

Abstract
During rotavirus entry, a virion penetrates a host cell membrane, sheds its outer capsid proteins, and releases a transcriptionally active subviral particle into the cytoplasm. VP5*, the rotavirus protein believed to interact with the membrane bilayer, is a tryptic cleavage product of the outer capsid spike protein, VP4. When a rotavirus particle uncoats, VP5* folds back, in a rearrangement that resembles the fusogenic conformational changes in enveloped-virus fusion proteins. We present direct experimental evidence that this rearrangement leads to membrane binding. VP5* does not associate with liposomes when mounted as part of the trypsin-primed spikes on intact virions, nor does it do so after it has folded back into a stably trimeric, low-energy state. But it does bind liposomes when they are added to virions before uncoating, and VP5* rearrangement is then triggered by addition of EDTA. The presence of liposomes during the rearrangement enhances the otherwise inefficient VP5* conformational change. A VP5* fragment, VP5CT, produced from monomeric recombinant VP4 by successive treatments with chymotrypsin and trypsin, also binds liposomes only when the proteolysis proceeds in their presence. A monoclonal antibody that neutralizes infectivity by blocking a postattachment entry event also blocks VP5* liposome association. We propose that VP5* binds lipid bilayers in an intermediate conformational state, analogous to the extended intermediate conformation of enveloped-virus fusion proteins.
The outer-layer proteins of rotaviruses are the molecular machinery of viral cell entry. The two components, VP4 and VP7, form a set of spike-like projections and an extended outer shell, respectively, as illustrated in Fig. 1A and B. VP4 is the principal agent of cell attachment and membrane penetration (16). Tryptic cleavage of VP4 into two fragments, designated VP8* and VP5* (Fig. 1A, B, and E), activates rotavirus particles for efficient infection (11). Prior to cleavage, the spike-like VP4 projections do not form, and its various domains are probably flexibly linked (6). The VP8* fragment, which forms the “heads” at the tips of a spike, is a lectin-like module that in many strains binds sialic acid (5, 10). The VP5* fragment, which forms the spike body, stalk, and foot, is probably the membrane-active element (9, 17). VP7 is a calcium sensor that constrains VP8*/VP5* in the spike-like conformation (1, 25). Loss of calcium ions leads to VP7 dissociation and in turn to VP5* conformational changes (8, 27).
FIG. 1.

Conformational states of rotavirus VP4. (A) Multilayered structure of a rotavirus particle, based on cryo-EM three-dimensional image reconstructions (4, 15). Color coding for the structural proteins is indicated by labels. (B) Detail of a VP4 spike. The VP5* fragment is in bold red; the VP8* fragment is in pink. (C) Structure of the dimer-clustered, projecting portion of VP5*, as determined by comparison of the crystal structure of the VP5Ag fragment (PDB accession code 2B4H) and cryo-EM images (9, 26). The presence of a third VP5Ag domain is indicated by the yellow oval. “L” labels the hydrophobic loops at the apex of the VP5Ag domain. Arrows suggest the transition to the folded-back conformation. (D) Folded-back structure of VP5CT (9) (PDB accession code 1SLQ). This conformation probably represents the final, post-membrane-penetration state of VP5*. A “foot” would be appended to each of the α-helices in the central coiled coil. (E) Position in the RRV VP4 polypeptide chain of various proteolytic fragments. Numbers above the bar are the initial and final residues in each segment. c-c, coiled-coil.
Our appreciation of the activities just summarized has come largely from biochemical and structural analyses. Particularly important have been X-ray crystal structures and nuclear magnetic resonance spectroscopy solution structures of the lectin-like core of VP8* (10, 19); crystal structures of large, N-terminal fragments of VP5* (9, 26); a crystal structure of VP7 (1); and cryo-electron microscopy (cryo-EM) structures of various virus-derived particles, including near-atomic resolution analyses of the inner capsid particle (known as the double-layer particle [DLP]) (Fig. 1A) and of the DLP recoated with VP7 (4, 28). The likely membrane-directed activities of VP5* have been inferred from the crystallographic studies of its N-terminal region and from comparison of these structures with cryo-EM reconstructions (9, 26). As illustrated in Fig. 1C and D, two states of the VP5* N-terminal region have been seen in crystal structures: a dimeric form, in crystals of VP5Ag (the “antigen domain,” residues 247 to 479 of VP4), and a trimeric form, in crystals of VP5Ag and VP5CT (residues 248 to 525 of VP4). The additional C-terminal residues in the latter fragment form a tight, three-chain coiled coil, indicating that the trimeric form is not a crystallographic artifact. Indeed, a proportion of the VP5* released from uncoating virions acquires biochemical and antigenic characteristics that match those of VP5CT (27). The VP5Ag dimer corresponds closely to the upright part of the spike seen by cryo-EM (26). A recent cryo-EM reconstruction confirms that the “foot” region of VP5*—the part anchored in the DLP—is trimeric (15). Thus, a conformational transition has been postulated, from the upright dimer, with the N-terminal region of an additional subunit elsewhere in the spike, to a folded-back trimer. The large, β-sandwich domain of VP5* (corresponding to VP5Ag) has a set of hydrophobic loops at one apex (9) (Fig. 1C). The sequence of one of them resembles the fusion loop sequence in the E1 protein of Semliki Forest virus (17). In the dimer state, these loops project outward, toward the target cell membrane, and they are masked by the receptor-binding VP8* heads. In the folded-back trimer, the VP8* heads have dissociated, and it can be assumed that the hydrophobic loops of the VP5* apex project away from the target membrane, since the C-terminal coiled coil must connect smoothly to the foot.
These structural features suggest that under appropriate circumstances, VP5* should interact with lipid bilayer membranes. Since release of VP7 is probably the mechanism by which the conformational change suggested in Fig. 1 is triggered (27), we chose to test whether loss of VP7 from virions leads to membrane insertion of VP5*. We show that if VP7 uncoats from virions in the presence of liposomes, VP5* associates with the lipid. Once it has reached its folded-back end state, however, VP5* appears not to bind liposomes, as might be expected from the geometry of its structure. The trimeric fragment shown in Fig. 1D (designated VP5CT) can be generated from monomeric, recombinant VP4 in vitro by successive chymotryptic and tryptic cleavages (7). If the cleavage occurs in the presence of liposomes, the resulting fragment associates with them; if liposomes are added later, it does not. We also show that a neutralizing monoclonal antibody (MAb) directed against VP5* blocks membrane binding, linking liposome interaction with functional rotavirus entry. We conclude from both the uncoating and the cleavage experiments that a transient intermediate is present during these processes that can insert into a membrane, probably through the hydrophobic loops at the apex of the antigen domain, but that the stable, folded-back product cannot do so.
MATERIALS AND METHODS
Virus.
Rhesus rotavirus (RRV), serotype G3, P5B[3], was propagated in MA-104 cells, which were grown in M199 medium (Invitrogen), supplemented with 7.5% fetal bovine serum (HyClone Laboratories, Inc.), 10 mM HEPES, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For virus inoculation and amplification, fetal bovine serum was omitted and M199 was supplemented to 1 μg/ml with porcine pancreatic trypsin (Sigma-Aldrich). At approximately 36 h postinfection, cells and medium were frozen for later purification of triple-layer particles (TLPs) and DLPs, as described in reference 25. Infectivity of virus preparations was determined by an infectious focus assay (25).
Recombinant VP4 and VP7.
Previously described baculovirus expression vectors encoding RRV VP4 or VP7 (8, 12, 17) were propagated in Sf9 insect cells grown in spinner cultures with Sf900 II serum-free medium (Invitrogen). VP4 was purified from the cells by anion exchange and size exclusion chromatography (7), and VP7 was purified from the medium by lectin affinity, immunoaffinity, and size exclusion chromatography as described previously (8). Concentrations of purified VP4 and VP7 were determined by measuring absorbance at 280 nm, using absorption coefficients of 95,100 M−1 cm−1 and 62,700 M−1 cm−1, respectively, calculated using the Vector NTI 7 software program (InforMax, Inc.).
Recoating.
DLPs were recoated with VP4 and VP7, essentially as described previously (25); VP7 was added in 2- to 3-fold molar excess over full occupancy, and VP4 was added at 0.2 mg/ml or more to achieve at least a 5-fold molar excess. Specific trypsin cleavage of VP4 to VP5* and VP8* requires its assembly into particles (7). By this criterion, VP4 recoated the DLPs to approximately 50% of its occupancy on authentic virions.
Protease treatment of rotavirus particles.
l-(Tosylamido-2-phenyl) ethyl chloromethyl ketone-treated trypsin (at least 10,350 BAEE units/mg; Worthington Biochemical Corp.) was freshly diluted from a 5-mg/ml stock and added to rotavirus TLPs or recoated particles to a final concentration of 1 μg/ml. One-chloro-3-tosylamido-7-amino-2-heptanone-treated chymotrypsin (at least 45 units/mg; Worthington Biochemical Corp.) was diluted from a 5-mg/ml stock and used at a final concentration of 1 μg/ml. After incubation at 37°C for 30 min, 100 mM ethanolic phenylmethylsulfonyl fluoride (PMSF) was added to a final concentration of 1 mM; the mixture was incubated on ice for 5 min before adding SDS-PAGE sample buffer.
Preparation of liposomes.
Phosphatidylcholine (PC) derived from chicken eggs (Avanti Polar Lipids) was dissolved in chloroform at 5 to 10 mg/ml and stored under nitrogen at −20°C. For each batch of liposomes, 2 mg PC was dried to a film with a dry nitrogen stream and purged under vacuum overnight in the dark. The dried PC was resuspended in HN buffer (20 mM HEPES, pH 7.3, 140 mM NaCl) with pulsed vortexing, followed by five freeze-thaw cycles using a dry ice-ethanol bath. The liposomes were then extruded 21 times through a 0.2-μm polycarbonate filter.
Liposome association of VP4 and its proteolytic fragments.
To assay liposome association of VP4 released from uncoating particles, 5 to 10 μg of TLPs (or recoated particles) were incubated for 1 h at 37°C with 12.5 μl liposome suspensions in 150 μl of HN. EDTA was added to 1 mM to uncoat the particles, or 1 mM CaCl2 was added as a control. To prevent degradation of VP5* after uncoating, PMSF was added to 1 mM. For density gradient centrifugation, samples were mixed with 70% (wt/vol) sucrose in HN to yield 350 μl of sample in 40% sucrose. This mixture was layered over a discontinuous sucrose gradient: 70%, 60%, and 50% (300 μl of each sucrose concentration) in HN. A 25% (150-μl) sucrose solution in HN was layered above the sample. For the other reported sucrose gradient experiments, samples in 40% sucrose were overlaid with 1 ml 25% sucrose and 150 μl 5% sucrose. For control experiments in which particles were maintained as TLPs, all sucrose solutions were supplemented with 1 mM CaCl2. Soluble, recombinant VP4 (5 to 10 μg) was incubated in 150 μl HN buffer (with or without liposomes) and digested with 1 μg/ml chymotrypsin for 30 min at 37°C, followed (in most cases) by adding trypsin to 1 μg/ml and incubating for an additional 30 min. Proteolysis was quenched by adding PMSF to 1 mM. Gradients were centrifuged at 180,000 × g in an SW-55 rotor (Beckman-Coulter) at 4°C for 2.5 h. Fractions were collected from the top using a wide-bore pipette tip and stored at −80°C.
Electrophoresis and immunoblots.
To detect VP4, samples were reduced with 5% β-mercaptoethanol, separated with 10 or 12% SDS-PAGE gels, electroblotted onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories), and detected by immunoblotting with MAb HS2 (20) or 4D8 (27) and peroxidase-conjugated goat anti-mouse IgG (Kirkegaard and Perry Laboratories, Inc.). VP7 was detected in nonreduced samples by immunoblotting with MAb M60 (22). VP6 was detected with an antiserum made in guinea pigs and peroxidase-conjugated goat anti-guinea pig IgG (Kirkegaard and Perry Laboratories, Inc.). All immunoblots were developed using a chemiluminescent peroxidase substrate (ECL Plus; GE Healthcare).
Neutralization of virions by MAbs.
MAbs 2G4 and 7A12 (22) were dialyzed into TNC (20 mM Tris [pH 8.0], 100 mM Nacl, 1 mM Cacl2). Five μg of virions were incubated overnight at 4°C with 100 μg/ml of MAb, corresponding to a 5-fold or greater excess of MAb over potential binding sites on the virions. Virion-antibody mixtures were used directly in liposome association experiments, as described above. Titers of infectious virus alone or mixed with MAb were determined by infectious focus assay as described previously (25). Briefly, virion-MAb samples were serially diluted into M199 containing 100 μg/ml MAb, inoculated onto MA-104 cells in 96-well plates, and incubated for 1 h at 37°C, after which the inocula were removed and replaced with M199 containing fetal bovine serum (FBS) and MAb 159 to inhibit spread.
RESULTS
Liposome association of VP5* during uncoating.
We used density gradient centrifugation to detect potential membrane interactions of VP5*. Figure 2 shows positions in the gradient of rotavirus proteins following uncoating of trypsin-cleaved virions by EDTA in the presence or absence of liposomes. Uncoating releases the outer-layer proteins, VP5* and VP7, from DLPs (which we located on the gradient with antibodies to VP6). Addition of liposomes together with EDTA shifts much of the released VP5* from fraction 4, of intermediate density, into fraction 1, the most buoyant. Liposome addition does not affect the position of VP7 or VP6.
FIG. 2.
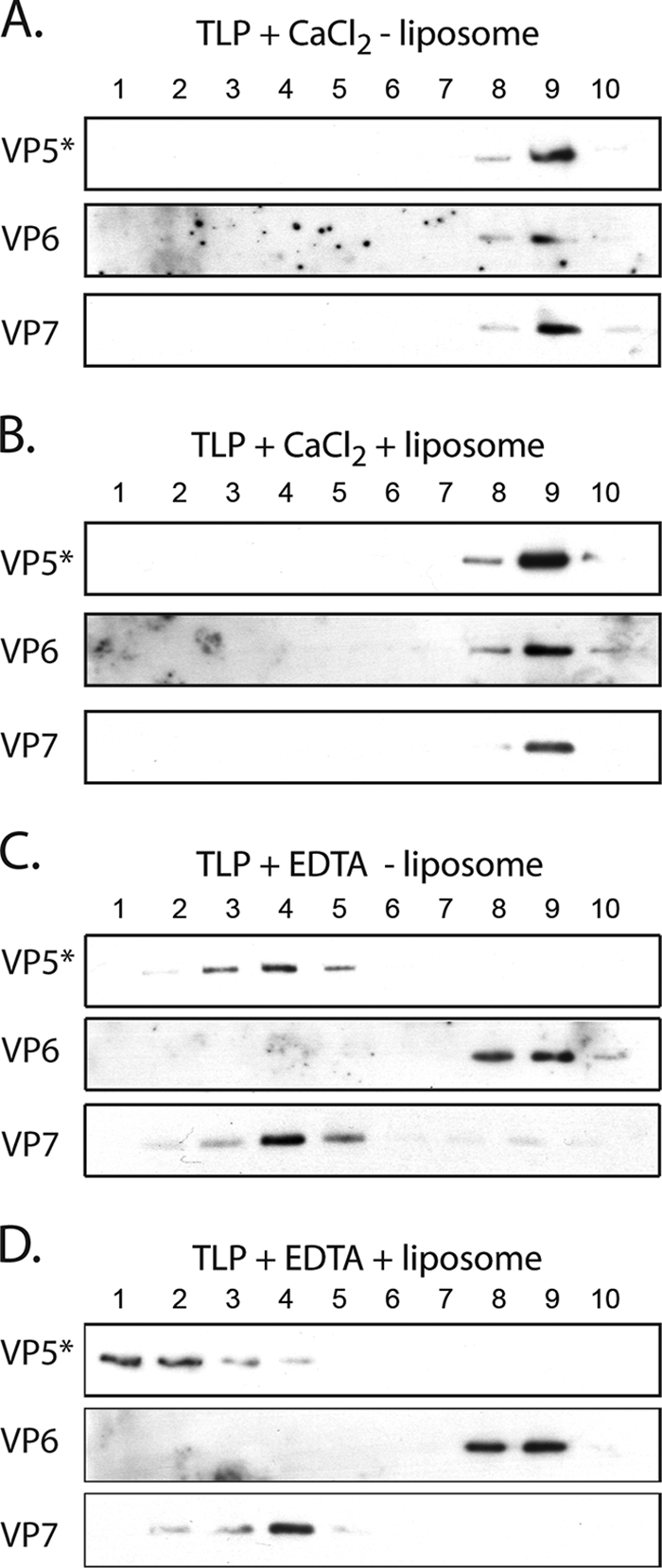
Interaction of rotavirus proteins with liposomes. Virions were incubated in 1 mM CaCl2 or in 1 mM EDTA in the presence or absence of liposomes, and the mixtures were analyzed on sucrose gradients (25% to 70% [wt/vol] in HN buffer). (A to D) Immunoblots of fractions from top (1) to bottom (10) of the gradients, using HS2 to detect VP5*, m60 to detect VP7, and guinea-pig anti-VP6 (1:5,000 dilution) to detect VP6 (DLPs). Refractive index measurements (not shown) for the four gradients in panels A to D confirmed that all four had indistinguishable density profiles.
Similar experiments show that uncleaved VP4 does not associate with liposomes when released from TLPs by addition of EDTA. To obtain TLPs with uncleaved VP4, we recoated DLPs with recombinant VP4 and VP7, avoiding partial VP4 cleavage during purification of virions. VP5* released from trypsin-treated recoated particles associated with liposomes to the same extent as VP5* from authentic cleaved virions when examined in parallel experiments (Fig. 3A). However, uncleaved VP4 did not move out of the most dense fractions. Cleavage of recoated particles with chymotrypsin yields a VP5*-like VP4 digestion product, designated VP5*C to distinguish it from authentic VP5*. Following uncoating of chymotrypsin-cleaved recoated particles, VP5*C also associated with liposomes, perhaps even more completely than VP5* itself (Fig. 3A). Chymotryptic cleavage did not produce infectious particles (Fig. 3B), consistent with published properties of chymotrypsin-cleaved authentic virions (2, 3).
FIG. 3.

Requirement of VP4 cleavage for membrane association. (A) Virions and recoated particles were treated as for Fig. 2 and analyzed on discontinuous sucrose gradients (5% to 40% [wt/vol]). Immunoblots with HS2 were used to detected VP5* in gradient fractions from top (1) to bottom (7). (B) Infectivity of recoated particles following treatment with trypsin (T), chymotrypsin (C), or chymotrypsin followed by trypsin (CT), normalized to trypsin-treated recoated particles as 100%. Uncl, mock digested.
VP5* associates with liposomes only if released in their presence from virions or recoated particles. In the experiment shown in Fig. 4, we varied the order of addition of virions, liposomes, and EDTA to the uncoating reaction. Adding EDTA 30 min before adding liposomes and separating the reaction products on a density gradient yielded minimal VP5* in the most buoyant (liposome-containing) fractions. The liposomes thus capture VP5* in a state that is no longer accessible 30 min after release is initiated.
FIG. 4.
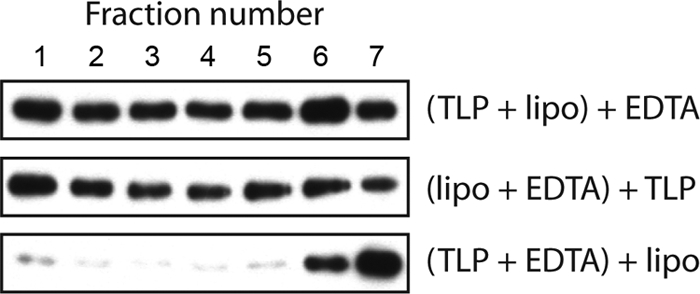
Liposome association of VP5* requires release in their presence. In each of the three experiments shown, the two components indicated in parentheses were mixed first, followed by addition of the third component 30 min later. Samples were then incubated for an additional 30 min (1 h total) before density gradient separation. Immunoblots with HS2 detected the positions of VP5* in fractions from top (1) to bottom (7) of discontinuous sucrose gradients (5% to 40% [wt/vol]).
The association of VP5* with liposomes is blocked when cleaved virions are bound by neutralizing MAb 2G4 before they are uncoated by addition of EDTA in the presence of liposomes (Fig. 5). MAb 2G4 selects a neutralization escape mutation in VP5* at residue 393, in the hydrophobic FG loop (17). Electron cryomicroscopy image reconstructions show that the Fab of 2G4 decorates the hydrophobic apex of VP5*, adjacent to the location where the receptor binding VP8* heads are mounted (24). Binding of neutralizing MAb 7A12, which selects an escape mutation in VP8* and decorates the VP8* heads (17, 24), does not block VP5* liposome association upon uncoating (Fig. 5). Incubation of virions with either MAb under the conditions of the liposome interaction experiment reduces their infectivity by approximately 10,000-fold (Fig. 5B).
FIG. 5.

MAb 2G4 blocks VP5* interaction with liposomes. (A) TLPs, either mock incubated or incubated with MAb 2G4 or 7A12, were uncoated in the presence of liposomes by the addition of 1 mM EDTA and separated over a discontinuous sucrose gradient. The fractions from top (1) to bottom (7) were heated to 95°C, analyzed by SDS-PAGE, and immunoblotted with horseradish peroxidase-conjugated HS2. (B) Neutralization of virus by the MAbs was determined by infectious focus assay. Error bars represent standard deviations of three titrations.
Correlation of VP5* conformational changes with liposome binding.
Tryptic cleavage of monomeric, recombinant VP4 does not give a uniform product, but successive treatment with chymotrypsin and trypsin yields the stable, trimeric species, VP5CT (7, 9). VP5CT and VP5* (the product of tryptic cleavage of VP4 on virions or recoated particles) have the same N termini, but the former lacks about 250 C-terminal residues. When examined by SDS-PAGE, VP5CT migrates to the position expected based on its calculated monomer molecular weight if the sample is heated to 95°C in SDS sample buffer before electrophoresis. VP5CT migrates more slowly, however, if the sample remains at room temperature, presumably because the trimer, stabilized by the extended, central coiled coil and by a 9-strand β-annulus (Fig. 1D), does not dissociate (7, 9). A substantial fraction of VP5* from uncoated virions also migrates more slowly if the sample is not heated, and this fraction reacts with a conformation-specific antibody raised against trimeric VP5CT (27). It is therefore likely that when released from virions, VP5* shifts into the trimeric conformation modeled by the crystal structure of VP5CT. The presence of liposomes during uncoating increases the yield of the slowly migrating species from about 30% to essentially 100% (Fig. 6A, lanes 4 and 8). Similar experiments with recoated particles showed that neither uncleaved VP4 nor VP5*C shifts to the slowly migrating form, even if released in the presence of liposomes (Fig. 6B). VP5*C reproducibly fails to enter the SDS-PAGE gel if the sample is not heated, presumably because dissolution of the liposome with SDS leads to aggregation rather than to formation of a VP5CT-like, folded-back trimer (Fig. 6B). Formation of a stable trimer thus appears to require the N terminus of VP5* produced by trypsin digestion.
FIG. 6.

Anomalous migration of VP5*. (A) Uncoating experiment. Virions were uncoated (by addition of 1 mM EDTA) in the presence or absence of liposomes, with addition of 1 mM CaCl2 as a control. The products were analyzed by SDS-PAGE and immunoblotting with HS2, with (+) or without (−) heating of the samples at 95°C for 10 min. When virions (lanes 2 and 6) are not uncoated by addition of EDTA or by heating in SDS-PAGE sample buffer, VP5* is not detected, presumably because it remains on intact virions and does not enter the gel. (B) Importance of VP4 proteolytic cleavage for SDS-resistant trimer formation. VP4 on recoated DLPs was cleaved with trypsin (T) or chymotrypsin (C) or left uncleaved (Un), and the particles were then uncoated with 1 mM EDTA in the presence or absence of liposomes. The products were analyzed by SDS-PAGE and immunoblotting with HS2, with (+) or without (−) heating of the samples at 95°C for 10 min. The slowly migrating VP5* band (lane 10) is seen only in unheated samples following conventional tryptic cleavage. In panel A (compare lanes 4 and 8) and panel B (compare lanes 4 and 10), the presence of liposomes enhances the yield of the SDS-resistant species.
Figure 7 shows that VP5CT associates with liposomes when prepared by serial digestion of VP4 in their presence but not if added to liposomes after cleavage and rearrangement to the slowly migrating form are complete. Together with the experiments shown in Fig. 4, these results suggest that VP5CT and VP5* associate with liposomes only in transient, intermediate conformations and that when the lipid bilayer is dissolved by detergent, they rearrange into very stable, folded-back trimers, as seen in the VP5CT crystal structure. If liposomes are added after VP5* or VP5CT has reached this stable conformation, no lipid binding can occur.
FIG. 7.
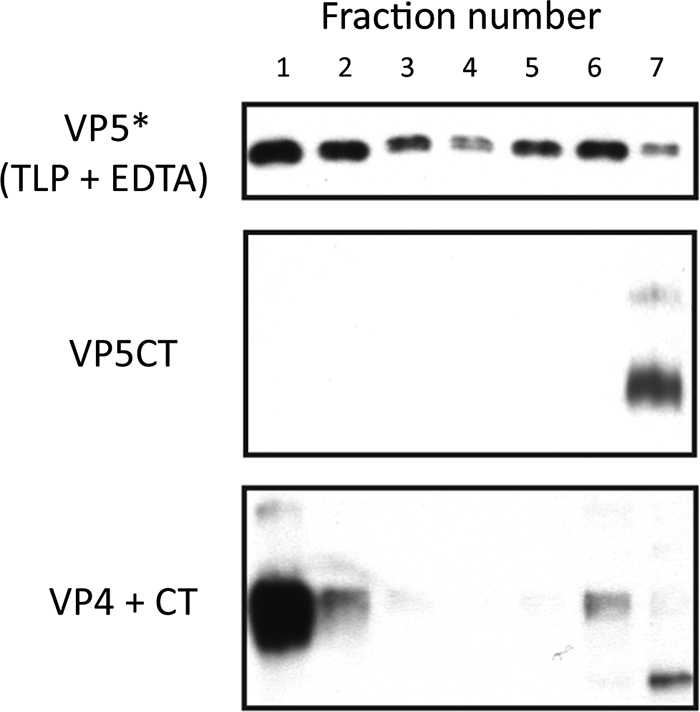
Association of VP5CT with liposomes, analyzed by immunoblotting of sucrose gradient fractions that have been separated by SDS-PAGE. Top panel: virions were uncoated with EDTA in the presence of liposomes before analysis. VP5* was detected by HS2 immunoblotting (as a standard). Middle panel: addition of purified VP5CT to liposomes, followed by analysis. Bottom panel: digestion of VP4 successively with chymotrypsin and trypsin in the presence of liposomes before analysis. Samples in middle and bottom panels were not heated and were probed with MAb 4D8.
In the experiments shown in Fig. 6 and 7, we detected VP5CT by immunoblotting with MAb 4D8 because the HS2 epitope is in the “foot” region that is missing from VP5CT (27; also unpublished data). MAb 4D8 detects only the slowly migrating, trimeric form of VP5* or VP5CT in immunoblots of samples that have not been heated (27). Therefore, we did not heat the electrophoresis samples for the experiment shown in Fig. 7.
DISCUSSION
Previous biochemical and structural analyses of VP4 and its cleavage fragments provided evidence of a conformational rearrangement that resembles the fold-back of enveloped virus fusion proteins (7, 9, 26, 24). None of the well-characterized stable conformers interacted with membranes in experimental systems, however. The experiments presented here connect VP4 conformational changes to lipid bilayer interactions and to likely stages of viral penetration in an endosome. We have evidence for at least four conformational states of the VP5* fragment: state 1, as part of VP4 in TLPs before tryptic activation; state 2, as VP5* in trypsin-treated TLPs; state 3, as an intermediate associated with a membrane bilayer; and state 4, as a folded-back trimer, with a structure that has an N-terminal region resembling VP5CT.
How does VP5* interact with a membrane? The antigen domain of VP5* is an elongated β-barrel with a cluster of three hydrophobic loops at one tip (26). Their location and the sequence similarity of one of them to the fusion loop of Semliki Forest virus (17) have led to the suggestion that a step in endosome disruption might involve insertion of the hydrophobic apex of the antigen domain into the membrane bilayer. In state 2, i.e., on the virion following activation by trypsin, the hydrophobic loops of two of these domains face away from the center of the virus particle, but they are covered by VP8* (9); the location of the third antigen domain is not evident from the published EM reconstructions. If the membrane association we detect indeed depends on insertion of these hydrophobic loops, then VP8* dissociation must be an additional consequence of EDTA-induced uncoating of VP7. We suggest in Fig. 8 (derived in part from the sequence of events proposed in Fig. 4 of reference 9) that exposure of the hydrophobic loops leads to membrane association of the antigen domains (state 3) and that subsequent formation of the stable trimer interface seen in VP5CT leads to the transition from state 3 to state 4. Aspects of this model—in particular, the importance of the hydrophobic loops for membrane insertion—can be tested by recoating rotavirus DLPs with VP7 and recombinant VP4 with mutations in these loops.
FIG. 8.

Model for membrane association of VP5*. The sequence of conformational rearrangements in the diagram begins with dissociation of VP8*, followed by formation of a transient, extended intermediate. The coiled coil (see Fig. 1) may form at this stage. The surface presented by the exposed hydrophobic loops allows the extended trimer to bind firmly to a membrane. This interaction enhances the yield of SDS-resistant VP5* trimers in our experiments. Folding back to the stable state represented by the VP5CT crystal structure (Fig. 1D)—the process proposed to drive membrane disruption (9)—results in a trimer conformation that can no longer associate with liposomes.
MAb 2G4, which binds the hydrophobic apex of VP5* and neutralizes virus (22, 24), prevents liposome binding by VP5* released from uncoating virions (Fig. 5). At neutralizing concentrations, 2G4 does not inhibit attachment to cells (21). It does prevent rotavirus-mediated 51Cr release from loaded cells (14), suggesting that it blocks membrane perforation. MAb 2G4 interference with VP5*-liposome interactions provides a biochemical correlate to observations of MAb 2G4 inhibition of steps during cell entry. The antibody inhibition data confirm that the interaction of VP5* with lipids observed in this cell-free system models events during cell entry and suggest that 2G4 may neutralize by interfering with VP5*-lipid interactions.
The proposed role of the membrane may also include potentiation or coordination of the VP5* rearrangement. The lower yield of slowly migrating (trimeric) VP5* when TLPs uncoat in the absence of membranes suggests that the lipid bilayer may stabilize a conformation that can undergo essentially quantitative foldback. Liposome binding also potentiates the fold-back rearrangement of flavivirus E glycoproteins (class II membrane fusion proteins of an enveloped-virus family) (18, 23).
Why does chymotryptic cleavage of VP4 not enhance infectivity? The data in Fig. 3 show that chymotrypsin digestion of VP4 incorporated into TLPs produces a VP5*-like fragment, VP5*C, which binds liposomes if uncoating occurs in their presence. If the bilayer is then dissolved, however, VP5*C cannot proceed to the stable trimeric end state and instead precipitates (unless solubilized by heating in SDS). The missing step in penetration of virions treated with chymotrypsin rather than trypsin is therefore likely to be the final rearrangement from state 3 to state 4. We further note that the apparently greater efficiency of liposome association of VP5*C (compared to the tryptic fragment, VP5*) is probably due either to a longer-lived intermediate or to precipitation (and hence disappearance from the assay) of any non-lipid-associated VP5*C species. These properties correlate with those of monomeric VP4 treated with chymotrypsin in solution. The VP5C fragment precipitates, but subsequent treatment with trypsin yields soluble VP5CT. The difference between VP5CT and VP5C (and between VP5* and VP5*C) is a short N-terminal segment (either residues 239 to 247 or residue 247 alone, depending on the thoroughness of chymotrypsin digestion), present in the latter but removed by trypsin (7). The N-terminal residues of VP5* probably need to reorganize during the dimer-to-trimer transition: they augment a sheet of the 2-fold partner in the dimer conformation and fold back against themselves in the trimer (26) (Fig. 1C and D). The extra residue(s) in VP5C and VP5*C may inhibit this rearrangement.
We have observed previously that the sequence of molecular events represented in Fig. 8 resembles the sequence of fusion-inducing conformational changes in the so-called “class II” fusion proteins of some enveloped viruses (e.g., flaviviruses and alphaviruses) (9, 13, 18, 23). In the present case, this sequence must produce a perforation of a bilayer rather than a fusion of two of them. To outline a specific mechanism for perforation, we will need to validate the proposed rearrangement and to determine whether it occurs with the foot of VP5* still anchored in the DLP or after VP5* has dissociated from the particle. Assuming that entry into the cytosol occurs following endocytosis into some sort of endosomal compartment, then either mechanism could in principle be effective.
Acknowledgments
We thank Marina Babyonyshev for technical assistance, John T. Patton for antibodies against VP6, Harry B. Greenberg for antibodies against VP4 and VP7, and Aaron Schmidt for helpful discussions.
This work was supported by NIH grant CA-13202 (to S.C.H.) and by NIH grant AI-053174 and an Ellison Medical Foundation New Scholars in Global Infectious Diseases Award (to P.R.D.). S.C.H. is an Investigator in the Howard Hughes Medical Institute.
Footnotes
Published ahead of print on 9 December 2009.
REFERENCES
- 1.Aoki, S. T., E. C. Settembre, S. D. Trask, H. B. Greenberg, S. C. Harrison, and P. R. Dormitzer. 2009. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 324:1444-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias, C. F., P. Romero, V. Alvarez, and S. Lopez. 1996. Trypsin activation pathway of rotavirus infectivity. J. Virol. 70:5832-5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnett, B. B., R. S. Spendlove, and M. L. Clark. 1979. Effect of enzymes on rotavirus infectivity. J. Clin. Microbiol. 10:111-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, J. Z., E. C. Settembre, S. T. Aoki, X. Zhang, A. R. Bellamy, P. R. Dormitzer, S. C. Harrison, and N. Grigorieff. 2009. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 106:10644-10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciarlet, M., J. E. Ludert, M. Iturriza-Gomara, F. Liprandi, J. J. Gray, U. Desselberger, and M. K. Estes. 2002. The initial interaction of rotavirus strains with N-acetyl-neuraminic (sialic) acid residues on the cell surface correlates with VP4 genotype, not species of origin. J. Virol. 76:4087-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford, S. E., S. K. Mukherjee, M. K. Estes, J. A. Lawton, A. L. Shaw, R. F. Ramig, and B. V. Prasad. 2001. Trypsin cleavage stabilizes the rotavirus VP4 spike. J. Virol. 75:6052-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dormitzer, P. R., H. B. Greenberg, and S. C. Harrison. 2001. Proteolysis of monomeric recombinant rotavirus VP4 yields an oligomeric VP5* core. J. Virol. 75:7339-7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dormitzer, P. R., H. B. Greenberg, and S. C. Harrison. 2000. Purified recombinant rotavirus VP7 forms soluble, calcium-dependent trimers. Virology 277:420-428. [DOI] [PubMed] [Google Scholar]
- 9.Dormitzer, P. R., E. B. Nason, B. V. Prasad, and S. C. Harrison. 2004. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430:1053-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dormitzer, P. R., Z.-Y. J. Sun, G. Wagner, and S. C. Harrison. 2002. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 21:885-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estes, M. K., D. Y. Graham, and B. B. Mason. 1981. Proteolytic enhancement of rotavirus infectivity: molecular mechanisms. J. Virol. 39:879-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiore, L., S. J. Dunn, B. Ridolfi, F. M. Ruggeri, E. R. Mackow, and H. B. Greenberg. 1995. Antigenicity, immunogenicity and passive protection induced by immunization of mice with baculovirus-expressed VP7 protein from rhesus rotavirus. J. Gen. Virol. 76:1981-1988. [DOI] [PubMed] [Google Scholar]
- 13.Gibbons, D. L., M. C. Vaney, A. Roussel, A. Vigouroux, B. Reilly, J. Lepault, M. Kielian, and F. A. Rey. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320-325. [DOI] [PubMed] [Google Scholar]
- 14.Kaljot, K. T., R. D. Shaw, D. H. Rubin, and H. B. Greenberg. 1988. Infectious rotavirus enters cells by direct cell membrane penetration, not by endocytosis. J. Virol. 62:1136-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li, Z., M. L. Baker, W. Jiang, M. K. Estes, and B. V. Prasad. 2009. Rotavirus architecture at subnanometer resolution. J. Virol. 83:1754-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ludert, J. E., N. Feng, J. H. Yu, R. L. Broome, Y. Hoshino, and H. B. Greenberg. 1996. Genetic mapping indicates that VP4 is the rotavirus cell attachment protein in vitro and in vivo. J. Virol. 70:487-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackow, E. R., R. D. Shaw, S. M. Matsui, P. T. Vo, M. N. Dang, and H. B. Greenberg. 1988. The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutralization and identification of a putative fusion region. Proc. Natl. Acad. Sci. U. S. A. 85:645-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313-319. [DOI] [PubMed] [Google Scholar]
- 19.Monnier, N., K. Higo-Moriguchi, Z. Y. Sun, B. V. Prasad, K. Taniguchi, and P. R. Dormitzer. 2006. High-resolution molecular and antigen structure of the VP8* core of a sialic acid-independent human rotavirus strain. J. Virol. 80:1513-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padilla-Noriega, L., R. Werner-Eckert, E. R. Mackow, M. Gorziglia, G. Larralde, K. Taniguchi, and H. B. Greenberg. 1993. Serologic analysis of human rotavirus serotypes P1A and P2 by using monoclonal antibodies. J. Clin. Microbiol. 31:622-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruggeri, F. M., and H. B. Greenberg. 1991. Antibodies to the trypsin cleavage peptide VP8* neutralize rotavirus by inhibiting binding of virions to target cells in culture. J. Virol. 65:2211-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw, R. D., P. T. Vo, P. A. Offit, B. S. Coulson, and H. B. Greenberg. 1986. Antigenic mapping of the surface proteins of rhesus rotavirus. Virology 155:434-451. [DOI] [PubMed] [Google Scholar]
- 23.Stiasny, K., S. L. Allison, J. Schalich, and F. X. Heinz. 2002. Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J. Virol. 76:3784-3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tihova, M., K. A. Dryden, A. R. Bellamy, H. B. Greenberg, and M. Yeager. 2001. Localization of membrane permeabilization and receptor binding sites on the VP4 hemagglutinin of rotavirus: implications for cell entry. J. Mol. Biol. 314:985-992. [DOI] [PubMed] [Google Scholar]
- 25.Trask, S. D., and P. R. Dormitzer. 2006. Assembly of highly infectious rotavirus particles recoated with recombinant outer capsid proteins. J. Virol. 80:11293-11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoder, J. D., and P. R. Dormitzer. 2006. Alternative intermolecular contacts underlie the rotavirus VP5* two- to three-fold rearrangement. EMBO J. 25:1559-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoder, J. D., S. D. Trask, P. T. Vo, M. Binka, N. Feng, S. C. Harrison, H. B. Greenberg, and P. R. Dormitzer. 2009. VP5* rearranges when rotavirus uncoats. J. Virol. 83:11372-11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, X., E. Settembre, C. Xu, P. R. Dormitzer, R. Bellamy, S. C. Harrison, and N. Grigorieff. 2008. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. U. S. A. 105:1867-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]