

Abstract
In the eukaryotic cell, DNA replication entails the interaction of multiple proteins with the DNA polymerase processivity factor PCNA. As the structure of the presumptive human cytomegalovirus (HCMV) DNA polymerase processivity factor UL44 is highly homologous to that of PCNA, we hypothesized that UL44 also interacts with numerous proteins. To investigate this possibility, recombinant HCMV expressing FLAG-tagged UL44 was generated and used to immunoprecipitate UL44 and associated proteins from infected cell lysates. Unexpectedly, nucleolin, a major protein component of the nucleolus, was identified among these proteins by mass spectrometry and Western blotting. The association of nucleolin and UL44 in infected cell lysate was confirmed by reciprocal coimmunoprecipitation in the presence and absence of nuclease. Western blotting and immunofluorescence assays demonstrated that the level of nucleolin increases during infection and that nucleolin becomes distributed throughout the nucleus. Furthermore, the colocalization of nucleolin and UL44 in infected cell nuclei was observed by immunofluorescence assays. Assays of HCMV-infected cells treated with small interfering RNA (siRNA) targeting nucleolin mRNA indicated that nucleolin was required for efficient virus production, viral DNA synthesis, and the expression of a late viral protein, with a correlation between the efficacy of knockdown and the effect on virus replication. In contrast, the level of neither global protein synthesis nor the replication of an unrelated virus (reovirus) was reduced in siRNA-treated cells. Taken together, our results indicate an association of nucleolin and UL44 in HCMV-infected cells and a role for nucleolin in viral DNA synthesis.
DNA polymerase is essential for the replication of DNA. Most replicative DNA polymerases include a catalytic subunit, required for DNA polymerization, and a processivity subunit that holds the catalytic subunit of the polymerase on DNA to permit continuous DNA synthesis and, in some cases, to interact with other proteins required for DNA synthesis as the need arises. For example, proliferating cell nuclear antigen (PCNA), the processivity factor of eukaryotic DNA polymerases δ and ɛ, is capable of numerous interactions with proteins that aid and abet DNA synthesis (33, 35). Human cytomegalovirus (HCMV) encodes a dimeric DNA polymerase, which includes the catalytic subunit UL54 and the presumptive processivity factor UL44. Previous studies of UL44 have revealed that UL44 forms a head-to-head homodimer (2) that has structural homology to PCNA (2, 3) and, like PCNA, can wrap around DNA (25). These results give rise to the hypothesis that UL44 can interact with multiple proteins involved in DNA synthesis.
Other than UL54 (12), to date, three viral proteins have been reported to associate with UL44 in the infected cell: the viral kinase UL97 (26, 34), the uracil DNA glycosylase UL114 (39, 40), and the DNA replication factor UL84 (14, 47). To investigate whether other viral and cellular proteins associate with UL44, a recombinant HCMV virus expressing FLAG-tagged UL44 was generated and used to immunoprecipitate UL44 and associated proteins from infected cell lysates. By using mass spectrometry (MS) analysis, a number of viral and cellular proteins were found to interact with FLAG-tagged UL44 in infected cell lysates. Unexpectedly, one of these proteins was nucleolin (Ncl), a DNA and RNA binding phosphoprotein found in the nucleolus of the cell that interacts with multiple cellular proteins and appears to have multiple functions in ribosome biogenesis, for example, ribosomal DNA (rDNA) transcription, rRNA maturation, and ribosome assembly (reviewed in reference 16). This led us to further investigate the UL44-nucleolin association and whether nucleolin is important for virus replication.
MATERIALS AND METHODS
Generation of the bacterial artificial chromosome AD169-BACFUL44 and virus FLAG44.
A single FLAG epitope (DYKDDDDK) was inserted between the first and second codons of the UL44 coding sequence in the bacterial artificial chromosome (BAC) AD169-BAC (20) by using the two-step Red recombination method, described previously by Tischer and coworkers (50), with Escherichia coli strain DY380 (29). Briefly, PCR primers FLAG44 Fw (5′-CGC CCG CTC CTT AGT CGA GAC TTG CAC GCT GTC CGG GAT GGA CTA CAA GGA TGA CGA CGA TAA GGA TCG CAA GTA GGG ATA ACA GGG TAA TCG ATT T-3′) and FLAG44 Rv (5′-GCG CCA GCG TCG GCG GCT CCG AGA GGC GCG TCT TGC GAT CCT TAT CGT CGT CAT CCT TGT AGT CCA TCC CGG GCC AGT GTT ACA ACC AAT TAA CC-3′) were used to amplify a DNA sequence from plasmid pEP-KanaS (50) consisting of an I-SceI-aphAI element flanked on either side by the part of the UL44 coding sequence containing the sequence for the FLAG tag. This PCR product was electroporated into DY380 cells harboring AD169-BAC, and Red recombination was induced to introduce the PCR product into AD169-BAC. Colonies were screened by restriction fragment analysis to confirm the recombination of the PCR product into the expected restriction fragment. The kanamycin resistance gene aphAI was removed by introducing plasmid pBAD-I-SceI (50) expressing I-SceI into bacteria. After heat shock, a second Red recombination event took place between the repeated UL44 sequences, which was stimulated by the presence of a double-strand break produced by I-SceI cleavage. Colonies were screened for their sensitivity to kanamycin. The resulting BAC AD169-BACFUL44 was sequenced to confirm the presence of the FLAG sequence in the UL44 coding sequence. AD169-BAC and AD169-BACFUL44 were electroporated into human foreskin fibroblast (HFF) cells (ATCC CRL-1684; American Type Culture Collection) as described previously (50), with plasmids pCGN71 (4), expressing the viral transcriptional transactivator pp71, and pBRep-Cre (20) to generate the viruses AD169rv and FLAG44, respectively.
Virus replication assays.
To assess HCMV replication, 1 × 105 HFF cells per well were seeded into 12-well plates 24 h before infection. At the time of infection, the indicated virus was added to each well at the multiplicity of infection (MOI) indicated in the text. After incubation for 1 h at 37°C, virus supernatant was removed and replaced with 1 ml of complete Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 5% fetal bovine serum (FBS) (Gibco). At the indicated time points, the medium from each well (virus supernatant) was taken from the cells and stored at −80°C until required. Dilutions of each virus supernatant were titrated simultaneously onto fresh monolayers of HFF cells to determine virus titers. To assess reovirus replication, purified reovirus type 1 Lang strain virions (particle/PFU ratio, ∼150) were diluted in 100 μl of attachment buffer (phosphate-buffered saline [PBS] with 2 mM MgCl2) and incubated with cell monolayers at the MOIs indicated in the text for 1 h at room temperature (RT). After the removal of unabsorbed virus by two washes with attachment buffer, cells were incubated in DMEM containing 10% FBS at 37°C for 16 h. Infected cells were lysed by freezing and thawing, and infectious titers of the lysates were measured by serial dilution onto L929 cell monolayers.
Western blotting.
Western blotting of proteins separated on 10% polyacrylamide gels was carried out as described elsewhere previously (48) by using monoclonal antibodies (MAbs) recognizing UL44 (1:1,000 dilution; Virusys), nucleolin (antibody 4E2, 1:200 dilution; Santa Cruz), Ku86 (antibody ab3107, 1:250 dilution; Abcam), Ku70 (antibody ab3114, 1:500 dilution; Abcam), pp28 (1:2,000 dilution; Virusys), IE86/72 (1:2,000 dilution; Virusys), and anti-UL97 antiserum (23) as primary antibodies. Goat anti-mouse-horseradish peroxidase (HRP)-conjugated antibody, goat anti-rabbit-HRP-conjugated antibody (both from Southern Biotech), and chemiluminescence solution (Pierce) were used to detect primary antibodies on film except for the experiment shown in Fig. 3, where anti-mouse TruBlot antibody conjugated to HRP (eBioscience), which recognizes the native but not the denatured form of the antibody, was used to detect primary antibodies recognizing UL44 and nucleolin. In all blots to detect FLAG-tagged protein, FLAG antibody M2 conjugated to HRP (1:1,000 dilution; Sigma) was used.
FIG. 3.

Reciprocal co-IP of UL44 and nucleolin. HFF cells were mock infected or infected with AD169 (MOI of 3). Cell lysates were prepared 72 h postinfection and precleared with immunoglobulin. IP of the lysate was then carried out with monoclonal antibodies recognizing either UL44 (A) or nucleolin (Ncl) (B) or immunoglobulin of the same isotype as the monoclonal antibody used. Proteins were analyzed by Western blotting using the monoclonal antibodies indicated to the right. The positions of molecular mass markers (kDa) are indicated to the left of A to C. (A) Lanes 1 and 2, uninfected cell lysate immunoprecipitated with immunoglobulin G (IgG) and anti-UL44 antibody (Ab), respectively; lanes 3 and 4, infected cell lysate immunoprecipitated with IgG and antibody (Ab), respectively; lane 5, infected cell lysate immunoprecipitated with MAb in the presence of benzonase (Ab+B); lane 6, infected cell lysate (Lys). (B) Lanes 1 and 2, uninfected cells immunoprecipitated with IgG and anti-nucleolin antibody (Ab), respectively; lanes 4 and 5, infected cells immunoprecipitated with IgG and antibody (Ab), respectively; lane 6, infected cell lysate immunoprecipitated with MAb in the presence of benzonase (Ab+B); lanes 3 and 7, uninfected and infected cell lysate (Lys), respectively. (C) Samples from lanes 5, 6, and 7 in B probed as described above with UL44 MAb. (D) The cell lysate used in the IP in A (lanes 4 and 5) was run on an ethidium bromide-stained 0.8% agarose gel. Lane 1, no sample; lane 2, IP in the absence of benzonase (A, lane 4); lane 3, IP in the presence of benzonase (A, lane 5). The position of the dye front, which corresponds to where a 0.5-kbp DNA would migrate, is indicated with an arrow.
IP of FLAG-tagged UL44 and analysis by MS and Western blotting.
Immunoprecipitation (IP) was carried out essentially as described previously (23). Briefly, 2.5 × 106 HFF cells were infected with AD169rv or FLAG44 in 100-mm dishes at an MOI of 3 PFU/cell in the presence or absence of 100 μM ganciclovir (GCV) diluted in dimethyl sulfoxide (DMSO). Where GCV was used, an equal volume of DMSO was added to control infections. At the indicated time points, plates were washed twice with PBS and lysed in 1 ml lysis buffer (50 mM HEPES-KOH [pH 7.4], 1% Triton X-100, 150 mM NaCl, 10% glycerol, 0.1 mM dithiothreitol [DTT], 2 mM EDTA) containing one Complete EDTA-free protease inhibitor tablet (Roche) per 50 ml at 4°C. For IP, 200 μl of clarified lysate was mixed with 40 μl of settled EZ-View anti-FLAG M2 affinity resin (Sigma) and incubated overnight at 4°C with rotation. Beads were then washed 4 times with 750 μl of lysis buffer for 20 min with rotation at 4°C. The protein was then eluted from beads by incubation in either 80 μl of 2× Laemmli buffer (27) at 95°C for 5 min or 100 μl Tris-buffered saline (TBS) containing 3× FLAG peptide (Sigma) at a final concentration of 150 ng/μl at 4°C with rotation for 30 min. The supernatant from the incubation with the FLAG peptide was mixed 1:1 with 2× Laemmli buffer and boiled at 95°C for 5 min.
For MS analysis, 30 μl of each sample was run on a 10% SDS-PAGE polyacrylamide gel. The gel was then stained with Simply Blue Safe stain (Invitrogen). Stained gel samples were submitted to the Taplin Mass Spectrometry Facility (Harvard Medical School) for liquid chromatography-tandem mass spectrometry (LC/MS/MS) analysis.
For Western blotting, 20 μl of protein eluted from the beads by boiling in Laemmli buffer was separated on a 10% SDS-PAGE gel, as was 20 μl of cell lysate mixed 1:1 with 2× Laemmli buffer, and boiled at 95°C for 5 min.
Reciprocal coimmunoprecipitation (co-IP).
A total of 3 × 105 HFF cells were infected at an MOI of 3 or mock infected and resuspended in 250 μl EBC2 buffer (50 mM Tris [pH 8.0], 300 mM NaCl, 0.5% NP-40). After clarification of the lysate, 20 μl protein A-Sepharose beads and 5 μg of the appropriate isotype control antibody (Bethyl Laboratories) were added, and the mixture was incubated at 4°C with rotation for 3 h. After centrifugation to remove beads, 20 μl of protein A-Sepharose beads was added with either 5 μg isotype control antibody, 5 μg UL44 monoclonal antibody (Virusys), or 5 μg nucleolin antibody 4E2 (Santa Cruz). After incubation overnight at 4°C with rotation, beads were spun down, and the supernatant was removed. Beads were washed 4 times with 1 ml EBC2 buffer and resuspended in 20 μl 2× Laemmli buffer. Where indicated, 400 U benzonase (Novagen) was added after clarification of the lysate by centrifugation. Twenty microliters of each sample was run on a 10% polyacrylamide gel for Western blotting. Where indicated, the supernatant used for the IP was mixed 1:1 (vol/vol) with 6× gel loading buffer, and 50 μl was run on a 0.8% agarose gel containing 100 μg/ml ethidium bromide.
Immunofluorescence.
A total of 5 × 104 HFF cells were plated onto glass coverslips. Cells were mock infected and fixed at room temperature with freshly made 4% paraformaldehyde in Dulbecco's phosphate-buffered saline (DPBS) and/or infected with AD169 (MOI of 3) and fixed with 4% paraformaldehyde at the time points indicated in the text. After washing in DPBS, cells were permeabilized with 0.3% Triton X-100 dissolved in DPBS at RT for 10 min. Once the cells were washed again with DPBS, they were incubated in 0.5% bovine serum albumin (BSA) dissolved in DPBS for 20 min at RT. Rabbit anti-nucleolin polyclonal antiserum (Abcam) or mouse anti-UL44 antiserum (Virusys) was applied as indicated in the text and incubated for 1 h at 37°C. Antiserum was removed by washing cells three times with DPBS, each time for 5 min with rocking. This procedure was repeated for the secondary antibodies in which fluorescently labeled Alexa Fluor 488 goat anti-rabbit or Alexa Fluor 594 goat anti-mouse antibody (Molecular Probes) was used. After incubation and washing, coverslips were mounted onto microscope slides with ProLong Antifade reagent (Invitrogen-Molecular Probes). Images were acquired by using an inverted spinning-disk confocal microscope (Axiovert 200 M; Carl Zeiss) linked to a computer-controlled spherical aberration correction device. Images shown were obtained by acquiring sequential optical planes spaced by 0.15 μm in the z axis and projecting those planes using Slidebook 4.2.
Transfection of siRNA into HFF cells.
The transfection of HFF cells with small interfering RNA (siRNA) was carried essentially as described elsewhere previously (55). Briefly, 1 × 105 HFF cells per well were seeded into 12-well plates 24 h before transfection. siControl nontargeting siRNA pool or ON-TARGETplus SMARTpool human Ncl (both from Dharmacon) was used. Where indicated, the four siRNAs that comprise the pool of siRNA in the ON-TARGETplus SMARTpool human Ncl pool were also transfected individually. Per well, 3 μl 20 μM siRNA and 3 μl Lipofectamine (Invitrogen) were diluted in 50 μl and 12 μl Optimem (Invitrogen), respectively. After 5 min at room temperature, both solutions were combined. After 20 min, 400 μl prewarmed Optimem was added to each transfection reaction mixture. The cell culture medium was removed, and the transfection mixture was added. At 6 h posttransfection, siRNA-containing medium was removed, and cells were washed twice with 1 ml of prewarmed complete DMEM containing 5% FBS. Transfected cells were then incubated in 1 ml of complete DMEM containing 5% FBS, and transfection was repeated twice more at 24-h intervals. Twenty-four hours after the third round of transfection, cells were either infected, pulsed with [35S]methionine, or prepared for analysis by Western blotting as described below.
Analysis of protein synthesis in siRNA-transfected HFF cells.
After transfection with siRNA, complete DMEM (Gibco) containing 5% FBS was removed, and cells were incubated in 1 ml minimal essential medium lacking methionine (Gibco) supplemented with l-cysteine and l-glutamine in the presence or absence of 200 μg/ml cycloheximide. After 1 h of incubation, 50 μCi [35S]methionine (Perkin-Elmer) was added to each well, and cells were incubated for a further 2 h. Media were removed, and cells were harvested in 100 μl 2× Laemmli buffer and boiled at 95°C for 5 min. Five microliters of each sample was used for analysis on a 10% polyacrylamide gel. Gels were then dried and exposed to a phosphorimager screen for 24 h.
Real-time quantitative PCR analysis of viral DNA synthesis.
Real-time PCR analysis of viral DNA was carried as described elsewhere previously (41). Briefly, DNA was isolated from infected siRNA-transfected HFF cells by using a DNeasy tissue kit (Qiagen) according to the manufacturer's instructions. Viral genomes were quantified with a primer pair (pp549s and pp812as) and a probe (pp770s) for UL83 (15), and the number of viral genomes was normalized to the number of cellular copies of β-actin with a previously described set of primers and probe (18). Unknown sample values were determined on the basis of a standard curve of known copy numbers of UL83 (AD169-BAC) and β-actin (pAB1-bactinPCRscript) (a kind gift from R. Kaletja, University of Wisconsin). PCR mixtures contained 5 μl of 200 μl extracted DNA, 50 nM primers, 50 nM probe, 12.5 μl TaqMan Universal PCR master mix (Applied Biosystems), and nuclease-free water (Ambion) to 25 μl. Real-time PCR was run by using an ABI 7900HT device, and data were analyzed by use of the SDS 2.2.1 program.
RESULTS
Generation and characterization of recombinant HCMV expressing FLAG-tagged UL44.
By use of MS, we previously identified a subset of proteins associated with UL44 by immunoprecipitating UL44 from lysates of cells infected with HCMV at a single time point postinfection (p.i.) using a monoclonal antibody (MAb) recognizing UL44 (47). In that study, we confined our MS analysis to only a few prominent bands in the immunoprecipitate. To more completely investigate what proteins associate with UL44, we first generated recombinant HCMV expressing FLAG-tagged UL44 so that protein complexes containing UL44 in the infected cell lysate could be isolated by immunoprecipitation (IP) utilizing an antibody recognizing the FLAG epitope. This method avoids interference between interacting proteins and an anti-UL44 MAb. By using Red two-step recombination (50), a single FLAG epitope (DYKDDDDK) was inserted between the first and second codons of the UL44 coding sequence in the bacterial artificial chromosome (BAC) AD169-BAC (20). This new BAC was termed AD169-BACFUL44. AD169-BAC and AD169-BACFUL44 were then transfected into HFF cells to generate the viruses AD169rv and FLAG44, respectively.
To determine if the insertion of the FLAG epitope into the UL44 sequence affects virus replication, one-step growth curves of AD169rv and FLAG44 infection were analyzed (Fig. 1A). The replication of FLAG44 in HFF cells was comparable to that of AD169rv. Levels of the UL44 protein in lysates of cells infected with either virus at different time points postinfection were assayed by Western blotting using a MAb recognizing UL44. The molecular mass of UL44 is predicted to be 46 kDa, and a similar amount of protein of this apparent molecular mass could be detected in lysates from cells infected with either virus (Fig. 1B). A slight decrease in the mobility of FLAG-tagged UL44 in the gel compared to that of the UL44 protein was also noted, presumably since the addition of the FLAG tag increases the molecular mass of UL44. The infected cell lysate was also analyzed by Western blotting using a monoclonal antibody recognizing FLAG to detect the FLAG-tagged UL44 protein (Fig. 1C). A single band corresponding to the molecular mass of FLAG-tagged UL44 could be observed in the FLAG44-infected cell lysate but not the AD169rv-infected cell lysate, confirming the addition of the FLAG epitope to UL44. Taken together, these results indicate that the incorporation of the FLAG epitope into UL44 does not affect virus replication or levels of the UL44 protein in infected cells.
FIG. 1.
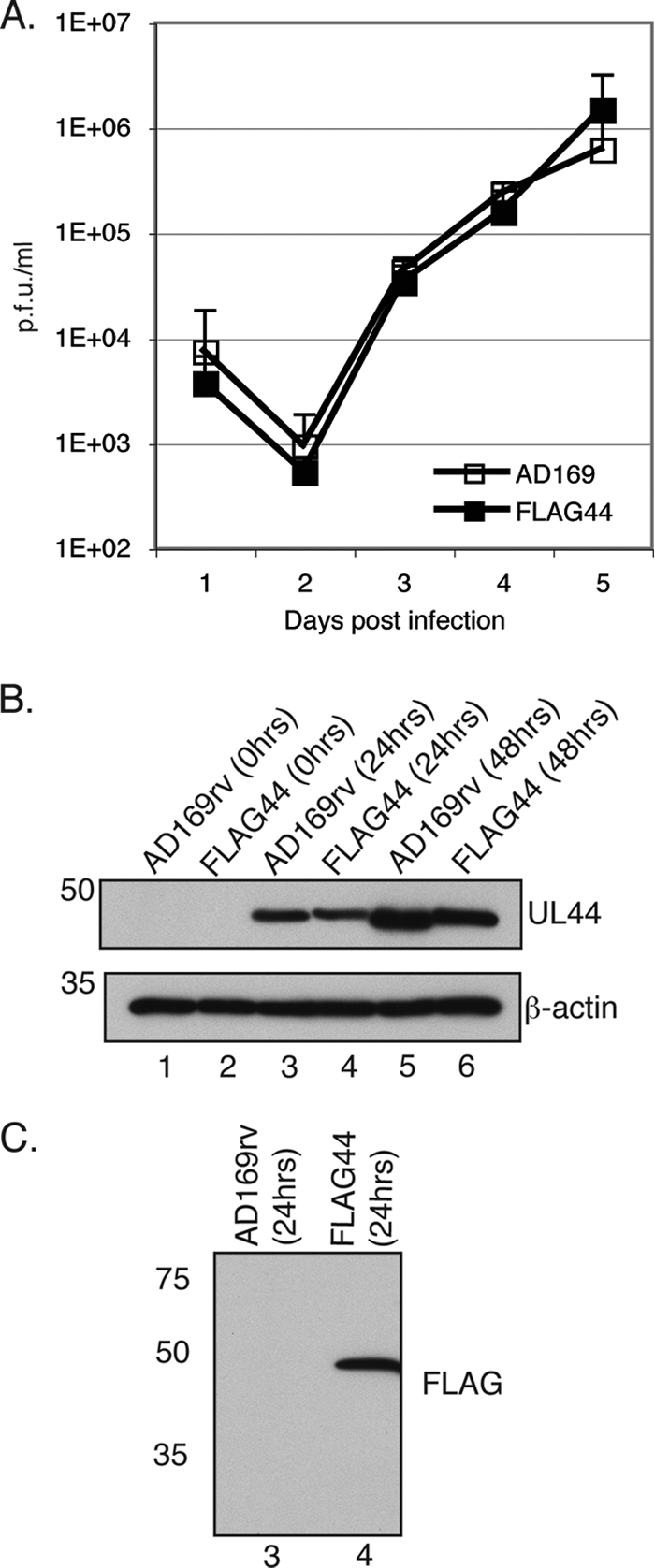
Characterization of FLAG44 virus. (A) Replication of AD169rv and FLAG44 viruses. HFF cells were infected at an MOI of 1, and virus supernatant was harvested at the indicated time points. The virus titer is represented as PFU/ml on HFF cells. Data points (open squares, AD169rv; closed squares, FLAG44) represent the mean titers for three experiments. Error bars represent the standard deviations for the titers from those experiments. (B) Western blotting of AD169rv- and FLAG44-infected cells. HFF cells were infected at an MOI of 1, and cell lysates were prepared at the indicated time points. UL44 (top) and β-actin (bottom) protein levels were assayed by Western blotting using antibodies recognizing these proteins. The positions of molecular mass markers (kDa) are indicated on the left. (C) Detection of FLAG-tagged UL44 by Western blotting. The cell lysate analyzed in lanes 3 and 4 of B was probed with an anti-FLAG antibody. The positions of molecular mass markers (kDa) are indicated on the left.
MS analysis of proteins associating with FLAG-tagged UL44 in infected cell lysates.
To identify proteins associating with FLAG-tagged UL44 in infected cell lysates, a series of experiments was conducted where proteins were immunoprecipitated from AD169rv- and FLAG44-infected cell lysates by using a MAb recognizing FLAG conjugated to Sepharose beads. Cell lysates were prepared at both 72 and 120 h p.i. to determine if different proteins associate with UL44 at different time points during viral DNA replication. Cell lysate was also prepared from infected cells treated with the nucleoside analogue ganciclovir (GCV), which inhibits HCMV DNA synthesis (51), to investigate if viral DNA synthesis is required for the association with UL44. Immunoprecipitated proteins were removed from the beads by boiling in Laemmli buffer or eluted from beads in the presence of the FLAG peptide and then separated by SDS-PAGE. Each lane was excised in its entirety and cut into fragments. After tryptic digestion, peptides were subjected to liquid chromatography-tandem MS (LC/MS/MS) analysis to determine their sequences and thus identify the proteins from which they were derived. In each experiment viral and cellular proteins could be detected in protein immunoprecipitated from the FLAG44-infected cell lysate, which were not found in protein immunoprecipitated from AD169rv-infected cell lysate; nearly all of these proteins were not found in our previous study (47). Of these proteins, those that could be identified in at least one experiment by the detection of 4 or more peptides are shown in Table 1. The full lists of all proteins immunoprecipitated from the FLAG44-infected cell lysate but not the AD169-infected cell lysate in experiments 1 to 4 are provided in supplemental Tables 1 to 4, respectively, at http://coen.med.harvard.edu.
TABLE 1.
Proteins identified by mass spectroscopy of proteins immunoprecipitated from FLAG44-infected cell lysates
| Protein identifieda | GenBank accession no. | No. of peptides identified in expt no. |
|||||
|---|---|---|---|---|---|---|---|
| 1b at 72 h p.i. | 2b |
3 at 72 h p.i. withb: |
4c at 72 h p.i. | ||||
| 72 h p.i. | 120 h p.i. | DMSO | GCV | ||||
| Viral | |||||||
| UL54 | NP_039988 | 30 | 22 | 21 | 25 | 42 | 13 |
| UL44 | NP_039978 | 15 | 22 | 22 | 17 | 15 | 10 |
| IRS1 | NP_040085 | 6 | 7 | 19 | 12 | ND | ND |
| UL25 | NP_039959 | 4 | 3 | 1 | 8 | ND | ND |
| UL84 | NP_040019 | 1 | 1 | 4 | 7 | ND | ND |
| Cellular | |||||||
| Nucleolin | P19338 | 18 | 18 | 19 | 20 | 6 | 7 |
| DEAH box protein 9 | Q08211 | NDd | 5 | 10 | 7 | ND | 2 |
| RNA helicase DDX3X | O00571 | ND | 9 | 4 | ND | 1 | ND |
| Cytoskeleton-associated protein 4 variant | Q53es6 | 12 | ND | 11 | ND | ND | 4 |
| DEAH box protein 36 | Q9h2u1 | ND | ND | ND | 4 | 1 | ND |
| ATP-dependent DNA helicase II (70-kDa subunit) (Ku70) | NP_001460 | 8 | 3 | 8 | 8 | ND | ND |
| 60S acidic ribosomal protein p0 | P05388 | 7 | 6 | 6 | 7 | ND | ND |
| 60S ribosomal protein l13 | P26373 | 7 | 4 | 6 | 6 | ND | ND |
| 40S ribosomal protein s9 | P46781 | 7 | 7 | 6 | 10 | ND | ND |
| Ribosome binding protein 1 | Q9p2e9 | 7 | ND | ND | ND | ND | ND |
| 60S ribosomal protein l7a | P62424 | 6 | 4 | 7 | 7 | ND | ND |
| 60S ribosomal protein l3 | P39023 | 6 | 5 | 7 | 7 | ND | ND |
| 60S ribosomal protein l4 | P36578 | 5 | 9 | 14 | 5 | ND | ND |
| 60S ribosomal protein l7 | P18124 | 6 | 5 | 6 | 8 | ND | ND |
| DEAH box protein 30 | Q7l2e3 | 6 | 6 | 5 | 4 | ND | ND |
| 60S ribosomal protein l6 | Q02878 | 5 | 6 | 8 | 4 | ND | ND |
| Prolyl 4-hydroxylase alpha-1 subunit precursor | P13674 | 5 | 4 | 1 | ND | 1 | ND |
| Interleukin enhancer-binding factor 3 | Q12906 | 4 | ND | ND | 6 | ND | ND |
| 40S ribosomal protein s2 | P15880 | 4 | 4 | 6 | 5 | ND | ND |
| 40S ribosomal protein s4 | P22090 | 4 | 2 | 2 | 5 | ND | ND |
| Ribosomal protein l8 | Q567q7 | ND | 3 | 3 | 4 | ND | ND |
| 60S ribosomal protein l5 | P46777 | 4 | ND | ND | 2 | ND | ND |
| 60S ribosomal protein l13a | P40429 | 4 | 2 | 6 | 3 | ND | ND |
| 60S acidic ribosomal protein p2 | P05387 | 4 | ND | 2 | ND | ND | ND |
| Insulin-like growth factor 2 mRNA binding protein 2 | Q9y6m1 | 4 | ND | 1 | ND | ND | ND |
| 40S ribosomal protein s6 | P62753 | 3 | 1 | 3 | 4 | ND | ND |
| Coatomer subunit alpha | P53621 | ND | 1 | 2 | ND | ND | 5 |
| Ig alpha-1 chain C region | P01876 | ND | ND | ND | ND | ND | 5 |
| Heat shock cognate 71 kDa | P11142 | ND | ND | ND | ND | ND | 4 |
| 40S ribosomal protein s15a | P62244 | 3 | 5 | 5 | 4 | ND | ND |
| Ribosomal protein s19 | Q8wvx7 | ND | ND | ND | 5 | ND | ND |
| RPS11 protein | Q498y6 | ND | ND | ND | 5 | ND | ND |
| ATP-dependent DNA helicase II (86-kDa subunit) (Ku86) | NP_066964 | 3 | 3 | 6 | 7 | ND | ND |
| RNA helicase DDX1 | Q92499 | ND | 4 | 2 | 3 | ND | ND |
| Nucleophosmin | P06748 | 3 | ND | 4 | 3 | ND | ND |
| Tubulin beta-1 chain | Q9h4h7 | ND | 5 | ND | ND | ND | ND |
| IgG1 light chain | S83373 | 1 | 5 | ND | 5 | ND | ND |
| DNA binding protein A | P16989 | ND | 4 | ND | 1 | ND | ND |
| Tubulin beta-6 chain | Q9buf5 | ND | 4 | ND | ND | ND | ND |
| Ubiquitin-associated protein 2-like CDMC5L | Q14157 | ND | ND | 4 | ND | ND | ND |
| Vimentin | NP_003371 | ND | 3 | ND | ND | 16 | ND |
| Ribosomal protein l17 | Q6nz54 | 2 | 4 | 3 | ND | ND | ND |
| 40S ribosomal protein s23 | P62266 | 1 | ND | ND | 5 | ND | ND |
| DNA topoisomerase II alpha subunit | AAI40792 | 1 | 1 | 5 | 5 | ND | ND |
| otthump00000016411 | Q5t8w0 | 1 | 1 | 1 | 6 | ND | ND |
| Plasminogen activator inhibitor 1 RNA binding protein | Q8nc51 | 1 | 4 | ND | ND | ND | ND |
Viral and cellular proteins identified by the detection of 4 or more peptides derived from them in any experiment.
Proteins were removed from beads for separation by SDS-PAGE by boiling.
Proteins were removed from beads for separation by SDS-PAGE by elution with the FLAG peptide.
ND, not detected.
As expected, peptides from UL44 and UL54 were abundantly detected in each experiment. UL44 peptides were found in three different sections of the gel, which could be due to posttranslational modifications of UL44, incomplete resolution during electrophoresis, or cross-contamination of gel fragments. Three other viral proteins, IRS1, UL25, and, as anticipated, UL84 (47), were detected in all experiments except when infected cells were treated with GCV (experiment 3) (Table 1) or when the protein was eluted by using FLAG peptides (experiment 4) (Table 1). Thus, similar repertoires of viral proteins were detected at 72 and 120 h p.i., but the association of UL84, IRS1, and UL25 with UL44 may require viral DNA synthesis. Why these proteins could not be detected when immunoprecipitated protein was eluted with the FLAG peptide (experiment 4) (Table 1) is unclear. The viral proteins UL97 and UL114 were previously reported to associate with UL44 (26, 34, 39, 40); however, peptides from UL114 could not be found in any experiment, and a single peptide from UL97 was detected in only one experiment (120 h p.i.) (see supplemental Table 2 at http://coen.med.harvard.edu).
In all experiments, peptides from a large number of cellular proteins could be detected (Table 1 and see supplemental Tables 1 to 4 at http://coen.med.harvard.edu). Again, there did not appear to be different repertoires of proteins associating with UL44 at 72 and 120 h p.i. The vast majority of the cellular proteins were not detected in IP of FLAG-tagged UL44 from lysates of GCV-treated cells (experiment 3) (Table 1). It is possible that that their association with UL44 may be dependent upon viral DNA synthesis; however, as the lack of viral DNA replication decreases UL44 levels in the infected cell (data not shown), it is possible that a number of these proteins are present below the level of detection of this assay. Also, these proteins were not identified when immunoprecipitated protein was eluted from beads by using the FLAG peptide. Peptides from only one protein, nucleolin, could be detected in all experiments, and in each experiment, more peptides derived from nucleolin could be detected than from any other cellular protein.
A number of peptides derived from both subunits of ATP-dependent helicase II (Ku70 and Ku86), which were previously reported to immunoprecipitate with UL44 and UL84 (14), could be also detected in the protein immunoprecipitated from the FLAG44-infected cell lysate (Table 1).
Identification of proteins immunoprecipitated from FLAG44-infected cells by Western blotting.
To further investigate the apparent associations of certain proteins with UL44, proteins immunoprecipitated from AD169rv- and FLAG44-infected cell lysates were examined by Western blotting (Fig. 2). By using a MAb recognizing nucleolin, a band with a molecular mass of approximately 100 to 110 kDa plus a band with a molecular mass of approximately 77 kDa (indicated with an arrow) in protein immunoprecipitated from the FLAG44-infected cell lysate (Fig. 2, lane 4), but not the AD169rv-infected cell lysate (lane 3), were detected. The molecular mass of nucleolin is predicted to be 77 kDa, but it is known to be extensively posttranslationally modified by phosphorylation and usually appears as a band with a molecular mass of approximately 100 to 110 kDa (16). The bands detected in the immunoprecipitated protein therefore most likely correspond to the unmodified and posttranslationally modified forms of nucleolin (16), which suggests that a posttranslational modification of nucleolin need not occur for the association with FLAG-tagged UL44.
FIG. 2.
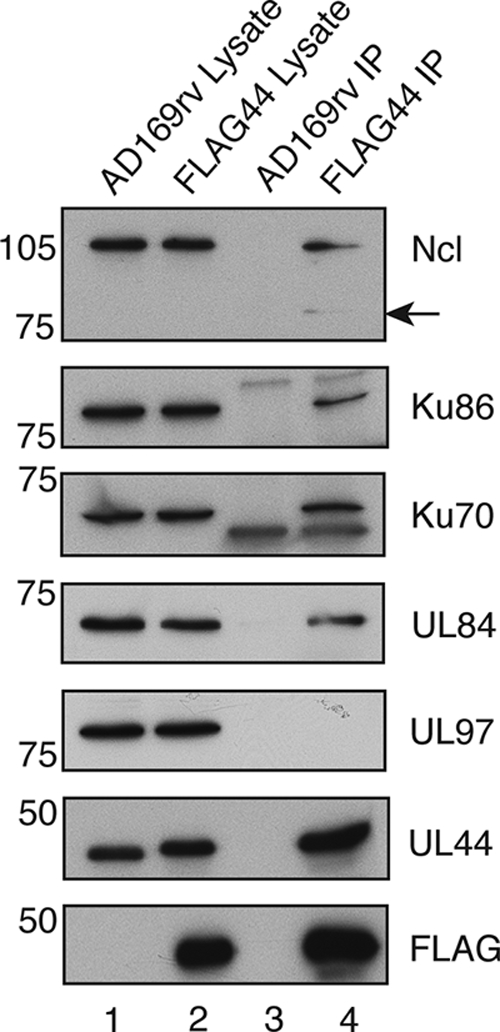
Detection of protein immunoprecipitated from FLAG44-infected cell lysates by Western blotting. Lysates from AD169rv (lane 1)-and FLAG44 (lane 2)-infected cells and protein immunoprecipitated using an anti-FLAG antibody from those lysates (lanes 3 and 4, respectively) were separated on a 10% polyacrylamide gel. Proteins in each lane were examined by Western blotting for the presence of UL44, FLAG, Ku86, Ku70, and nucleolin (Ncl) by using antibodies recognizing these proteins as indicated to the right. The positions of molecular mass markers (kDa) are indicated to the left. Arrow indicates the detection of a 77-kDa band.
The presence of Ku70 and Ku86 in the FLAG44-infected cell lysate was also confirmed by using a MAb that recognizes these proteins. Both proteins could be detected in protein immunoprecipitated from the FLAG44-infected cell lysate (Fig. 2, lane 4) but not the AD169rv-infected cell lysate (lane 3).
The blot was also probed with MAbs recognizing UL84 and UL97. UL84 could be detected with UL84 MAb in protein immunoprecipitated from the FLAG44-infected cell lysate (Fig. 2, lane 4) but not in protein immunoprecipitated from the AD169rv-infected cell lysate (lane 3), further confirming the UL44/UL84 association in infected cell lysates that was observed previously (14, 47). UL97, which was not reliably detected in immunoprecipitated protein by MS (Table 1), could not be detected in protein immunoprecipitated from either FLAG44- or AD169rv-infected cell lysates, confirming the data shown in Table 1 and indicating that in our assays, proteins most likely do not nonspecifically bind FLAG-tagged UL44 or beads bearing FLAG antibody.
Reciprocal co-IP of UL44 and nucleolin in the presence and absence of nuclease.
To investigate whether nucleolin can associate with UL44 lacking a FLAG tag in infected cell lysates, IP was carried out on lysates from HFF cells infected with HCMV strain AD169 or uninfected HFF cell lysates using either a MAb recognizing UL44 (Fig. 3A), a MAb recognizing nucleolin (Fig. 3B), or a control antibody of the same isotype as the accompanying MAb (Fig. 3A and B). Immunoprecipitated protein was examined by Western blotting using antibodies recognizing UL44 and nucleolin (Fig. 3). UL44 could be observed in infected cell lysates and in the proteins immunoprecipitated from infected cell lysates with UL44 MAb (Fig. 3A, bottom, lanes 4 and 6) but not in uninfected cell lysates, the proteins immunoprecipitated from uninfected cell lysates, or lysates immunoprecipitated with isotype control antibody. The 100- to 110-kDa band of nucleolin could be seen in infected cell lysates and in the proteins immunoprecipitated from infected cell lysates with UL44 MAb (Fig. 3A, top, lanes 4 and 6) but not in the proteins immunoprecipitated from uninfected cell lysates or lysates immunoprecipitated with isotype control antibody (Fig. 3A, top, lanes 2 and 3). The 77-kDa form of the protein shown in Fig. 2B could not be detected (not shown), perhaps due to its low abundance.
When IP was carried out using nucleolin MAb, both the posttranslationally modified (approximately 100- to 110-kDa) and unmodified (77-kDa) forms of nucleolin could be observed in proteins immunoprecipitated from uninfected and infected cell lysates with nucleolin MAb (Fig. 3B, top, lanes 2 and 5), while only the posttranslationally modified (approximately 100- to 110-kDa) form was observed in uninfected and infected cell lysate (Fig. 3B, top, lanes 3 and 7). Nucleolin could not be found in protein immunoprecipitated from uninfected and infected cell lysates with isotype control antibody (Fig. 3B, top, lanes 1and 4). It was also noted that larger amounts of nucleolin could be seen in cell lysates prepared from infected cells (lane 7) than in lysates prepared from uninfected cells (lane 3). When the blot was probed with UL44 MAb, UL44 could be found only in protein immunoprecipitated from infected cell lysates with nucleolin MAb (Fig. 3B, bottom, lane 5). Taken together, the data shown in Fig. 3A and B demonstrate reciprocal co-IP of UL44 and nucleolin, confirming their association in infected cell lysates from AD169-infected cells in the absence of the FLAG tag.
UL44 and nucleolin are both known to bind DNA (16, 30, 31, 54). DNA binding proteins can associate during IP due to their adjacent binding on DNA rather than due to protein-protein interactions (28, 49). To determine if nucleic acid is required for the association of UL44 and nucleolin in cell lysates during IP, IP was also carried out in the presence of the nonspecific nuclease benzonase. To confirm the action of benzonase on nucleic acid, the cell lysates analyzed in Fig. 3A were examined on an ethidium bromide-stained agarose gel (Fig. 3D). In the absence of benzonase, a robust staining of nucleic acid could be seen (Fig. 3D, lane 2), whereas in the presence of benzonase (lane 3), there was little, if any, more staining than that for a no-sample control (lane 1), indicating that benzonase had efficiently degraded the nucleic acid in the cell lysate. The presence of benzonase did not appear to alter the levels of UL44 or nucleolin that could be detected by Western blotting of protein immunoprecipitated from infected cell lysates with UL44 MAb (Fig. 3A, lanes 4 and 5) but did decrease the amount of UL44 that could be detected by Western blotting of proteins immunoprecipitated from infected cell lysates using nucleolin MAb (Fig. 3B, lanes 5 and 6). To ensure that the UL44 protein seen in lane 6 of Fig. 3B is not due to spillage from neighboring lanes of the gel, samples were blotted again with empty lanes between samples (Fig. 3C). UL44 could be observed in the protein immunoprecipitated in the presence of benzonase (Fig. 3C, lane 6) albeit less than in the absence of benzonase (lane 5). These data may indicate that during IP of nucleolin from infected cell lysates, some UL44 associates with nucleolin by binding to nucleic acid, while during IP of UL44, little, if any, nucleolin is bound adventitiously via interactions with nucleic acid. Nevertheless, taken together, the results presented in Fig. 3 indicate that there is an association between UL44 and nucleolin in infected cell lysates that is not due to the simultaneous binding of these proteins to DNA during IP.
We went on to investigate the association of UL44 with Ku70 and Ku86 in AD169-infected cell lysates. The samples shown in Fig. 3A were probed with MAbs recognizing either Ku86 or Ku70. Ku86 and Ku70 could be readily detected in infected cell lysates (Fig. 3A, middle, lane 6) but not in immunoprecipitated protein (Fig. 3A, middle, lanes 4 and 5), thus failing to confirm the association of UL44 and Ku70 or Ku86 under these conditions. It is possible that the association between FLAG-tagged UL44 and the Ku proteins observed in Fig. 2 occurs via the FLAG tag on UL44. We therefore confined our subsequent investigations to nucleolin.
Nucleolin levels increase during infection.
As noted above, larger amounts of nucleolin could be seen in cell lysates prepared from infected cells (Fig. 3B, lane 7) than in lysates prepared from uninfected cells (Fig. 3B, lane 3). To confirm this observation, lysates prepared from infected and uninfected cells at different time points were examined by Western blotting using MAbs recognizing nucleolin and UL44 (Fig. 4A). UL44 could also be detected in increasing amounts from 24 to 72 h p.i. (lanes 2, 4, and 6). Similar levels of nucleolin were observed in both infected and uninfected cells at 24 h p.i. (lanes 1 and 2); however, at 48 and 72 h p.i. (lanes 3 to 6), higher levels of nucleolin were detected in infected cells than in uninfected cells, confirming the observations made from the data shown in Fig. 3.
FIG. 4.
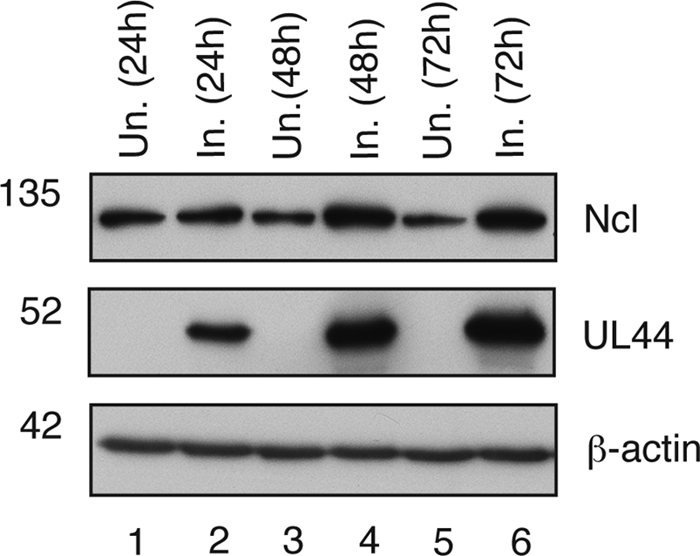
Analysis of nucleolin levels in infected and uninfected cells. Shown are nucleolin levels at different time points postinfection. Lysates from HFF cells uninfected (Un.) and infected (In.) with AD169 (MOI of 1) were prepared at the indicated time points and analyzed by Western blotting using antibodies recognizing nucleolin (Ncl), UL44, or β-actin, as indicated to the right. The positions of molecular mass markers (kDa) are indicated to the left.
Localization of nucleolin in HCMV-infected cells.
Nucleolin is found predominantly in the nucleolus of the cell but is dispersed from the nucleolus upon herpes simplex virus type 1 (HSV-1) infection (5, 10, 32). The localization of nucleolin and UL44 during HCMV infection was assayed by immunofluorescence (IF) (Fig. 5). Images were obtained by acquiring and merging sequential optical planes in the z axis. In uninfected cells (Fig. 5, panel 1), concentrated foci of nucleolin (green staining) in nucleoli (white arrows) were observed in the nucleus of the cell, with fainter staining elsewhere in the nucleus and even less staining in the cytoplasm. By 48 h p.i., the intensity of nucleolin staining increased throughout the cell (Fig. 5, panel 4). By 72 and 96 h p.i. (Fig. 5, panels 7 and 10, respectively), the cytoplasmic staining of nucleolin was less than that observed at 48 h p.i.; however, levels of nucleolin throughout the nucleus remained considerably higher than those seen for uninfected cells, consistent with our observations that nucleolin levels increase in HCMV-infected cells (Fig. 3 and 4). Anti-UL44 staining of uninfected cells yielded only occasional punctate staining in the cytoplasm of cells (Fig. 5, panel 2). UL44 staining could be observed by 48 h p.i. and persisted through 96 h p.i, marking the nucleus where it was found predominantly in viral replication compartments. Cells that had not been exposed to antiserum recognizing nucleolin or UL44 at all time points showed no obvious fluorescence (not shown), indicating that the staining seen in uninfected and infected cells is not due to a nonspecific cross-reaction of the secondary antibody with a viral or cellular protein. When the images of infected cells stained for nucleolin and UL44 were merged (Fig. 5, panels 6, 9, and 12), speckles of colocalized nucleolin and UL44 could be observed scattered within viral replication compartments from 48 h p.i. onwards. In HCMV-infected cells, therefore, nucleolin is distributed throughout the nucleus of the cell and not restricted to one particular compartment of the nucleus. The colocalization of nucleolin with UL44 occurs within viral replication compartments in the nucleus.
FIG. 5.

Localization of nucleolin in HCMV-infected cells. HFF cells uninfected (panels 1 to 3) or infected with AD169 (MOI of 3) (panels 4 to 12) were fixed at different time points postinfection (indicated to the right) and stained with antisera recognizing nucleolin (Ncl) and UL44 and secondary antibody conjugated to green (Ncl) or red (UL44) fluorophores. Images were obtained by acquiring and merging sequential optical planes on the z axis. (Left) Cells stained to determine the positions of nucleolin at the indicated time points. (Middle) Cells stained to determine the positions of UL44 at the indicated time points. (Right) Images from the left and middle columns merged. The positions of nucleoli are indicated by arrows.
FIG. 10.

Virus production and viral DNA synthesis in HFF cells transfected with siRNA. In three separate experiments, HFF cells were transfected with nucleolin (Ncl) siRNA or control siRNA and then infected with AD169 (MOI of 1). (A) At 96 h postinfection, the virus titer was determined by titrating virus on HFF cells. Virus titer is represented as PFU/ml. (B) Viral DNA synthesis in each experiment was determined by quantitative real-time PCR. The amount of viral DNA assayed is represented as copies of the viral gene UL83 per copy of the cellular gene β-actin.
FIG. 9.

Levels of viral and cellular proteins during infection of cells containing nucleolin siRNA. (A) Time course of protein levels. HFF cells were transfected with pools of nucleolin siRNA or control siRNA and then infected with AD169 (MOI of 1). Cell lysates were prepared at the indicated time points, and proteins from these lysates were separated on a 10% SDS-polyacrylamide gel. Levels of viral and cellular proteins were assayed by Western blotting using antibodies recognizing immediate-early proteins, UL44, pp28, nucleolin (Ncl), and β-actin, as indicated to the right. (B) Determination of relative protein levels in infected cells at 72 h p.i. A 2-fold dilution series of protein from lane 5 of A (lanes 1 to 3) was analyzed by Western blotting using antibodies recognizing nucleolin (Ncl) and pp28, as indicated to the right, compared to undiluted protein from lane 6 of A. The positions of molecular mass markers (kDa) are indicated to the left.
Efficient HCMV replication in HFF cells requires nucleolin.
To investigate whether nucleolin is important for the replication of HCMV, HFF cells were transfected with either a pool of 4 siRNA molecules recognizing nucleolin mRNA or a pool of 4 siRNA molecules designed to not recognize any viral or cellular transcripts (control siRNA). Western blotting of transfected cell lysates (Fig. 6A) indicated that nucleolin siRNA had reduced the level of nucleolin protein (Fig. 6A, lane 2) compared to that in cells transfected with control siRNA (lane 1). Dilutions of protein from cells transfected with control siRNA (Fig. 6A, lane 1) were compared to undiluted protein from cells transfected with nucleolin siRNA by Western blotting (Fig. 6A, lane 2), indicating that nucleolin protein levels had been knocked down by at least 4-fold compared to the control (Fig. 6B). Transfected cells were infected with AD169, and up to a 10-fold decrease in virus titers was observed in nucleolin siRNA-treated cells compared to virus production from cells transfected with control siRNA (Fig. 6C), suggesting that nucleolin is important for HCMV replication. Similar results were observed when cells were infected at a higher MOI (MOI of 10) (data not shown).
FIG. 6.

Knockdown of nucleolin with siRNA in HFF cells. (A) Western blotting of HFF cells transfected with siRNA. Proteins from lysates of cells transfected with a pool of siRNA recognizing nucleolin mRNA (lane 2) or control siRNA (lane 1) were assayed by Western blotting using antibodies recognizing nucleolin (Ncl) or β-actin, as indicated to the right. The positions of molecular mass markers (kDa) are indicated to the left. (B) Determination of relative protein levels in cells transfected with siRNA pools. A 2-fold dilution series of protein from lane 1 of A (lanes 1 to 3) was analyzed by Western blotting using antibodies recognizing nucleolin (Ncl) or β-actin, as indicated to the right, compared to undiluted protein from lane 2 of A. The positions of molecular mass markers (kDa) are indicated to the left. (C) Virus production from siRNA-transfected cells. Cells were infected with AD169 at an MOI of 1, and virus was harvested 96 h postinfection. Data points indicate the average virus titers (PFU/ml) from two experiments. Error bars indicate the standard errors of the means of those results. (D) Nucleolin levels from cells transfected with individual nucleolin siRNA. Lysates of cells transfected with control siRNA (lane 1) and each of the 4 individual siRNAs targeting nucleolin (lanes 2 to 5, siRNAs 5 to 8, respectively) were assayed by Western blotting using antibodies recognizing nucleolin (Ncl) or β-actin, as indicated to the right. The positions of molecular mass markers (kDa) are indicated to the left. (E) Determination of relative protein levels in cells transfected with individual siRNAs. A 2-fold dilution series of protein from lane 2 of D (lanes 1 to 3) was analyzed by Western blotting using antibodies recognizing nucleolin (Ncl) compared to undiluted protein from lane 3 of D. The positions of molecular mass markers (kDa) are indicated to the left. (F) Virus production from cells transfected with individual siRNAs. HFF cells transfected with control siRNA or siRNAs 5 to 8 were infected with AD169 at an MOI of 1. Titers of virus (PFU/ml) from these cells were determined in duplicate at 96 h postinfection. Data points indicate the average virus titers (PFU/ml) from titrations of virus performed in duplicate. These data are representative of data from 2 independent experiments.
As a further test of whether the observed reduction in virus replication was due to the specific knockdown of the nucleolin protein and not an off-target effect of any siRNA in the pool, the ability of the virus to replicate in cells containing each of the four siRNAs used in the pool was tested. Western blotting of the transfected cells (Fig. 6D) showed that, compared to cells transfected with control siRNA (Fig. 6D, lane 1), relatively little reduction in nucleolin levels was evident in cells transfected with siRNAs 5 and 8 (lanes 2 and 5, respectively), but a much more notable decrease in nucleolin levels could be observed in cells transfected with siRNAs 6 and 7 (lanes 3 and 4, respectively). Dilutions of protein from cells transfected with control siRNA (Fig. 6D, lane 1) were compared to undiluted protein from cells transfected with siRNA 5 by Western blotting (Fig. 6D, lane 2), which indicated that nucleolin protein levels had been knocked down by less than 2-fold in siRNA 5-treated cells compared to cells treated with control siRNA (data not shown). Dilutions of protein from cells transfected with siRNA 5 (Fig. 6D, lane 2) were compared to undiluted protein from cells transfected with siRNA 6 by Western blotting (Fig. 6D, lane 3), which indicated that nucleolin was present at a level between 2-fold and 4-fold lower for siRNA 6-treated cells than for siRNA 5-treated cells (Fig. 6E). AD169 replication in cells transfected in parallel with individual siRNAs was also assayed (Fig. 6F). No notable reduction in levels of virus production from cells transfected with siRNA 5 or 8 could be observed; however, a reduction in the level of virus production (3- to 4-fold) was observed for cells transfected with siRNA 6 or 7. As the reduction in the level of virus production correlates with the levels of nucleolin that we observed for siRNA-transfected cells, it is likely that the reduction in virus production shown in Fig. 6C is due to the knockdown of nucleolin levels in the cell and not an off-target effect of any one of the siRNAs in the pool of siRNAs used. Notably, the knockdown of nucleolin by siRNAs 5 and 8 did not result in a notable decrease in the level of virus production. Given the increase in nucleolin levels that we observed for infected cells, it is possible that the knockdown by siRNAs 5 and 8 observed is insufficient to affect virus production.
Nucleolin was previously reported to have multiple roles in ribosome biogenesis (16). The defect in virus production that we observed for siRNA-treated cells may therefore be the result of the host cell's inability to synthesize protein when nucleolin levels are reduced. Cells transfected with pools of nucleolin siRNA or control siRNA were pulsed, in the presence or absence of the translation inhibitor cycloheximide, with [35S]methionine to radiolabel newly translated protein. Proteins in lysates of these cells were separated on an SDS-PAGE gel and stained (Fig. 7). Similar levels of protein could be observed in each lane of the stained gel (Fig. 7A, lanes 1 to 4). The gel was then exposed to a phosphorimager screen. Similar levels of protein were synthesized in cells containing either nucleolin siRNA or control siRNA (Fig. 7B, lanes 1 and 2), and in the presence of cycloheximide, no labeling of protein was observed (lanes 3 and 4). Western blotting of transfected cells was performed in parallel by using a nucleolin MAb, and results similar to those shown in Fig. 6A were observed (data not shown). This experiment indicates that the knockdown of nucleolin protein levels with siRNA did not globally affect protein synthesis.
FIG. 7.

Protein synthesis in siRNA-transfected cells. HFF cells were transfected with either nucleolin (Ncl) siRNA or control siRNA and were pulsed with [35S]methionine in the presence or absence of 200 μg/ml cycloheximide (CHX), and cell lysates were prepared. Proteins in each cell lysate were separated on a 10% SDS-polyacrylamide gel, which was stained, dried, and digitally scanned (A) and then exposed to a phosphorimager screen (B). The positions of molecular mass markers (kDa) are indicated to the left.
To further assess if the defect in HCMV replication that we observed for cells containing nucleolin siRNA is due to a requirement for nucleolin during HCMV replication or due to a nonspecific effect on the ability of cells to support virus replication in general, HFF cells were transfected as described above with pools of nucleolin siRNA or control siRNA and infected with either HCMV or reovirus (Fig. 8). As described above, levels of HCMV virus replication in cells transfected with nucleolin siRNA decreased by approximately 10-fold compared to levels of virus replication in cells transfected with control siRNA. In contrast, the knockdown of nucleolin protein levels did not appear to affect reovirus replication, as similar amounts of virus could be recovered from cells containing either nucleolin siRNA or control siRNA. Similar results were obtained by infecting cells with a lower MOI of reovirus (MOI of 0.01) or vesicular stomatitis virus (VSV) (MOI of 0.1) (data not shown). With the caveat that reovirus and VSV are RNA viruses that replicate in the cytoplasm over a shorter time period than that required for the replication of HCMV, which is a DNA virus whose genome replicates in the nucleus, the results indicate that the knockdown of nucleolin does not nonspecifically decrease the ability of cells to support virus replication.
FIG. 8.
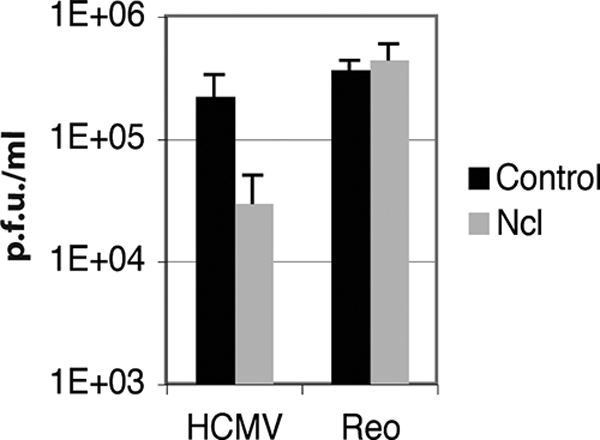
Replication of HCMV and reovirus in siRNA-transfected cells. HFF cells transfected with either nucleolin siRNA or control siRNA were infected with HCMV (MOI of 1) or reovirus (Reo) (MOI of 0.1). Virus was harvested at 96 h p.i. for HCMV and at 16 h postinfection for reovirus and titrated onto HFF and L cells, respectively, to determine HCMV and reovirus titers (PFU/ml). The mean titers from three experiments are shown. Error bars represent the standard deviations.
As the knockdown of nucleolin did not appear to affect global protein production or abrogate the ability of cells to support the replication of an unrelated virus, it is likely that the reduction in the level of HCMV replication that we observed for cells treated with nucleolin siRNA is due to a requirement of nucleolin during HCMV replication rather than due to a nonspecific effect.
Expression of viral proteins and viral DNA synthesis during knockdown of nucleolin.
To determine when nucleolin is required during HCMV replication, we first assayed the production of viral proteins of different kinetic classes during replication in cells where nucleolin had been knocked down. Cell lysates of HFF cells transfected with pools of nucleolin siRNA or control siRNA and infected with AD169 were prepared at several time points, and protein levels were assayed by Western blotting (Fig. 9). A reduction in nucleolin protein levels in cells transfected with nucleolin siRNA could be seen at early time points compared to cells transfected with control siRNA (Fig. 9, lanes 1 to 4); however, at 72 h p.i, increased levels of nucleolin could be seen in cells transfected with either siRNA pool compared to earlier time points (lanes 5 and 6). Consistent with data shown in Fig. 3B and 4, higher levels of nucleolin were seen in infected cell lysates compared to uninfected cell lysates. Analysis of diluted protein from lane 5 compared to undiluted protein from lane 6 of Fig. 9A by Western blotting (Fig. 9B) indicated that at 72 h p.i., nucleolin protein levels were knocked down approximately 3- to 4-fold.
The knockdown of nucleolin protein levels did not appear to reduce the protein levels of either the immediate-early virus gene products IE86 and IE72 or the UL44 protein, an early gene product. However, a decrease in the level of the pp28 protein could be detected in cells transfected with nucleolin siRNA (Fig. 9A, lane 5) compared to cells transfected with control siRNA (lane 6) at 72 h p.i. Analysis of diluted protein from lane 5 compared to undiluted protein from lane 6 of Fig. 9A by Western blotting (Fig. 9B) indicates that pp28 levels were reduced by approximately 3-fold in cells transfected with nucleolin siRNA compared to cells transfected with control siRNA.
As pp28 is the product of a viral gene whose expression is dependent upon viral DNA synthesis (11), we hypothesized that its reduction might be an effect of reduced viral DNA synthesis. In three experiments, virus production and viral DNA synthesis were determined by use of HFF cells transfected with pools of nucleolin siRNA or control siRNA and infected with AD169. In each experiment an approximately 10-fold reduction in the level of virus production from cells transfected with nucleolin siRNA could again be observed compared to virus production from cells transfected with control siRNA (Fig. 10A). The number of viral genomes present in each sample was determined by quantitative real-time PCR, specifically, normalizing the copy number of a viral locus (UL83) to the copy number of a cellular locus (β-actin). In each experiment a 3- to 4-fold reduction in the number of copies of UL83/β-actin was observed for infected cells transfected with nucleolin siRNA compared to infected cells transfected with control siRNA (Fig. 10B). These results indicate, therefore, that nucleolin is necessary for efficient viral DNA synthesis, and the reduction in pp28 levels in cells transfected with nucleolin siRNA observed in Fig. 9 is most likely due to a reduction in the level of viral DNA synthesis in cells transfected with nucleolin siRNA.
DISCUSSION
In this study we report the identification by MS of numerous viral and cellular proteins that associate with a FLAG-tagged version of UL44 in infected cell lysates. We have previously identified a subset of proteins associated with UL44 by MS by immunoprecipitating UL44 from lysates of cells infected with HCMV at a single time point postinfection using a MAb recognizing UL44 (47). With the exception of UL84 and DEAH box protein 9, the proteins identified here differ from those identified by our previous study. This is most likely due to differing experimental procedures between the two studies, in particular the more comprehensive approach used here, and may possibly reflect different proteins interacting with UL44 under different biochemical conditions. It should be stressed that of all the proteins found to associate with FLAG-tagged UL44 in this study, only a few will be functionally significant. Indeed, it is likely that the large number of ribosomal and RNA-modifying proteins immunoprecipitating with FLAG-tagged UL44 nonspecifically bind to UL44 and/or nucleolin during IP.
In this study we uncovered a potential association of UL44 and nucleolin in infected cell lysates. Nucleolin is a major protein component of the nucleolus, the site of ribosome biogenesis in the cell (7). This protein is known to be a DNA and RNA binding phosphoprotein that interacts with multiple cellular proteins and appears to have multiple functions in ribosome biogenesis, including rDNA transcription, rRNA maturation, and ribosome assembly (reviewed in reference 16). We observed an association of nucleolin and UL44 in infected cell lysates by reciprocal co-IP in the presence and absence of nuclease. The colocalization of UL44 and nucleolin within viral replication compartments was detected by immunofluorescence assays. Interestingly, the colocalization of the two proteins was observed as speckles throughout the viral replication compartment, which is reminiscent of the immunofluorescence observed when viral replication compartments are stained with bromodeoxyuridine to identify the location of replicating viral DNA (38). Further investigations will be required to assess the significance of this pattern of colocalization. We also found that the level of nucleolin increases during infection and, based on siRNA knockdown experiments, that nucleolin is required for viral DNA synthesis and efficient virus production. Although only a 10-fold decrease in the level of virus production was seen upon the knockdown of nucleolin, it is possible that a more complete knockdown of nucleolin protein levels and inhibition of the mechanism that increases nucleolin levels upon infection could have a more profound effect on virus replication. Taken together, our results indicate an association of nucleolin with the DNA replication protein UL44 and a role for nucleolin in HCMV replication during viral DNA synthesis. While it has yet to be demonstrated that an association of UL44 and nucleolin in the infected cell is required for efficient viral DNA synthesis, we speculate that it is likely that nucleolin either influences UL44 function or provides a function during viral DNA synthesis that is dependent upon its interaction, directly or indirectly, with UL44.
Nucleolin was previously implicated in the DNA replication of other viral DNA genomes and eukaryotic DNA. Nucleolin, topoisomerase I, and simian virus 40 (SV40) T antigen can form a complex that is thought to unwind the double-stranded DNA of the SV40 genome (44). Additionally, nucleolin was previously shown to interact with proteins involved in cellular DNA synthesis such as replication protein A (RPA) (24, 53) and topoisomerase I (6). This might raise the possibility that nucleolin could help recruit these cellular replication proteins to the HCMV replication fork. However, we do not favor this possibility, as neither these proteins nor others required for the replication of cellular DNA were detected in protein immunoprecipitated with UL44 in our current study.
An intriguing possibility is that nucleolin is present at the viral replication fork to participate in chromatin remodeling. Nucleolin was previously reported to interact directly with histone H2-H3 dimers (17) and to act as an H2-H3 histone chaperone and enhance the activity of the chromatin modifiers SWI/SNF and ACF to remodel nucleosomes, facilitating transcription (1). These results led to the proposition that nucleolin has a function similar to that of the cellular FACT (facilitates chromatin transcription) complex (1, 36), which is a histone chaperone that interacts with H2-H3 (37). Studies of the Saccharomyces cerevisiae homologue of FACT (yFACT) have shown that it is required for DNA replication and is present at the replication fork via its interaction with polymerase α (9, 13, 43, 56). Like nucleolin, yFACT interacts with RPA (52), which led to the suggestion that FACT is responsible for the construction of nucleosomes during DNA replication. It was proposed, therefore, that nucleolin, like FACT, can act in DNA replication, potentially remodeling nucleosomes during DNA replication, and may have functions in DNA repair and recombination, again related to the interaction with histones (36). Further study is required to determine if nucleolin acts like FACT at the HCMV replication fork.
It must also be noted that the data that we present here do not exclude the possibility that nucleolin plays multiple roles in HCMV replication. Although a 10-fold decrease in virus titers was typically observed when nucleolin was knocked down with siRNA, only a 3- to 4-fold reduction in the level of viral DNA replication was detected. Thus, it is possible that nucleolin may also be required for HCMV replication at a step in the virus life cycle after viral DNA synthesis, potentially late viral gene expression, in which UL44 was previously suggested to have a role (21, 22).
It is unknown whether the increase in nucleolin levels in infected cells is due to a cellular response to infection or to a virally encoded mechanism to ensure that a cellular protein required for virus replication is present at high levels in the infected cell. It was previously reported that there is no change in nucleolin mRNA levels during infection (8, 19, 45, 57). It is possible, therefore, that the accumulation of nucleolin is the result of translational regulation or increasing stability due to a posttranslational modification or interaction with another viral or cellular protein, possibly UL44.
It was recently reported that nucleolin is necessary for the efficient replication of herpes simplex virus type 1 (HSV-1) (10). It remains unclear at what stage of the HSV-1 replication cycle nucleolin is required, and it is unknown which, if any, HSV-1 proteins associate with nucleolin in the infected cell. As there are notable structural and functional similarities between UL44 and the HSV-1 DNA polymerase accessory subunit UL42 (3, 58), it will be interesting to determine if nucleolin associates with UL42 during HSV-1 replication. Also, it was previously demonstrated that the expression of HSV-1 UL24 is both necessary and sufficient to induce the dispersal of nucleolin from the nucleolus (5, 32). It is unknown if any HCMV protein possesses this ability. It is also unknown if the dispersal of nucleolin from the nucleolus occurs or is required for HCMV replication and/or the association with UL44 in the HCMV-infected cell. Also, we note that there is some disagreement as to whether nucleolin levels increase during HSV-1 infection (10, 32).
Recently, Stow and coworkers (46) demonstrated that another nucleolar protein, upstream binding factor (UBP), is recruited to sites of HSV-1 replication in the nucleus and suggested that this protein is required for virus replication, while Salsman et al. (42) reported that a number of viral proteins from HSV-1, HCMV, and Epstein-Barr virus (EBV) localize to the nucleolus (although no specific relocalization of HSV-1 UL42, HCMV UL44, or the EBV DNA polymerase accessory factor BMRF1 to the nucleolus was observed). Our current study reinforces the idea that the interaction between viral and nucleolar proteins is likely to be important for herpesvirus replication. A key question that will have to be addressed in future work is how this occurs.
Acknowledgments
We thank Max Nibert (Harvard Medical School) for encouragement and Robert Kalteja (University of Wisconsin) and Sean Whelan (Harvard Medical School) for generously providing advice and reagents. We also thank Tomas Kirchhausen (Harvard Medical School) for the use of microscopy facilities. We also gratefully acknowledge the assistance and invaluable advice received from members of the Coen laboratory, in particular Jeremy Kamil and Laurie Silva, and Ross Tomaino (Taplin Biological Mass Spectrometry Facility, Harvard Medical School).
This work was supported in part by NIH grants AI19838 and AI26077 to D.M.C. and an award from the William Randolph Hearst Fund to B.L.S.
Footnotes
Published ahead of print on 9 December 2009.
REFERENCES
- 1.Angelov, D., V. A. Bondarenko, S. Almagro, H. Menoni, F. Mongelard, F. Hans, F. Mietton, V. M. Studitsky, A. Hamiche, S. Dimitrov, and P. Bouvet. 2006. Nucleolin is a histone chaperone with FACT-like activity and assists remodeling of nucleosomes. EMBO J. 25:1669-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appleton, B. A., J. Brooks, A. Loregian, D. J. Filman, D. M. Coen, and J. M. Hogle. 2006. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. Differences in structure and function relative to unliganded UL44. J. Biol. Chem. 281:5224-5232. [DOI] [PubMed] [Google Scholar]
- 3.Appleton, B. A., A. Loregian, D. J. Filman, D. M. Coen, and J. M. Hogle. 2004. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol. Cell 15:233-244. [DOI] [PubMed] [Google Scholar]
- 4.Baldick, C. J., Jr., A. Marchini, C. E. Patterson, and T. Shenk. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 71:4400-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertrand, L., and A. Pearson. 2008. The conserved N-terminal domain of herpes simplex virus 1 UL24 protein is sufficient to induce the spatial redistribution of nucleolin. J. Gen. Virol. 89:1142-1151. [DOI] [PubMed] [Google Scholar]
- 6.Bharti, A. K., M. O. Olson, D. W. Kufe, and E. H. Rubin. 1996. Identification of a nucleolin binding site in human topoisomerase I. J. Biol. Chem. 271:1993-1997. [DOI] [PubMed] [Google Scholar]
- 7.Boisvert, F. M., S. van Koningsbruggen, J. Navascues, and A. I. Lamond. 2007. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8:574-585. [DOI] [PubMed] [Google Scholar]
- 8.Browne, E. P., B. Wing, D. Coleman, and T. Shenk. 2001. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 75:12319-12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budd, M. E., A. H. Tong, P. Polaczek, X. Peng, C. Boone, and J. L. Campbell. 2005. A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genet. 1:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calle, A., I. Ugrinova, A. L. Epstein, P. Bouvet, J. J. Diaz, and A. Greco. 2008. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J. Virol. 82:4762-4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Depto, A. S., and R. M. Stenberg. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241-3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ertl, P. F., and K. L. Powell. 1992. Physical and functional interaction of human cytomegalovirus DNA polymerase and its accessory protein (ICP36) expressed in insect cells. J. Virol. 66:4126-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Formosa, T., S. Ruone, M. D. Adams, A. E. Olsen, P. Eriksson, Y. Yu, A. R. Rhoades, P. D. Kaufman, and D. J. Stillman. 2002. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics 162:1557-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao, Y., K. Colletti, and G. S. Pari. 2008. Identification of human cytomegalovirus UL84 virus- and cell-encoded binding partners by using proteomics analysis. J. Virol. 82:96-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gault, E., Y. Michel, A. Dehee, C. Belabani, J. C. Nicolas, and A. Garbarg-Chenon. 2001. Quantification of human cytomegalovirus DNA by real-time PCR. J. Clin. Microbiol. 39:772-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginisty, H., H. Sicard, B. Roger, and P. Bouvet. 1999. Structure and functions of nucleolin. J. Cell Sci. 112:761-772. [DOI] [PubMed] [Google Scholar]
- 17.Godfrey, J. E., A. D. Baxevanis, and E. N. Moudrianakis. 1990. Spectropolarimetric analysis of the core histone octamer and its subunits. Biochemistry 29:965-972. [DOI] [PubMed] [Google Scholar]
- 18.Hanfler, J., K. A. Kreuzer, K. Laurisch, N. Rayes, P. Neuhaus, C. A. Schmidt, and H. Oettle. 2003. Quantitation of cytomegalovirus (hCMV) DNA and beta-actin DNA by duplex real-time fluorescence PCR in solid organ (liver) transplant recipients. Med. Microbiol. Immunol. 192:197-204. [DOI] [PubMed] [Google Scholar]
- 19.Hertel, L., and E. S. Mocarski. 2004. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J. Virol. 78:11988-12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hobom, U., W. Brune, M. Messerle, G. Hahn, and U. H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isomura, H., M. F. Stinski, A. Kudoh, T. Murata, S. Nakayama, Y. Sato, S. Iwahori, and T. Tsurumi. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isomura, H., M. F. Stinski, A. Kudoh, S. Nakayama, S. Iwahori, Y. Sato, and T. Tsurumi. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 81:6197-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamil, J. P., and D. M. Coen. 2007. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J. Virol. 81:10659-10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, K., D. D. Dimitrova, K. M. Carta, A. Saxena, M. Daras, and J. A. Borowiec. 2005. Novel checkpoint response to genotoxic stress mediated by nucleolin-replication protein A complex formation. Mol. Cell. Biol. 25:2463-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komazin-Meredith, G., R. J. Petrella, W. L. Santos, D. J. Filman, J. M. Hogle, G. L. Verdine, M. Karplus, and D. M. Coen. 2008. The human cytomegalovirus UL44 C clamp wraps around DNA. Structure 16:1214-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krosky, P. M., M. C. Baek, W. J. Jahng, I. Barrera, R. J. Harvey, K. K. Biron, D. M. Coen, and P. B. Sethna. 2003. The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J. Virol. 77:7720-7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 28.Lai, J. S., and W. Herr. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U. S. A. 89:6958-6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, E. C., D. Yu, J. Martinez de Velasco, L. Tessarollo, D. A. Swing, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56-65. [DOI] [PubMed] [Google Scholar]
- 30.Loregian, A., B. A. Appleton, J. M. Hogle, and D. M. Coen. 2004. Specific residues in the connector loop of the human cytomegalovirus DNA polymerase accessory protein UL44 are crucial for interaction with the UL54 catalytic subunit. J. Virol. 78:9084-9092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loregian, A., E. Sinigalia, B. Mercorelli, G. Palu, and D. M. Coen. 2007. Binding parameters and thermodynamics of the interaction of the human cytomegalovirus DNA polymerase accessory protein, UL44, with DNA: implications for the processivity mechanism. Nucleic Acids Res. 35:4779-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lymberopoulos, M. H., and A. Pearson. 2007. Involvement of UL24 in herpes-simplex-virus-1-induced dispersal of nucleolin. Virology 363:397-409. [DOI] [PubMed] [Google Scholar]
- 33.Maga, G., and U. Hubscher. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051-3060. [DOI] [PubMed] [Google Scholar]
- 34.Marschall, M., M. Freitag, P. Suchy, D. Romaker, R. Kupfer, M. Hanke, and T. Stamminger. 2003. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 311:60-71. [DOI] [PubMed] [Google Scholar]
- 35.Moldovan, G. L., B. Pfander, and S. Jentsch. 2007. PCNA, the maestro of the replication fork. Cell 129:665-679. [DOI] [PubMed] [Google Scholar]
- 36.Mongelard, F., and P. Bouvet. 2007. Nucleolin: a multiFACeTed protein. Trends Cell Biol. 17:80-86. [DOI] [PubMed] [Google Scholar]
- 37.Orphanides, G., W. H. Wu, W. S. Lane, M. Hampsey, and D. Reinberg. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400:284-288. [DOI] [PubMed] [Google Scholar]
- 38.Penfold, M. E., and E. S. Mocarski. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46-61. [DOI] [PubMed] [Google Scholar]
- 39.Prichard, M. N., H. Lawlor, G. M. Duke, C. Mo, Z. Wang, M. Dixon, G. Kemble, and E. R. Kern. 2005. Human cytomegalovirus uracil DNA glycosylase associates with ppUL44 and accelerates the accumulation of viral DNA. Virol. J. 2:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ranneberg-Nilsen, T., H. A. Dale, L. Luna, R. Slettebakk, O. Sundheim, H. Rollag, and M. Bjoras. 2008. Characterization of human cytomegalovirus uracil DNA glycosylase (UL114) and its interaction with polymerase processivity factor (UL44). J. Mol. Biol. 381:276-288. [DOI] [PubMed] [Google Scholar]
- 41.Saffert, R. T., and R. F. Kalejta. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salsman, J., N. Zimmerman, T. Chen, M. Domagala, and L. Frappier. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 4:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlesinger, M. B., and T. Formosa. 2000. POB3 is required for both transcription and replication in the yeast Saccharomyces cerevisiae. Genetics 155:1593-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seinsoth, S., H. Uhlmann-Schiffler, and H. Stahl. 2003. Bidirectional DNA unwinding by a ternary complex of T antigen, nucleolin and topoisomerase I. EMBO Rep. 4:263-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slobedman, B., J. L. Stern, A. L. Cunningham, A. Abendroth, D. A. Abate, and E. S. Mocarski. 2004. Impact of human cytomegalovirus latent infection on myeloid progenitor cell gene expression. J. Virol. 78:4054-4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stow, N. D., V. C. Evans, and D. A. Matthews. 2009. Upstream-binding factor is sequestered into herpes simplex virus type 1 replication compartments. J. Gen. Virol. 90:69-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strang, B. L., E. Sinigalia, L. A. Silva, D. M. Coen, and A. Loregian. 2009. Analysis of the association of the human cytomegalovirus DNA polymerase subunit UL44 with the viral DNA replication factor UL84. J. Virol. 83:7581-7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strang, B. L., and N. D. Stow. 2005. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 79:12487-12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor, T. J., and D. M. Knipe. 2004. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78:5856-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191-197. [DOI] [PubMed] [Google Scholar]
- 51.Tocci, M. J., T. J. Livelli, H. C. Perry, C. S. Crumpacker, and A. K. Field. 1984. Effects of the nucleoside analog 2′-nor-2′-deoxyguanosine on human cytomegalovirus replication. Antimicrob. Agents Chemother. 25:247-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.VanDemark, A. P., M. Blanksma, E. Ferris, A. Heroux, C. P. Hill, and T. Formosa. 2006. The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol. Cell 22:363-374. [DOI] [PubMed] [Google Scholar]
- 53.Wang, Y., J. Guan, H. Wang, D. Leeper, and G. Iliakis. 2001. Regulation of DNA replication after heat shock by replication protein A-nucleolin interactions. J. Biol. Chem. 276:20579-20588. [DOI] [PubMed] [Google Scholar]
- 54.Weiland, K. L., N. L. Oien, F. Homa, and M. W. Wathen. 1994. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 34:191-206. [DOI] [PubMed] [Google Scholar]
- 55.Wiebusch, L., M. Truss, and C. Hagemeier. 2004. Inhibition of human cytomegalovirus replication by small interfering RNAs. J. Gen. Virol. 85:179-184. [DOI] [PubMed] [Google Scholar]
- 56.Zhou, Y., and T. S. Wang. 2004. A coordinated temporal interplay of nucleosome reorganization factor, sister chromatin cohesion factor, and DNA polymerase alpha facilitates DNA replication. Mol. Cell. Biol. 24:9568-9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu, H., J. P. Cong, G. Mamtora, T. Gingeras, and T. Shenk. 1998. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:14470-14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuccola, H. J., D. J. Filman, D. M. Coen, and J. M. Hogle. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5:267-278. [DOI] [PubMed] [Google Scholar]