

Abstract
In this study a number of herpes simplex virus type 1 (HSV-1) proteins were screened, using a yeast-two-hybrid assay, for interaction with the tegument protein pUL48 (VP16). This approach identified interactions between pUL48 and the capsid proteins pUL19 (VP5), pUL38 (VP19C), and pUL35 (VP26). In addition, the previously identified interaction of pUL48 with the major tegument protein pUL36 (VP1/2) was confirmed. All of these interactions, except that of pUL35 and pUL48, could be confirmed using an in vitro pulldown assay. A subsequent pulldown assay with intact in vitro-assembled capsids, consisting of pUL19, pUL38, and pUL18 (VP23) with or without pUL35, showed no binding of pUL48, suggesting that the capsid/pUL48 interactions initially identified were more then likely not biologically relevant. This was confirmed by using a series of HSV-1 mutants lacking the gene encoding either pUL35, pUL36, or pUL37. For each HSV-1 mutant, analysis of purified deenveloped C capsids indicated that only in the absence of pUL36 was there a complete loss of capsid-bound pUL48, as well as pUL37. Collectively, this study shows for the first time that pUL36 is a major factor for addition of both pUL48 and pUL37, likely through a direct interaction of both with nonoverlapping sites in pUL36, to unenveloped C capsids during assembly of HSV-1.
The herpes simplex virion has four components: an electron-dense core containing the double-stranded DNA genome (152 kb), the capsid, the tegument, and an outer envelope containing glycoprotein (55). The most widely accepted pathway for herpes viral assembly and egress involves a process of envelopment, deenvelopment, and reenvelopment (39). After assembly in the nucleus, the mature herpes simplex virus type 1 (HSV-1) nucleocapsid or C capsid (capsid-containing DNA genome) undergoes primary envelopment through the inner nuclear membrane into the perinuclear space. The enveloped particles are deenveloped at the outer nuclear membrane. Acquisition of inner tegument onto the capsid precedes secondary or reenvelopment, which involves acquisition of outer tegument and glycoprotein envelope in or close to the Golgi. Fully assembled virions are finally released by exocytosis.
The HSV-1 protein pUL48 (VP16, α-transinducing factor [TIF], or Vmw65), which is a major component of the virion tegument (1,000 to 1,500 copies per virion), has a number of functions (29). First, it promotes the transcription of immediate-early (IE) viral genes, including infected cell protein 0 (ICP0), ICP4, and ICP22 and ICP27, through formation of a transcription complex with two cellular proteins, Oct-1 and HCF (21, 26, 59, 69). This complex forms on regulatory motifs upstream of each IE viral gene. For HSV-1 pUL48, the transactivation activity depends on a highly acidic C-terminal 80-amino-acid transcriptional activation domain (9). Recently, de novo synthesis of HSV-1 pUL48 in neurons has also been shown to coordinate the expression of viral IE genes, with resulting reactivation from latency (61). Previous studies on HSV-1 have suggested that both the tegument proteins pUL46 (VP11/12) and pUL47 (VP13/14) also have a role in enhancing pUL48-induced IE viral gene expression possibly through a direct interaction (65, 70, 71). Second, pUL48 has a structural role during assembly. Deletion of the UL48 gene in HSV-1 and the related Alphaherpesvirinae subfamily members equine herpesvirus 1 (EHV-1) and pseudorabies virus (PrV) blocks secondary envelopment in the cytoplasm (19, 44, 66). Interaction between the capsid and capsidless tegument structures appears to be essential to drive secondary envelopment of the virus. The role of pUL48 appears to be in linking the capsid/inner tegument and outer tegument/glycoprotein complexes (19, 44, 65). Additional evidence for pUL48 in inner and outer tegument layers has come from detergent solubilization/salt extractions of purified HSV-1 virions (45, 56, 70). The documented interactions of pUL48 with other herpes viral structural proteins include interactions with the tegument proteins pUL49 (VP22) (16, 65), pUL36 (VP1/2) (65), pUL41 (58), pUL46 (28, 65), and pUL3.5 (18) and interactions with the viral glycoproteins gH (24), gB, and gD (73).
HSV-1 pUL48 might also have an additional role during assembly in the nucleus. This is based on a report that it appears, like the tegument proteins pUS3 (23, 53), pUL11 (3), and pUL49 (50), to be a component of primary enveloped virions (46). This contrasts with PrV, where pUL48 has been shown not to be a component of primary virions (19). The distribution of HSV-1 pUL48 in the nucleus has been shown to partially overlap with the capsid protein pUL38 (VP19C) in structures termed assemblons, which appear to be sites of immature capsid assembly (67). Furthermore, HSV-1 intranuclear capsids were shown to label for pUL48 antigen in rat neurons (42). In addition, several other tegument proteins, including pUL41 (52), pUL36, and pUL37 (6) have also been shown to be associated with intranuclear HSV-1 B and/or C capsids. In contrast, two studies on HSV-1 (63, 68) and one study on PrV (43) indicate pUL36 is not associated with intranuclear capsids. The presence of pUL36, and other tegument proteins, on intranuclear capsids and the possibility that HSV-1 and PrV differ in tegument acquisition on capsids remains an area of contention.
The present study set out to define determinants for the addition of pUL48 to HSV-1 capsids during the course of viral assembly. A combination of yeast two-hybrid and in vitro pulldown assays identified and confirmed a number of interactions between pUL48 and both capsid proteins and the tegument protein pUL36. Further analysis of in vitro-assembled capsids and capsids from cells infected with HSV-1 lacking pUL35 (VP26), pUL36 or pUL37 highlighted the specific requirement for pUL36 in the addition of pUL48 to C capsids.
MATERIALS AND METHODS
Expression constructs.
The constructs displayBait pUL6, pUL11, pUL18, pUL19(VP5)UD (upper domain; amino acids 451 to 1054), pUL35, pUL36N (amino acids 1 to 767), pUL38, and pUL38N (amino acids 1 to 90) and displayTarget pUS10, pUL16, and pUL48N (amino acids 1 to 411) have been previously described (15, 33, 65). The expression constructs pGEX-5X-1 DYNLT3 (dynein light chain rp3) and pCMV-myc kinesinC (kinesin-1 heavy chain amino acids 814 to 963) have been previously described (15, 65). The pUL19UD gateway entry clone (33) was used to introduce the pUL19UD into pCMV-myc (Clontech) that had been made compatible with the gateway system using a Gateway vector conversion system (Invitrogen). DNA fragments encoding pUL35, pUL38, or pUL36N were removed from displayBait by an EcoRI/XhoI or an EcoRI digest and inserted into EcoRI/XhoI- or EcoRI-digested pCMV-myc (vector modified as described in reference 65). A PCR fragment corresponding to pUL38C (amino acids 91 to 465) was generated by using oligonucleotide primers that incorporated EcoRI in the forward primer and XhoI along with a stop codon in the reverse primer for directional cloning into pCMV-myc. A DNA fragment which encodes pUL48N was removed from displayTarget by an EcoRI/XhoI digest and inserted into EcoRI/XhoI-digested pGEX-5X-1 (Amersham). Genes or fragments thereof were amplified by PCR using a Gene-Amp XL PCR kit (Applied Biosystems). All constructs were sequenced to confirm gene sequence and correct reading frame.
Viruses and cells.
HSV-1 parental strain 17 and HSV-1 containing single deletions of genes encoding pUL35 (dmVP26-minus), pUL36 (ARΔUL36), or pUL37 (FRΔUL37) in a strain 17 background were kindly provided by Frazer Rixon (MRC Virology Unit, Glasgow, United Kingdom) (7, 54). Complementing stable cell lines HAUL36-1 and 80C02, expressing HSV-1 pUL36 or pUL37, respectively, for propagation of pUL36 and pUL37 deletion viruses were also kindly provided by Frazer Rixon (54).
The cell lines HeLa and Vero, along with HAUL36-1 and 80C02, were maintained at 37°C (5% CO2) in T-75 Falcon flasks (BD Biosciences) in 20 ml of maintenance medium consisting of Dulbecco modified Eagle Medium (DMEM; Invitrogen) supplemented with 9% (vol/vol) fetal calf serum (FCS; JRH Biosciences), 100 U of penicillin/ml, and 100 μl of streptomycin/ml (Sigma). For the HAUL36-1 and 80C02 cell lines, the medium was supplemented with 1% (vol/vol) nonessential amino acids (NEAA; Invitrogen) and 1% (vol/vol) G418 (Invitrogen).
Vero or complementing cells were infected at a multiplicity of infection (MOI) of 0.01 PFU/ml with a low passage of HSV-1 strain 17, dmVP26-minus, ARΔUL36, or FRΔUL37 in DMEM supplemented with 1% FCS (plus 1% NEAA and 0.5 mg of G418/ml for HAUL36-1 and 80C02). Virus inocula were added to cell monolayers, and cells were incubated at 37°C for 1 h to allow binding of the virus, before additional medium was added. Cells were incubated at 37°C until 80% displayed cytopathic effect (48 to 72 h). The cells were then frozen and thawed three times and subjected to sonication (three times for 20 s each time), and cellular debris was removed by centrifugation at 800 × g for 20 min at 4°C. The supernatant was filtered through a disposable 0.22-μm-pore-size Millex-GP filter (Millipore), divided into 1-ml aliquots, and stored at −80°C.
Virus titers were determined by plaque assay. For HSV-1 strain 17 and dmVP26-minus, plaque assays were performed with Vero cells. For ARΔUL36 or FRΔUL37, plaque assays were performed with complementing cell lines HAUL36-1 and 80C02. Respective cell lines were grown to confluence in 24-well Falcon tissue culture plates in DMEM supplemented with 9% FCS, plus 1% NEAA and 0.5 mg of G418/ml when we used HAUL36-1 and 80C02 cell lines. Serial dilutions were prepared in DMEM with 1% FCS. Cells were infected and incubated at 37°C for 2 h. Virus was then removed, and cells were overlaid with 1 ml per well of DMEM supplemented with 1% FCS and 1.6% (wt/vol) carboxymethyl cellulose (Sigma), followed by incubation at 37°C. At 48 h postinfection, the medium was removed, and the cells were fixed with 100% methanol and stained with 0.1% (wt/vol) crystal violet dissolved in 20% (vol/vol) ethanol to allow visualization of the plaques.
Yeast two-hybrid assay.
The use of the LexA-based yeast two-hybrid assay to determine protein-protein interactions has been previously described (65). The protocols for qualitative assessment of protein-protein interactions, quantification of each positive interaction using a liquid β-galactosidase assay, and determination of protein expression in yeast were as previously described (65).
Transfection of HeLa cells.
Recombinant myc-tagged proteins were expressed in HeLa cells in six-well plates as previously described (65) with the exception that the transfection reagent was FuGENE-6 (Roche). Lysates were harvested 48 h posttransfection in 200 μl of lysis buffer that consisted of phosphate-buffered saline (PBS) containing 0.1% (vol/vol) Triton X-100 and mammalian protease inhibitors (Sigma).
In vitro-assembled capsids.
Prior to binding assays in vitro-assembled capsids, isolated from recombinant baculovirus-infected insect cells, with or without pUL35 (60) (kindly provided by Frazer Rixon) were diluted to 0.01 mg of total protein/ml in PBS containing 0.1% (vol/vol) Triton X-100.
In vitro pulldown assay.
Glutathione S-transferase (GST)-tagged pUL48N was expressed and harvested as previously described (15) with the exception that induction was at 30°C for 3 h. Conditions for binding and washing were as previously described (15), except that 5 mM dithiothreitol was omitted from all buffers and 100 μl of GST-bind beads (Novagen) was used. Immobilized GST or GST pUL48N was then incubated with rocking at 4°C for 3 h with transfected HeLa cell lysates or in vitro-assembled capsids. Beads were pelleted by spinning at 1,000 × g for 5 min and then washed, as previously described (15), to remove unbound proteins. Bound protein complexes were then eluted by heating in 50 μl of reducing Laemmli buffer for 5 min at 96°C.
Purification of C capsids.
Vero cells were grown to a confluence of 80% in 10x T-150 Falcon flasks before infection. Virus was added to each flask in 15 ml of DMEM with 1% (vol/vol) FCS. Cells were infected with HSV-1 parental strain 17 or dmVP26-minus at an MOI of 1 PFU/ml and harvested 48 h postinfection or infected with ARΔUL36 or FRΔUL37 at an MOI of 10 PFU/ml and harvested 24 h postinfection. Capsids were purified as previously described (62) but with some modifications. Briefly, cells were harvesting by scraping and then spun down at 800 × g for 10 min at 4°C. Cell pellets were resuspended in 2× capsid lysis buffer (40 mM Tris-HCl [pH 7.4], 1 M NaCl, 2 mM EDTA, 2% [vol/vol] Triton X-100, and mammalian protease inhibitors). Cell lysates were freeze-thawed three times and sonicated twice for 20 s. Cell debris was then removed by centrifugation at 10,000 × g for 20 min at 4°C. Supernatant containing capsids was then layered over a 20 to 65% (wt/vol) continuous sucrose gradient in TNE (20 mM Tris-HCl [pH 7.6], 500 mM NaCl, 1 mM EDTA). Capsids were isolated by spinning at 24,000 × g for 60 min at 4°C.
Three bands were present in the gradient corresponding to A, B, and C capsids, respectively. The faster sedimenting viral genomic DNA-containing C capsid band was then harvested from the gradient by using a syringe and then further purified by spinning at 24,000 × g for 60 min at 4°C through a 35% (wt/vol) sucrose cushion in PBS. C capsids were resuspended in 1 ml of capsid storage buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% [vol/vol] Triton X-100, and mammalian protease inhibitors) overnight at 4°C. For long-term storage capsids were stored at −80°C.
Amplification of viral genomic DNA from C capsids.
Purified C capsids (50 μl) were lysed in 10 mM Tris-HCl buffer (pH 7.5), 2.5% (wt/vol) sodium dodecyl sulfate (SDS), 15 mM EDTA, and 500 mM NaCl at 37°C for 5 min. Viral genomic DNA was extracted twice with phenol-chloroform and then ethanol precipitated. The DNA pellet was resuspended in 20 μl of water supplemented with 50 μg of RNase A (Sigma)/ml. A set of previously described primers (33) was then used for PCR amplification of the HSV-1 viral gene US10 using Phusion High Fidelity Polymerase (Finnzymes) and the isolated viral genomic DNA as a template.
Analysis of protein complexes.
Proteins were separated by SDS-PAGE and, for analysis of expression in yeast or for subsequent binding experiments, immunoblots were processed as previously described (65). Detection was performed with an Odyssey Infra-Red imaging system (LI-COR Biosciences).
Antibodies.
The following antibodies were used for immunoblots. The primary antibodies to detect expression of yeast two-hybrid displayBait fusion and displayTarget fusion, respectively, included mouse monoclonal antibodies against LexA and hemagglutinin (HA) (both from Santa Cruz). Monoclonal antibody against c-myc (Santa Cruz) was used for detection of myc-tagged fusion proteins. The primary antibodies to HSV-1 proteins included mouse monoclonal against pUL19 (1:3,500 dilution; kindly provided by Frazer Rixon) (37), and rabbit polyclonal antibodies against pUL37 (1:10,000 dilution; kindly provided by Thomas Mettenleiter, Institute of Molecular Biology, Friedrich-Loeffler-Institut, Greifswald-Insel Riems) (34), pUL48 (1:3,000 dilution; Abcam), and gD (1:5,000 dilution; Abcam). An additional rabbit polyclonal antibody raised against purified HSV-1 nuclear C capsids (PTNC) was used to detect tegument protein pUL36, as well as the capsid proteins pUL18 (VP23) and pUL35 (1:5,000 dilution; kindly provided by Frazer Rixon) (51). Secondary antibodies for detection using the Odyssey system included goat anti-mouse IRDye 680-conjugated IgG (1:5,000 dilution; LI-COR) and goat anti-rabbit IRDye 800-conjugated IgG (1:5,000 dilution; LI-COR).
RESULTS
Yeast two-hybrid assay to identify pUL48 binding proteins.
Initially, a fragment of HSV-1 pUL48 corresponding to amino acids 1 to 411 (which lacked the C-terminal activation domain and was previously referred to as pUL48-AD [33] but is referred to here as pUL48N) was screened by using the yeast two-hybrid assay against HSV-1 outer capsid proteins and the HSV-1 major tegument protein pUL36 (VP1/2). Amino acids 1 to 411 contain the core of pUL48, whose structure has been identified as containing regions involved in both virion assembly and DNA-induced complex formation with host cell proteins HCF and Oct-1 (35). The outer HSV-1 capsid proteins tested included pUL6, pUL18 (VP23), pUL19 (VP5), pUL35 (VP26), and pUL38 (VP19C) (27). In some cases, only protein fragments were tested. In particular, the pUL19 upper domain (UD) (amino acids 451 to 1054), whose structure has been previously determined, was tested since it has been shown to be the most accessible for interaction with other proteins, including pUL35 and tegument proteins (4, 5, 72). The fragment corresponding to amino acids 1 to 90 of pUL38 was chosen as both full-length and fragment 91-465 autoactivate as Bait in the yeast two-hybrid assay (15). Furthermore, this region of pUL38 is not directly involved in capsid triplex formation with pUL18 and is likely to be accessible for interaction with other proteins (1, 49). Screening with the outer capsid protein pUL25 in Bait or in the reverse orientation with pUL48N in Bait was not possible due to autoactivation (15, 65).
Screening of Target pUL48N against various Bait capsid constructs identified a number of positive interactions including with pUL36N (amino acids 1 to 767), pUL19UD, pUL38N (amino acids 1 to 90), and pUL35 (Fig. 1A). No interaction was observed with pUL18 or pUL6 (Fig. 1A). A positive control consisted of the previously reported interaction of pUL11 and pUL16 (65). These interactions, with the exception of pUL38, were previously reported using yeast two-hybrid screens from our laboratory with the distinction being that the previous screens were conducted with pUL48 full-length as a target (33, 65) rather then with pUL48N as in the present study. Expression of pUL11, pUL19UD, and pUL38N in Bait (Fig. 1B), along with pUL16 and pUL48N in Target (Fig. 1C), was also confirmed. The expression of Bait pUL6, pUL18, pUL35, and pUL36N in yeast has been previously described (15, 41).
FIG. 1.

Yeast two-hybrid analysis of the interactions of HSV-1 tegument protein pUL48. (A) Target pUL48N (amino acids 1 to 411) was screened against a number of Bait fusion constructs using the LexA yeast two-hybrid assay. Summary of activity values obtained from a liquid β-galactosidase assay. The activity was calculated from the following equation: β-galactosidase activity = 1,000 × A420/(t × V × OD660), where t = time (in minutes) of incubation, V is the volume of cells (ml) used in the assay, and OD600 is the optical density at 600 nm. The given activity values are the averages of measurements from at least three separate colonies. Positive control corresponds to Bait pUL11 plus Target pUL16. (B) Immunoblot with monoclonal anti-LexA showing expression of Bait fusion constructs in yeast. (C) Immunoblot with monoclonal anti-HA showing expression of Target fusion constructs in yeast. For both panels B and C, samples (from left to right) consisted of yeast cotransformed with the combination of Bait pUL11/Target pUL16, Bait pUL19UD/Target pUL48N, or Bait pUL38N/Target pUL48N. For determination of protein expression, samples were subjected to protein extraction before separation by SDS-PAGE (12%). The position of each fusion protein is indicated (*). pUL19UD (amino acids 451 to 1054), pUL36N (amino acids 1 to 767), and pUL38N (amino acids 1 to 90).
In vitro pulldown assay to confirm yeast two-hybrid analysis.
An in vitro pulldown assay was undertaken to confirm the pUL48 interactions identified by the yeast two-hybrid screen. This involved addition of HeLa cell lysates containing recombinantly expressed myc-tagged pUL19UD, pUL36N, pUL38, pUL38C (amino acids 91 to 465), or kinesinC (amino acids 814 to 963) to GST or GST-tagged pUL48N immobilized on beads. Beads were then washed, and bound complexes were eluted and analyzed by SDS-PAGE and Western blotting. Testing of myc-tagged pUL35 was not feasible due to low expression levels in HeLa cells (not shown). Binding of myc-tagged pUL19UD, pUL36N, and pUL38 to GST-tagged pUL48N was observed with no binding to GST only (Fig. 2A and C). The specificity of binding was demonstrated with no binding of myc-tagged pUL38C (Fig. 2A) or kinesinC (Fig. 2C) to GST-tagged pUL48N. This confirms the yeast two-hybrid observations for the binding of pUL19UD, pUL36N, and pUL38 to pUL48N (Fig. 1A). In addition, this indicates that the major binding site in pUL38 for pUL48 maps to the first 90 amino acids. The presence of comparable amounts of GST and GST-tagged pUL48N eluting from beads in each case was also demonstrated (Fig. 2B and D).
FIG. 2.

In vitro pulldown assay to assess the interaction between individual recombinant viral proteins and recombinant tegument protein pUL48. Lysates from HeLa cells expressing recombinant myc-tagged pUL19UD (amino acids 451 to 1054), pUL36N (amino acids 1 to 767), pUL38, pUL38C (amino acids 91 to 456), and kinesinC (amino acids 814 to 963) were added to GST or GST-tagged pUL48N (amino acids 1 to 411) immobilized on glutathione-Sepharose beads. Interacting complexes were subsequently eluted and separated by SDS-PAGE (12%). (A) Immunoblot with monoclonal anti-myc antibody shows the presence of myc-tagged pUL19UD and pUL38 coeluting with GST-tagged pUL48N. The presence of each expressed myc-tagged capsid protein in HeLa cell lysates was also confirmed. (B) Coomassie blue stain gel confirming presence of GST-tagged proteins eluted from glutathione-Sepharose beads. (C) Immunoblot with monoclonal anti-myc antibody shows the presence of myc-tagged pUL36N coeluting with GST-tagged pUL48N. The presence of each expressed myc-tagged protein in HeLa cell lysates was also confirmed. (D) Coomassie blue stain gel confirming presence of GST-tagged proteins eluted from glutathione-Sepharose beads.
Pulldown assay using in vitro-assembled capsids.
To establish whether pUL48 can bind to capsids in the absence of tegument proteins, an in vitro pulldown assay was used using in vitro-assembled capsids. These capsids consist of the outer capsid proteins pUL18, pUL19, and pUL38, as well as the scaffold proteins VP21, VP24, and VP22a, expressed and assembled in recombinant baculovirus-infected insect cells (60). These capsids resemble HSV-1 B capsids (contain scaffolding proteins but lack DNA) of HSV-1 (27, 60). In vitro-assembled capsids with or without the 12-kDa outer capsid protein pUL35, which is not required for capsid assembly (60, 62), showed no discernible binding to GST-tagged pUL48N (Fig. 3A). In comparison, the positive control consisting of GST-tagged DYNLT3 (rp3) showed specific binding to capsids containing pUL35 (Fig. 3A), as previously reported (15). Although not directly shown this also indicates that pUL35 was present in the pUL35 plus capsids. The presence of equivalent amounts of input capsid preparations was confirmed by both anti-pUL19 antibody (Fig. 3A) and Coomassie blue stain (Fig. 3B). The diffuse capsid input pUL19 bands observed in the upper panel reflects the fact that detection with antibody is saturating due to high input levels of pUL19, as confirmed by the Coomassie blue stain of pUL19 in the lower panel. The Coomassie blue stain also confirmed the presence of the major constituents pUL19 (149 kDa), pUL38 (50 kDa), pUL18 (34 kDa), and VP22a (cleaved form of scaffold protein) in the input in vitro-assembled capsids (Fig. 3B), as documented previously (60). The bands for pUL38, VP22a, and pUL18 are less intense then pUL19. The predicted copy number per HSV-1 B capsid is 960 for pUL19, 375 for pUL38, 1153 for VP22a, and 572 for pUL18 (27). The abundance of VP22a does decrease depending on storage of assembled capsids and a similar relative staining intensity of capsid constituents, as observed in the present study has been reported previously (48). In addition, the Coomassie blue stain confirmed the presence of GST-tagged proteins coeluting from beads (Fig. 3B). These results indicate that, in the contrast to the yeast two-hybrid and in vitro pulldown assays with individually expressed viral proteins, in the context of an intact capsid that neither pUL19, pUL38, or pUL35 are sufficient for binding of pUL48. Therefore, additional factors such as the presence of other capsid proteins, pUL6, pUL17, pUL25, or tegument, such as pUL36, is required for the addition of pUL48. Alternatively, given the capsids resemble B capsids a conformational change from B to C capsids, which contain viral genomic DNA but lack scaffolding proteins, may confer the ability of capsids to engage pUL48.
FIG. 3.
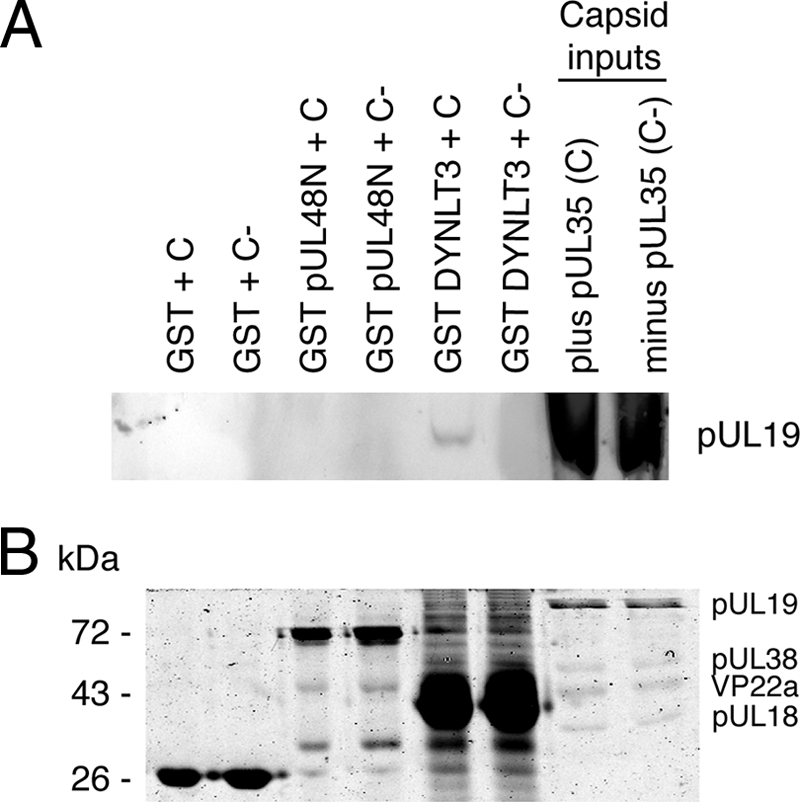
Pulldown assay to assess interaction of in vitro-assembled capsids with recombinant tegument protein pUL48. In vitro-assembled capsids were added to GST, GST-tagged pUL48N (amino acids 1 to 411), or GST-tagged DYNLT3 (dynein light chain rp3) immobilized on glutathione-Sepharose beads. Interacting complexes were subsequently eluted and separated by SDS-PAGE (12%). (A) Immunoblot with monoclonal anti-pUL19 antibody as a marker for capsids plus pUL35 (C) and capsids minus pUL35 (C−) shows the presence of pUL35 containing capsids coeluting with GST-tagged DYNLT3 but not GST or GST-tagged pUL48N. (B) Coomassie blue stain gel confirming presence of GST-tagged proteins eluted from glutathione-Sepharose beads and also equivalent amounts of input C and C− capsids. The position of capsid proteins in the input capsid lanes are indicated.
Composition of purified C capsids.
To establish what are the major determinants for addition of pUL48 to biologically relevant intact capsids the composition of isolated C capsids from total infected cell lysates was undertaken. C capsids were isolated from Vero cells lysates after at least 24 h postinfection with either HSV-1 parental strain 17, pUL35-minus (dmVP26-minus [7]), pUL36-minus (ARΔUL36 [54]), or pUL37-minus (FRΔUL37 [54]) viruses. It was not possible to determine the composition of extracellular virions as loss of pUL36 or pUL37 results in accumulation of unenveloped C capsids with no release of extracellular virions (12, 13, 34, 54). Deletion of pUL35 only has a minor effect on viral assembly (32). The specificity of the observed phenotype for ARΔUL36 and FRΔUL37 has been previously verified by the generation of rescuants, which display wild-type growth kinetics (54).
The composition of purified C capsids was then analyzed by immunoblotting for viral proteins (Fig. 4A). Confirmation that intact C capsids, and not just monomeric proteins, were isolated in each case was determined by ascertaining the presence of capsid proteins pUL18 and pUL19 (Fig. 4A). The presence of viral genomic DNA in each case also indicates that intact DNA-containing C capsids, and not just monomeric viral proteins, were isolated (Fig. 4B). The validity of such an analysis for the presence of intact C capsids has been previously described (45). Preparation of capsids was performed with a lysis buffer containing 0.5 M NaCl and 1% Triton X-100, which is known to remove most of the viral envelope and outer tegument layers (68). The presence of pUL48 in both inner and outer tegument layers has been documented using similar detergent solubilization/salt extractions of purified HSV-1 virions (45, 56, 70). The outer tegument layer containing a proportion of pUL48 that interacts with the viral envelope would be removed by treatment with 0.5 M NaCl and 1% Triton X-100. The remaining pUL48 in the inner tegument layer associated with the capsid would not be removed by this treatment, as confirmed for the parental strain (Fig. 4A). The presence of viral envelope was ascertained by blotting for viral glycoprotein gD with only a minor amount present in the parental strain capsid as observed previously (68) (Fig. 4A).
FIG. 4.

Consequence of the loss of pUL35, pUL36, or pUL37 on the addition of pUL48 to C capsids. C capsids were purified from Vero cell lysates harvested after at least 24 h postinfection with either HSV-1 parental strain 17, pUL35-minus (dmVP26-minus), pUL36-minus (ARΔUL36), or pUL37-minus (FRΔUL37) viruses. (A) Immunoblot identifying constituents of purified C capsids with mock-infected and HSV-1 parental strain 17-infected cell lysates as controls for antibody specificity. Primary antibodies to HSV-1 proteins included mouse monoclonal against pUL19, and rabbit polyclonal antibodies against pUL37, pUL48, and gD. An additional rabbit polyclonal antibody raised against purified HSV-1 nuclear C capsids (PTNC) was used to detect tegument protein pUL36, as well as the capsid proteins pUL18 and pUL35. (B) Agarose gel stained with ethidium bromide verifying the presence of viral genomic DNA in isolated C capsids. DNA was extracted from isolated capsids and amplified by using specific primers to the gene encoding HSV-1 tegument protein pUS10 (939 bp). The positive control contained as DNA template the construct displayTarget/pUS10, while the negative control contained no DNA template.
Analysis of isolated C capsids for the presence of additional structural viral proteins was then performed. Further indication that intact capsids were purified was confirmed by the presence of the capsid protein pUL35 in all cases, except as expected in the pUL35-minus capsids (Fig. 4A). The presence of pUL36 was confirmed in all cases, except as expected in the pUL36-minus capsids. This indicates that deletion of pUL37 does not alter the addition of pUL36 to capsids, in support of previous observations with PrV (30). The presence of pUL37 in isolated capsids was confirmed for the parental strain and pUL35-minus viruses although there was less pUL37 in the pUL35-minus virus compared to the parental strain (Fig. 4A). As expected, pUL37 was not present in the pUL37-minus virus and, in addition, was not present in the pUL36-minus capsids (Fig. 4A). This supports previous observations that the direct interaction of pUL37 with pUL36 (30, 64, 65) is required for pUL37 incorporation into the tegument during assembly (11, 54). Furthermore, the previously identified yeast two-hybrid interaction of pUL35 and pUL37 in HSV-1 (33) appears to have no significant biological role since deletion of pUL35 decreases but does not block incorporation of pUL37 into capsids (Fig. 4A). Analysis of the presence of pUL48 indicated only in the case of pUL36-minus capsids an almost total loss of pUL48 (Fig. 4A). There was no apparent effect on addition of pUL48 to capsids lacking pUL37 or pUL35, confirming the lack of biological relevance for the interaction of pUL48 with pUL35 (Fig. 4A). No previous interactions between pUL48 and pUL37 have been detected in previous yeast two-hybrid screens of HSV-1 (33, 65). This strongly supports the notion that the direct interaction of pUL48 with pUL36, as determined in the present study and previously (65), is the major determinant for addition of pUL48 to C capsids.
DISCUSSION
The accepted model for herpes viral assembly dictates that capsids are formed in the nucleus and later acquire tegument proteins in the cytoplasm (39). Three forms of capsids are found in the nucleus: A capsids (lacking DNA and scaffolding proteins), B capsids (containing scaffolding proteins but lacking DNA), and C capsids (containing DNA but lacking scaffolding proteins) (27). The C capsids are formed from B capsids, while A capsids appear to be capsids that have not packaged DNA correctly. The C capsids then undergo tegumentation in the cytoplasm to form mature virions (39). However, there are recent reports that suggest that, at least for HSV-1, tegumentation of the capsid might actually commence in the nucleus, as a number of tegument proteins have been shown to be associated with purified nuclear B and/or C capsids, including pUL36, pUL37, pUL41, and pUL46 (6, 45, 52).
The use of cryo-electron microscopy/tomography and electron microscopy has started to unravel the nature of interactions between tegument and the outer capsid shell of HSV-1. Two independent studies suggest an asymmetric distribution of tegument around the capsid shell (25, 47). Cryo-electron microscopy and tomography of HSV-1 C capsids has shown the presence of protein density associated with capsid pentons, which was attributed to tegument and in particular the major tegument protein pUL36 (7, 25, 72). A more recent cryo-electron microscopy study suggests that the identity of this protein density is more likely a heterodimer of the capsid proteins pUL17 and pUL25 (63). The inner tegument proteins, which include pUS3, pUL36, and pUL37 (22), are the most likely to engage in direct links with outer capsid proteins during viral assembly. The nature of pUS3 incorporation has not been defined but deletion of PrV pUS3 has no effect on the incorporation of pUL36 (40). Addition of pUL36 to capsids depends on an interaction with pUL25 (8, 51) and possibly pUL19 in capsid pentons (4, 7, 38). Subsequent addition of pUL37 proceeds through a direct interaction with pUL36 (11, 20, 30, 65).
From viral gene deletion studies it is known that tegument proteins pUL36, pUL37, and pUL48, the latter of which appears to span the inner and outer tegument layers (45, 56, 70), are critical for secondary envelopment in the cytoplasm during assembly of HSV-1 and PrV (12, 13, 19, 20, 31, 34, 44, 54). Less well defined is the interactions and interplay between these three proteins during viral assembly. In the present study, we sought to further define this relationship by first clarifying whether pUL48 interacts directly with capsid proteins. Using a yeast two-hybrid assay, we identified direct interactions between HSV-1 pUL48N, lacking the C-terminal activation domain, and the capsid proteins pUL19, pUL35, and pUL38, along with the tegument protein pUL36. Previously, we identified interactions of pUL19, pUL35, and pUL36 with pUL48 full-length also by using a yeast two-hybrid screen (33, 65). The identified interactions involving pUL48N, except the interaction involving pUL35, importantly were confirmed in the present study using an in vitro pulldown assay. A further pulldown assay using an in vitro-assembled capsid that resembled B capsids and contains the outer capsid proteins pUL18, pUL19, pUL35, and pUL38 but lacks tegument (60, 62) indicated that pUL48N does not bind to intact capsids in the absence of tegument. This is supported by two independent studies which failed to identify pUL48 on purified nuclear B or C capsids (52, 68). Collectively, this suggests that the direct interactions of capsid proteins pUL19, pUL35, or pUL38 with pUL48N identified in the present study and with pUL48 full-length in a previous study (33) are not biologically relevant, at least in the context of addition of pUL48 to capsids during assembly. The inherent nonspecific binding properties of pUL48 has been highlighted in a previous study (17). Given that most of the pUL48N/capsid protein interactions identified in the present study using a yeast two-hybrid screen could be independently confirmed, also in the present study, with an in vitro pulldown assay does suggest that they may have biological relevance but probably not in the assembly of HSV-1.
For HSV-1 or PrV, the loss of pUL36 or pUL37 blocks secondary envelopment in the cytoplasm (11-13, 20, 31, 54), whereas the loss of pUL35 has a minor effect on viral replication (2, 10, 14, 32). In the present study, analysis of the composition of C capsids purified from cells infected with HSV-1 constructs lacking the gene for either pUL35, pUL36, or pUL37 illustrated for the first time that pUL48 requires the presence of pUL36 for addition to capsids. The absence of pUL36 also resulted in no addition of pUL37 to capsids. Previous studies with HSV-1 pUL36-null constructs indicated that the interaction of pUL37 with pUL36 is required for incorporation into the tegument (11, 54). In addition, a PrV construct consisting of a pUL36 mutant which lacked the binding site for pUL37 showed a similar phenotype to a pUL37-null virus (20), i.e., the accumulation of unenveloped pUL36-containing C capsids. The absence of pUL35 decreased the levels of pUL37 on capsids but had no effect on the addition of pUL48 to capsids. This suggests that the interaction of capsid protein pUL35 with pUL48, identified in the present study, and the interaction of pUL35 with pUL37 identified in a previous study (33) are not significant in the context of intact capsids. This is not unexpected given pUL35 is not essential for replication of HSV-1 (10). The absence of pUL37 had no effect on the addition of pUL36 or pUL48 to capsids. A previous study on PrV has shown that pUL36 is still added to capsids in the absence of pUL37 (30). No direct interaction between pUL48 and pUL37 has been reported, and we were unable to detect an interaction between these two proteins in our yeast two-hybrid screens (33, 65).
This is the first study that clearly shows in HSV-1 that capsid-bound pUL36 is required for the subsequent addition of pUL37 and pUL48, most likely through direct interactions. A previously identified direct interaction between HSV-1 pUL36 and pUL48 (33, 65) was also confirmed in the present study. We have previously mapped the pUL48 binding site in HSV-1 pUL36 to residues 124 to 511 (41). The direct interaction of pUL36 and pUL37 has been confirmed in HSV-1 (65), PrV (30), and varicella-zoster virus (64), and the pUL37 binding site in pUL36 has been mapped to pUL36 residues 512 to 608 (30, 41). This site is distinct from the pUL48 binding site in pUL36, and independent binding of HSV-1 pUL48 and pUL37 to pUL36 has been confirmed in a previous study (41).
An alternative explanation for our findings is that the loss of pUL36 indirectly impairs incorporation of pUL48. It may be that pUL36 directs capsids to the site of pUL48 incorporation, which is then mediated by interactions with other viral proteins. Viral gene deletion studies on both PrV and HSV-1 have shown that pUL36 is important for microtubule-dependent transport of capsids during egress (36, 57). Although the present study does not rule out a trafficking defect in the pUL36-minus virus that is responsible for lack of pUL48 incorporation, there is additional evidence to suggest otherwise. Certainly, numerous deletion studies with PrV and HSV-1 suggest C capsids start to acquire tegument in the cytoplasm (39) (for HSV-1 this process may even begin earlier in the nucleus [6]) without any requirement for trafficking to any particular cellular compartment such as the Golgi compartment. In further support of this, it has been shown in the cell body of neurons that a number of HSV-1 tegument proteins, including pUL48, are incorporated into unenveloped C capsids in the cytoplasm prior to secondary envelopment at the Golgi body (42).
In conclusion, pUL48 is a multifunctional protein that not only has a regulatory role in IE viral gene transcription but also has a structural role during assembly, where it appears to be involved in a number of interactions. In HSV-1, there is most likely a tripartite complex, similar to that described for the tegument proteins pUL11, pUL16, and pUL21 (39), consisting of the essential tegument proteins pUL36, pUL37, and pUL48, which mediates secondary envelopment of C capsids during viral egress (Fig. 5). The interaction of both pUL37 and pUL48 with nonoverlapping binding sites in pUL36 is most likely required for secondary envelopment of HSV-1. Although pUL37 and pUL48 are critical for secondary envelopment of PrV, they are not absolutely essential (19, 31, 34). It may be in PrV that pUL48 (or other tegument) compensates for pUL37 and vice versa. It would be of interest to construct a PrV mutant lacking both pUL48 and pUL37 to establish that these are the only links, via pUL36, which facilitate addition of outer tegument during viral assembly. Future studies in our laboratory will focus on fine mapping the interacting domains of HSV-1 pUL36 and pUL48, which will guide the construction of mutant viruses to specifically define the role of this interaction during viral assembly.
FIG. 5.

Diagram of proposed HSV-1 pUL48 interactions that link capsid/inner tegument proteins with outer tegument/glycoproteins during viral assembly.
Acknowledgments
This study was supported by grants 402457 and 457315 from the Australian National Health and Medical Research Council (to A.L.C. and R.J.D.) and an Endeavor International Postgraduate Research Scholarship and International Postgraduate Award (to D.K.).
We thank A. Aggarwal for assistance in isolation of capsids. We also thank C. Fraefel and B. Kelly for critically reading the manuscript.
Footnotes
Published ahead of print on 18 November 2009.
REFERENCES
- 1.Adamson, W. E., D. McNab, V. G. Preston, and F. J. Rixon. 2006. Mutational analysis of the herpes simplex virus triplex protein VP19C. J. Virol. 80:1537-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antinone, S. E., G. T. Shubeita, K. E. Coller, J. I. Lee, S. Haverlock-Moyns, S. P. Gross, and G. A. Smith. 2006. The herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J. Virol. 80:5494-5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines, J. D., and B. Roizman. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker, M. L., W. Jiang, W. J. Wedemeyer, F. J. Rixon, D. Baker, and W. Chiu. 2006. Ab initio modeling of the herpesvirus VP26 core domain assessed by CryoEM density. PLoS Comput. Biol. 2:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowman, B. R., M. L. Baker, F. J. Rixon, W. Chiu, and F. A. Quiocho. 2003. Structure of the herpesvirus major capsid protein. EMBO J. 22:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bucks, M. A., K. J. O'Regan, M. A. Murphy, J. W. Wills, and R. J. Courtney. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, D. H., J. Jakana, D. McNab, J. Mitchell, Z. H. Zhou, M. Dougherty, W. Chiu, and F. J. Rixon. 2001. The pattern of tegument-capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J. Virol. 75:11863-11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coller, K. E., J. I. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alpha herpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cousens, D. J., R. Greaves, C. R. Goding, and P. O'Hare. 1989. The C-terminal 79 amino acids of the herpes simplex virus regulatory protein, Vmw65, efficiently activate transcription in yeast and mammalian cells in chimeric DNA-binding proteins. EMBO J. 8:2337-2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai, P., N. A. DeLuca, and S. Person. 1998. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 247:115-124. [DOI] [PubMed] [Google Scholar]
- 11.Desai, P., G. L. Sexton, E. Huang, and S. Person. 2008. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 82:11354-11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai, P., G. L. Sexton, J. M. McCaffery, and S. Person. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259-10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dohner, K., K. Radtke, S. Schmidt, and B. Sodeik. 2006. Eclipse phase of herpes simplex virus type 1 infection: efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 80:8211-8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Douglas, M. W., R. J. Diefenbach, F. L. Homa, M. Miranda-Saksena, F. J. Rixon, V. Vittone, K. Byth, and A. L. Cunningham. 2004. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 279:28522-28530. [DOI] [PubMed] [Google Scholar]
- 16.Elliott, G., G. Mouzakitis, and P. O'Hare. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932-7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farnsworth, A., T. W. Wisner, and D. C. Johnson. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81:319-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs, W., H. Granzow, B. Klupp, A. Karger, K. Michael, C. Maresch, R. Klopfleisch, and T. Mettenleiter. 2007. Relevance of the interaction between alphaherpesvirus UL3.5 and UL48 proteins for virion maturation and neuroinvasion. J. Virol. 81:9307-9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs, W., H. Granzow, B. G. Klupp, M. Kopp, and T. C. Mettenleiter. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729-6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs, W., B. G. Klupp, H. Granzow, and T. C. Mettenleiter. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879-11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerster, T., and R. Roeder. 1988. A herpesvirus transactivating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc. Natl. Acad. Sci. U. S. A. 85:6347-6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2004. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 78:1314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gross, S. T., C. A. Harley, and D. W. Wilson. 2003. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1-12. [DOI] [PubMed] [Google Scholar]
- 25.Grunewald, K., P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister, and A. C. Steven. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302:1396-1398. [DOI] [PubMed] [Google Scholar]
- 26.Herr, W. 1998. The herpes simplex virus VP16-induced complex: mechanisms of combinatorial transcriptional regulation. Cold Spring Harbor Symp. Quant. Biol. 63:599-607. [DOI] [PubMed] [Google Scholar]
- 27.Homa, F. L., and J. C. Brown. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7:107-122. [DOI] [PubMed] [Google Scholar]
- 28.Kato, K., T. Daikoku, F. Goshima, H. Kume, K. Yamaki, and Y. Nishiyama. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149-2162. [DOI] [PubMed] [Google Scholar]
- 29.Kelly, B. J., C. Fraefel, A. L. Cunningham, and R. J. Diefenbach. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 145:173-186. [DOI] [PubMed] [Google Scholar]
- 30.Klupp, B. G., W. Fuchs, H. Granzow, R. Nixdorf, and T. C. Mettenleiter. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klupp, B. G., H. Granzow, E. Mundt, and T. C. Mettenleiter. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 75:8927-8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krautwald, M., C. Maresch, B. Klupp, W. Fuchs, and T. Mettenleiter. 2008. Deletion or green fluorescent protein tagging of the pUL35 capsid component of pseudorabies virus impairs virus replication in cell culture and neuroinvasion in mice. J. Gen. Virol. 89:1346-1351. [DOI] [PubMed] [Google Scholar]
- 33.Lee, J., V. Vittone, E. Diefenbach, A. Cunningham, and R. Diefenbach. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347-354. [DOI] [PubMed] [Google Scholar]
- 34.Leege, T., H. Granzow, W. Fuchs, B. G. Klupp, and T. C. Mettenleiter. 2009. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 90:1560-1568. [DOI] [PubMed] [Google Scholar]
- 35.Liu, Y., W. Gong, C. C. Huang, W. Herr, and X. Cheng. 1999. Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein VP16. Genes Dev. 13:1692-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luxton, G. W., J. I. Lee, S. Haverlock-Moyns, J. M. Schober, and G. A. Smith. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClelland, D. A., J. D. Aitken, D. Bhella, D. McNab, J. Mitchell, S. M. Kelly, N. C. Price, and F. J. Rixon. 2002. pH reduction as a trigger for dissociation of herpes simplex virus type 1 scaffolds. J. Virol. 76:7407-7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNabb, D. S., and R. J. Courtney. 1992. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology 190:221-232. [DOI] [PubMed] [Google Scholar]
- 39.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2009. Herpesvirus assembly: an update. Virus Res. 143:222-234. [DOI] [PubMed] [Google Scholar]
- 40.Michael, K., B. G. Klupp, T. C. Mettenleiter, and A. Karger. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 80:1332-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mijatov, B., A. L. Cunningham, and R. J. Diefenbach. 2007. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368:26-31. [DOI] [PubMed] [Google Scholar]
- 42.Miranda-Saksena, M., R. A. Boadle, P. Armati, and A. L. Cunningham. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J. Virol. 76:9934-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohl, B. S., S. Bottcher, H. Granzow, J. Kuhn, B. G. Klupp, and T. C. Mettenleiter. 2009. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 83:9641-9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mossman, K. L., R. Sherburne, C. Lavery, J. Duncan, and J. R. Smiley. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 74:6287-6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy, M., M. Bucks, K. O'Regan, and R. Courtney. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279-289. [DOI] [PubMed] [Google Scholar]
- 46.Naldinho-Souto, R., H. Browne, and T. Minson. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80:2582-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newcomb, W. W., and J. C. Brown. 2009. Time-dependent transformation of the herpesvirus tegument. J. Virol. 83:8082-8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newcomb, W. W., F. L. Homa, D. R. Thomsen, F. P. Booy, B. L. Trus, A. C. Steven, J. V. Spencer, and J. C. Brown. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol. 263:432-446. [DOI] [PubMed] [Google Scholar]
- 49.Okoye, M. E., G. L. Sexton, E. Huang, J. M. McCaffery, and P. Desai. 2006. Functional analysis of the triplex proteins (VP19C and VP23) of herpes simplex virus type 1. J. Virol. 80:929-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Padula, M. E., M. L. Sydnor, and D. W. Wilson. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 83:4757-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasdeloup, D., D. Blondel, A. L. Isidro, and F. J. Rixon. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610-6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Read, G. S., and M. Patterson. 2007. Packaging of the virion host shutoff (Vhs) protein of herpes simplex virus: two forms of the Vhs polypeptide are associated with intranuclear B and C capsids, but only one is associated with enveloped virions. J. Virol. 81:1148-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts, A., F. Abaitua, P. O'Hare, D. McNab, F. Rixon, and D. Pasdeloup. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. Herpes simplex viruses, p. 2501-2601. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed. Lippincott/The Williams & Wilkins Co., Philadelphia, PA.
- 56.Schmitz, J. B., A. G. Albright, P. R. Kinchington, and F. J. Jenkins. 1995. The UL37 protein of herpes simplex virus type 1 is associated with the tegument of purified virions. Virology 206:1055-1065. [DOI] [PubMed] [Google Scholar]
- 57.Shanda, S., and D. Wilson. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388-7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein Vhs. J. Virol. 68:2339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stern, S., M. Tanaka, and W. Herr. 1989. The Oct-1 homoeodomain directs formation of a multiprotein-DNA complex with the HSV transactivator VP16. Nature 341:624-630. [DOI] [PubMed] [Google Scholar]
- 60.Tatman, J. D., V. G. Preston, P. Nicholson, R. M. Elliott, and F. J. Rixon. 1994. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J. Gen. Virol. 75:1101-1113. [DOI] [PubMed] [Google Scholar]
- 61.Thompson, R. L., C. M. Preston, and N. M. Sawtell. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 5:e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomsen, D. R., L. L. Roof, and F. L. Homa. 1994. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J. Virol. 68:2442-2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol. Cell 26:479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uetz, P., Y. A. Dong, C. Zeretzke, C. Atzler, A. Baiker, B. Berger, S. V. Rajagopala, M. Roupelieva, D. Rose, E. Fossum, and J. Haas. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239-242. [DOI] [PubMed] [Google Scholar]
- 65.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Einem, J., D. Schumacher, D. J. O'Callaghan, and N. Osterrieder. 2006. The alpha-TIF (VP16) homologue (ETIF) of equine herpesvirus 1 is essential for secondary envelopment and virus egress. J. Virol. 80:2609-2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ward, P. L., W. O. Ogle, and B. Roizman. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623-4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolfstein, A., C. H. Nagel, K. Radtke, K. Dohner, V. J. Allan, and B. Sodeik. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227-237. [DOI] [PubMed] [Google Scholar]
- 69.Wysocka, J., and W. Herr. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28:294-304. [DOI] [PubMed] [Google Scholar]
- 70.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang, Y., D. A. Sirko, and J. L. McKnight. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 73:3210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu, Q., and R. J. Courtney. 1994. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology 204:590-599. [DOI] [PubMed] [Google Scholar]