B virus capsids: quasi-equivalent variations in spike epitopes and binding of different antibodies to the same epitope. J. Mol. Biol. 355:562-576. [DOI] [PubMed] [Google Scholar]
- 25.Johnson, J. M., J. Tang, Y. Nyame, D. Willits, M. J. Young, and A. Zlotnick. 2005. Regulating self-assembly of spherical oligomers. Nano Lett. 5:765-770. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, M. C., H. M. Scobie, Y. M. Ma, and V. M. Vogt. 2002. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J. Virol. 76:11177-11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katen, S. P., and A. Zlotnick. 2009. Thermodynamics of virus capsid assembly. Methods Enzymol. 455:395-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koschel, M., D. Oed, T. Gerelsaikhan, R. Thomssen, and V. Bruss. 2000. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J. Virol. 74:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanman, J., T. T. Lam, M. R. Emmett, A. G. Marshall, M. Sakalian, and P. E. Prevelige, Jr. 2004. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11:676-677. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 31.Ma, Y. M., and V. M. Vogt. 2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J. Virol. 76:5452-5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy, A. J. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63:32-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240-255. [DOI] [PubMed] [Google Scholar]
- 34.Nassal, M. 1992. Conserved cysteines of the hepatitis B virus core protein are not required for assembly of replication-competent core particles nor for their envelopment. Virology 190:499-505. [DOI] [PubMed] [Google Scholar]
- 35.Nassal, M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235-249. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 37.Salfeld, J., E. Pfaff, M. Noah, and H. Schaller. 1989. Antigenic determinants and functional domains in core antigen and e antigen from hepatitis B virus. J. Virol. 63:798-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seeger, C., F. Zoulim, and W. S. Mason. 2007. Hepadnaviruses, p. 2977-3029. In D. M. Knipe, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 39.Singh, S., and A. Zlotnick. 2003. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J. Biol. Chem. 278:18249-18255. [DOI] [PubMed] [Google Scholar]
- 40.Stray, S. J., C. R. Bourne, S. Punna, W. G. Lewis, M. G. Finn, and A. Zlotnick. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. U. S. A. 102:8138-8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stray, S. J., P. Ceres, and A. Zlotnick. 2004. Zinc ions trigger conformational change and oligomerization of hepatitis B virus capsid protein. Biochemistry 43:9989-9998. [DOI] [PubMed] [Google Scholar]
- 42.Stray, S. J., J. M. Johnson, B. G. Kopek, and A. Zlotnick. 2006. An in vitro fluorescence screen to identify antivirals that disrupt hepatitis B virus capsid assembly. Nat. Biotechnol. 24:358-362. [DOI] [PubMed] [Google Scholar]
- 43.Virgin, H. W., E. J. Wherry, and R. Ahmed. 2009. Redefining chronic viral infection. Cell 138:30-50. [DOI] [PubMed] [Google Scholar]
- 44.WHO. 2004. WHO position on the use of hepatitis B vaccines. Wkly. Epidemiol. Rec. 28:255-263. [PubMed] [Google Scholar]
- 45.Wingfield, P. T., S. J. Stahl, J. Kaufman, A. Zlotnick, C. C. Hyde, A. M. Gronenborn, and G. M. Clore. 1997. The extracellular domain of immunodeficiency virus gp41 protein: expression in Escherichia coli, purification, and crystallization. Protein Sci. 6:1653-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wingfield, P. T., S. J. Stahl, R. W. Williams, and A. C. Steven. 1995. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 34:4919-4932. [DOI] [PubMed] [Google Scholar]
- 47.Winn, M. D., G. N. Murshudov, and M. Z. Papiz. 2003. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374:300-321. [DOI] [PubMed] [Google Scholar]
- 48.Wynne, S. A., R. A. Crowther, and A. G. Leslie. 1999. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 3:771-780. [DOI] [PubMed] [Google Scholar]
- 49.Yeh, C. T., Y. F. Liaw, and J. H. Ou. 1990. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J. Virol. 64:6141-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan, T. T., M. H. Lin, S. M. Qiu, and C. Shih. 1998. Functional characterization of naturally occurring variants of human hepatitis B virus containing the core internal deletion mutation. J. Virol. 72:2168-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan, T. T., G. K. Sahu, W. E. Whitehead, R. Greenberg, and C. Shih. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 73:5731-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou, S., and D. N. Standring. 1992. Cys residues of the hepatitis B virus capsid protein are not essential for the assembly of viral core particles but can influence their stability. J. Virol. 66:5393-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zlotnick, A. 2007. Distinguishing reversible from irreversible virus capsid assembly. J. Mol. Biol. 366:14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zlotnick, A. 2005. Theoretical aspects of virus capsid assembly. J. Mol. Recognit. 18:479-490. [DOI] [PubMed] [Google Scholar]
- 55.Zlotnick, A. 1994. To build a virus capsid. An equilibrium model of the self assembly of polyhedral protein complexes. J. Mol. Biol. 241:59-67. [DOI] [PubMed] [Google Scholar]
- 56.Zlotnick, A., R. Aldrich, J. M. Johnson, P. Ceres, and M. J. Young. 2000. Mechanism of capsid assembly for an icosahedral plant virus. Virology 277:450-456. [DOI] [PubMed] [Google Scholar]
- 57.Zlotnick, A., N. Cheng, S. J. Stahl, J. F. Conway, A. C. Steven, and P. T. Wingfield. 1997. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc. Natl. Acad. Sci. U. S. A. 94:9556-9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zlotnick, A., J. M. Johnson, P. W. Wingfield, S. J. Stahl, and D. Endres. 1999. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 38:14644-14652. [DOI] [PubMed] [Google Scholar]
- 59.Zlotnick, A., A. Lee, C. R. Bourne, J. M. Johnson, P. L. Domanico, and S. J. Stray. 2007. In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions). Nat. Protoc. 2:490-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.