

Abstract
The epothilones are naturally occurring, cytotoxic macrolides that function through a paclitaxel (Taxol)-like mechanism. Although structurally dissimilar, both classes of molecules lead to the arrest of cell division and eventual cell death by stabilizing cellular microtubule assemblies. The epothilones differ in their ability to retain activity against multidrug-resistant (MDR) cell lines and tumors where paclitaxel fails. In the current account, we focus on the relationship between epothilone and paclitaxel in the context of tumors with multiple drug resistance. The epothilone analogue Z-12,13-desoxyepothilone B (dEpoB) is >35,000-fold more potent than paclitaxel in inhibiting cell growth in the MDR DC-3F/ADX cell line. Various formulations, routes, and schedules of i.v. administration of dEpoB have been tested in nude mice. Slow infusion with a Cremophor-ethanol vehicle proved to be the most beneficial in increasing efficacy and decreasing toxicity. Although dEpoB performed similarly to paclitaxel in sensitive tumors xenografts (MX-1 human mammary and HT-29 colon tumor), its effects were clearly superior against MDR tumors. When dEpoB was administered to nude mice bearing our MDR human lymphoblastic T cell leukemia (CCRF-CEM/paclitaxel), dEpoB demonstrated a full curative effect. For human mammary adenocarcinoma MCF-7/Adr cells refractory to paclitaxel, dEpoB reduced the established tumors, markedly suppressed tumor growth, and surpassed other commonly used chemotherapy drugs such as adriamycin, vinblastine, and etoposide in beneficial effects.
Paclitaxel (Taxol**), Fig. 1, is currently used as the front-line therapeutic agent in a variety of solid forms of cancer including ovarian, breast, colon, lung, and liver neoplasms (1). Acquired resistance to paclitaxel and other commonly used cancer chemotherapy agents may be mediated by a number of mechanisms, including overexpression of the energy-dependent drug-transport protein P-glycoprotein (ref. 2, and references therein). Broad-spectrum resistance to structurally and mechanistically diverse anticancer agents constitutes the multidrug-resistance (MDR) phenotype. A search for paclitaxel analogues with improved performance in vitro and in vivo has met with limited success (3, 4), although certain MDR reversal agents appear promising when coadministered with the anticancer agent (5).
Figure 1.

Structural representations of paxlitaxel (Taxol) and dEpoB.
Although the 16-membered ring structure of the epothilones bears little structural resemblance to paclitaxel, the two agents share a common cellular mechanism of action (6–8). Both the epothilones and paclitaxel exert their biological effects by stabilizing microtubule assemblies, thus leading to the arrest of cell division and eventual cell death. By far the most intriguing property of the epothilones at the in vitro level is their lack of cross resistance to MDR cell lines when compared with major antitumor agents, including paclitaxel, vinblastine, adriamycin, camptothecin, and etoposide, all of which are currently used in clinical settings (6–9). The epothilones are also more water-soluble (6, 10–12) and more readily available through chemical synthesis (6, 13–20) than is paclitaxel. The solubility advantage could allow for less cumbersome administration and increased bioavailability of the chemotherapy agent than paclitaxel. The synthetic advantage would be useful in generating epothilone congeners more advantageous than the natural product.
We recently reported that Z-12,13-desoxyepothilone (dEpoB; Fig. 1), a synthetic intermediate en route to epothilone B,‡‡ demonstrated promising activity both in vitro and in murine models harboring tumor xenografts (6, 21). In the previous report, we also showed that dEpoB was >35,000-fold more potent than paclitaxel in inhibiting cell growth of DC-3F/ADX in vitro. During the earlier in vivo pharmacologic evaluations of the epothilones, both dEpoB and paclitaxel were administered with dimethyl sulfoxide (DMSO) as a solvent by using i.p. injection (6, 22). However, i.v. administration is more appropriate for paclitaxel than for dEpoB, resulting in higher efficacy and lower toxicity. Because the primary focus of this research was to compare dEpoB and paclitaxel, we adjusted the administration of dEpoB to correspond to conditions optimal for paclitaxel.
For this report, we used i.v. injection and adopted Cremophor/ethanol (1:1) as a solvent for drug administration, conditions of administration that correspond closely to those optimal for paclitaxel. The use of Cremophor/EtOH solvent for prolonged and/or repeated i.v. infusion of dEpoB led to markedly improved therapeutic effects against MDR, refractory tumors, and non-MDR human tumor xenografts in which full curative effects have been achieved for MX-1 and CCRF/CEM.
METHODS
Chemicals.
dEpoB (NSC-703147) used in this study was obtained in our laboratory through a modified, practical total synthesis as described (20). For in vivo studies, dEpoB was dissolved in Cremophor/EtOH (1:1) vehicle unless otherwise indicated. Cremophor EL was purchased from Sigma. Paclitaxel in Cremophor/EtOH formulation was obtained from Bristol-Myers Squibb. Vinblastine sulfate (VBL, Velban; Eli Lilly), etoposide (VP-16, Vepisid; Bristol-Myers Squibb), and adriamycin (DX or Adr, Doxorubicin⋅HCl; Astra Pharmaceutical, Worcester, MA) were used in the manufacturer’s formulation and diluted with saline or DMSO as needed.
Animals.
Athymic nude mice bearing the nu/nu gene were used for all human tumor xenografts. Outbred, Swiss-background mice were obtained from Taconic Farms. Male mice 6–10 weeks old weighing 20–26 g were used for most experiments. For i.v. injection or i.v. infusion, the drug was administered via the tail vein. Each individual mouse was confined in a restrainer for a 30- to 120-min drug-administration period, whereas for 6- to 24-hr drug administration by i.v. infusion, the mouse was free to move about in a cage with the needle secured by tape. Tumor volume was assessed by measuring length × width × height (or width) using a caliper. The Harvard PHD2000 syringe pump (Harvard Apparatus) with multitrack was used for i.v. infusion. Typically, the infusion volume for dEpoB in Cremophor/EtOH (1:1) was 100 μl + 2.5 ml of saline for a 2- or 6-hr infusion and 100 μl + 4 ml of saline for a 24-hr infusion. All animal studies were conducted in accordance with the guidelines of the National Institutes of Health “Guide for the Care and Use of Animals” and protocol reviewed by the Memorial Sloan-Kettering Cancer Center’s Institutional Animal Care and Use Committee. In keeping with the policy of this committee for the humane treatment of tumor-bearing animals, mice were euthanized when tumors reached ≥10% of their total body weight.
Tumor and Cell Lines.
The MX-1 human breast carcinoma xenograft used in this study has been used for many years in the National Cancer Institute screening panel of experimental tumors and thus is well characterized (23). The CCRF-CEM human T cell acute lymphoblastic leukemia cell line and its vinblastine-resistant subline (CCRF-CEM/VBL) were obtained from W. T. Beck (University of Illinois, Chicago, IL). CCRF-CEM/paclitaxel cell line was developed in this laboratory (T.-C.C.) using continuous exposure of CCRF-CEM cells with increasing and sublethal (IC50 ≈ IC90) concentrations of paclitaxel for 10 months. The fresh medium with paclitaxel was replenished every 7–12 days. The CCRF-CEM/paclitaxel cell line exhibited 57-fold resistance to paclitaxel (IC50 = 0.12 μM) when compared with original CCRF-CEM cells at the beginning of the experiment (IC50 = 0.0021 μM; see Table 1). Human mammary adenocarcinoma MCF-7 cells and their doxorubicin-resistant subline (MCF-7/Adr) and murine lymphoid leukemic cells (P388 and P388/Adr) were obtained from the cell bank of the Pharmacology Core Laboratory of the Sloan-Kettering Institute for Cancer Research. P388/Adr cells were selected in P388 ascites tumor-bearing B6D2 F1 mice by repeated challenge with sublethal doses of DX. To maintain the drug-resistant phenotypes, the resistant cell lines were cultured once every month in the presence of the selecting agent (DX, VBL, or paclitaxel) at approximately their IC50 concentrations. The cells then were resuspended in fresh media for a minimum of 4 days before each assay or transplantation. The following human cancer cells were obtained from American Type Culture Collection: mammary carcinoma, MX-1; ovarian adenocarcinoma, SK-OV-3; lung carcinoma, A549; colon adenocarcinoma, HT-29; and prostate adenocarcinoma, PC-3.
Table 1.
Relative potency of dEpoB and paclitaxel in inhibiting cell growth in vitro
| Tumor cells | dEpoB | Paclitaxel |
|---|---|---|
| IC50, μM | ||
| Human tumors | ||
| MX-1 | 0.088 | 0.177 |
| MCF-7 | 0.0029 | 0.0033 |
| MCF-7/Adr | 0.0071 (2.4x) | 0.150 (46x) |
| CCRF-CEM | 0.0095 | 0.0021 |
| CCRF-CEM/paclitaxel | 0.0162 (1.7x) | 0.120 (57x) |
| SK-OV-3 | 0.0069 | 0.0024 |
| A-549 | 0.0035 | 0.0012 |
| PC-3 | 0.0128 | 0.0464 |
| HT-29 | 0.0081 | 0.0018 |
| Murine tumors | ||
| P388 | 0.0068 | 0.0029 |
| P388/Adr | 0.0042 (0.62x) | 0.326 (111x) |
Cell growth inhibition in vitro was measured by SRB assay (24) for MX-1, MCF-7, MCF-7/Adr, SK-OV-3, A-549, PC-3, and HT-29 cells after a 72-hr incubation, whereas for CCRF-CEM, CCRF-CEM/paclitaxel, P388, and P388/Adr cells, XTT tetrazonium assay (25) was used. The IC50 values were determined for six to seven concentrations of each drug using a computer program (26, 27). The numbers in parenthesis are folds of resistance based on the IC50 ratio when compared with the corresponding parent cell lines. The data for MCF-7, CCRF-CEM, and P388 and their sublines are in ref. 20.
Cytoxicity Assays.
The cells were cultured at an initial density of 5 × 104 cells per ml. They were maintained in a 5% CO2-humidified atmosphere at 37°C in RPMI medium 1640 (GIBCO/BRL) containing penicillin (100 units/ml), streptomycin (100 μg/ml) (GIBCO/BRL), and 10% heat-inactivated fetal bovine serum. For solid tumor cells growing in a monolayer (such as MX-1 or MCF-7/Adr), cytotoxicity of the drug was determined in 96-well microtiter plates by using the sulforhadamine B method as described by Skehan et al. (24) for measuring the cellular protein content. For cells that were grown in suspension (such as CCRF-CEM or P388 and their sublines), cytotoxicity was measured by using the 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-carboxanilide)-2H terazodium hydroxide (XTT)-microculture tetrazonium method (25) in duplicate in 96-well microtiter plates. For both methods, the absorbance of each well was measured with a microplate reader (EL-340, Bio-Tek, Burlington, VT). Each run entailed six to seven concentrations of the tested drugs. Dose-effect relationship data were analyzed with the median-effect plot (26) by using a previously described computer program (27).
RESULTS
Relative Potency of Cell Growth Inhibition in Vitro.
The human tumor cells used in this report for the xenograft studies of dEpoB and paclitaxel in vivo also were compared for the relative susceptibility to these drugs for cell growth inhibition in vitro (Table 1). For the parent tumor cell lines such as MX-1, MCF-7, and CCRF-CEM, both dEpoB and paclitaxel showed similar potency. With drug-resistant cell lines MCF-7/Adr and CCRF-CEM/paclitaxel, paclitaxel showed 46-fold and 57-fold resistance, respectively, when compared with the corresponding parent lines. By contrast, dEpoB showed only 2.4-fold and 1.7-fold resistance against the same drug-resistant cell lines, respectively. Extended in vitro studies for susceptibility of other tumor cells indicated that human ovarian SK-OV-3, lung A-549, and colon HT-29 tumor cells were similarly sensitive to paclitaxel and dEpoB, with paclitaxel being 3- to 4.5-fold more potent than dEpoB (Table 1). In contrast, human prostate PC-3 adenocarcinoma cells have higher IC50 values for both dEpoB and paclitaxel, with dEpoB being 3.6-fold more potent than paclitaxel. Murine P388 lymphoid leukemic cells selected in vivo for DX resistance were found to be 4-fold more resistant to doxorubicin and 111-fold resistant to paclitaxel, but dEpoB showed no cross resistance (Table 1).
Optimal Mode of Treatment.
Table 2 shows the effects of different doses, formulations, routes, and schedules of drug administration on toxicity and therapeutic effect of dEpoB against MX-1 tumor in nude mice. In general, i.p. administration of dEpoB with DMSO showed good therapeutic effect and only moderate toxicity. At 35 mg/kg every other day (Q2D) × 5 i.p., dEpoB markedly reduced tumor size and 3 of 10 mice were cured. Shifting from DMSO to Cremophor/EtOH (1:1), Q2D × 5 i.v. injection at 15 or 20 mg/kg, i.p. injection at 20 mg/kg, i.v. 2-hr infusion at 30 mg/kg still was lethal (Table 2).
Table 2.
Comparison of therapeutic effect and toxicity of dEpoB with different doses, solvents, schedules, and routes of administration in nude mice bearing MX-1 xenografts
| Dose, mg/kg | Schedule | Solvent* | Route | Average tumor size (T/C)† | Tumor free mice/total mice | Toxicity death/total mice |
|---|---|---|---|---|---|---|
| 15 | Q2D × 5 | DMSO | i.p. injection | 0.41 | 0/5 | 0/5 |
| 20 | Q2D × 8 | DMSO | i.p. injection | 0.33‡ | 0/5 | 0/5 |
| 25 | Q2D × 5 | DMSO | i.p. injection | 0.04 | 0/6 | 0/6 |
| 35 | Q2D × 5 | DMSO | i.p. injection | 0.02 | 3/10 (6, 8, 10)¶ | 0/10 |
| 15 | Q2D × 5 | C/EtOH | i.v. 30-min infusion | ND | ND | 4/6 (15, 15, 15, 17)¶ |
| 20 | Q2D × 2 | C/EtOH | i.v. 1-min injection | ND | ND | 1/1 (4) |
| 20 | Q2D × 5 | C/EtOH | i.v. 30-min infusion | ND | ND | 6/6 (10, 10, 11, 11, 11, 11) |
| 20 | Q2D × 1 | C/EtOH | i.p. injection | ND | ND | 1/1 (3) |
| 30 | Q2D × 5 | C/EtOH | i.v. 2-hr infusion | ND | ND | 2/3 (10, 10) |
| 30 | Q2D × 6 | C/EtOH | i.v. 6-hr infusion | 0 | 5/5 (8, 12, 12, 12, 12) | 0/5 |
| 30 | Q1D × 4 | C/EtOH | i.v. 6-hr infusion | ND | ND | 1/1 (6) |
| 30 | Q2D × 4 | C/EtOH | i.v. 24-hr infusion | 0, ND | 1/2 (10) | 1/2 (12) |
| 60 | Q4D × 2 | C/EtOH | i.v. 24-hr infusion | 0, ND | 1/2 (12) | 1/2 (16) |
| 50 | Q2D × 5 | PEG§ | oral | 0.68 | 0/1 | 0/1 |
ND, not determined (mice died). MX-1, human mammary carcinoma, 50 mg per mouse implanted s.c. into mice on day 0. Treatments started on day 10. Q2D, every other day.
DMSO: Dimethylsulfoxide 40 μl per mouse; C/EtOH: Cremophor/EtOH (1:1), 100 μl for i.p. injection; 100 μl + saline 0.2 ml for 1-min i.v. or 30-min injection; 100 μl + saline (3–5 ml) for 2–24 hr i.v. infusions.
Average tumor size of treated group/average tumor size of the control group at 2 days after the last dose.
B-16 melanoma tumor xenograph for this group; all other groups were for MX-1 xenografts.
Polyethylene glycol-400/ethanol (10:1).
The numbers in parenthesis refer to the number of days that mice, after the beginning of treatment, were tumor-free or the number of days before death due to toxicity after the beginning of treatment.
Interestingly, 30 mg/kg Q2D × 6 doses with drug administration by i.v. infusion over a 6-hr period achieved total cure in five of five mice with no deaths. The same dosage given daily for 4 consecutive days proved lethal. dEpoB administration at 30 mg/kg Q2D × 4 doses but by i.v. infusion over a 24-hr period showed good therapeutic effect but appreciable toxicity. At 60 mg/kg, Q4D × 2 doses, administration of dEpoB over a 24 hr period by i.v. infusion led to both cure and lethality. Thus, dEpoB in Cremophor/EtOH (1:1) 30 mg/kg and Q2D × 6 doses with-6 hr i.v. infusion appeared to be the optimal mode of treatment.
Therapeutic Efficacy Against Human Tumor Xenografts in Nude Mice.
Although dEpoB performed similarly to paclitaxel against MX-1 tumor xenografts when administered i.p. in DMSO solvent (22), marked toxicity and rather poor therapeutic effects were observed when dEpoB was administered i.p. in Cremophor/EtOH (1:1). Likewise, far superior therapeutic effects were observed with i.p. administration of dEpoB in DMSO than i.p. administration of paclitaxel in Cremophor/EtOH formulation (21). In light of these discoveries, we felt it was important to focus on conditions that are optimal for paclitaxel. When i.v. injection was used with either DMSO or Cremophor/EtOH as a solvent, paclitaxel toxicity was markedly reduced and showed good therapeutic effects. Whereas dEpoB showed considerable toxicity in i.v. bolus injection (Table 2), both dEpoB and paclitaxel (6-hr i.v. infusion, Q2D × 5 doses, Cremophor/EtOH solvent, at tolerable doses for each) showed marked therapeutic effects and achieved full cure for the non-MDR MX-1 tumor xenograft (Fig. 2). Likewise, identical administration of dEpoB and paclitaxel against human colon HT-29 xenograft in athymic nude mice significantly diminished the established tumor.
Figure 2.

Therapeutic effect of dEpoB and paclitaxel in nude mice bearing MX-1 xenografts after Q2D × 5 6-hr i.v. infusion. Human mammary carcinoma (MX-1) tissue (40 mg) was implanted s.c. into nude mice on day 0. I.v. infusions were given on days 8, 10, 12, 14, and 16. ⋄, control with vehicle only; ○, Taxol 15 mg/kg; ▵, Taxol 24 mg/kg, and ×, dEpoB 30 mg/kg. The average tumor volumes of the control group on days 14, 16, and 18 were 170 ± 10, 246 ± 29, and 345 ± 42 mm3 (mean ± SEM; n = 5), respectively. The vehicle for 6-hr, i.v. infusion was 100 μl of Cremophor/EtOH (1:1) + 3.5 ml of saline solution.
For MDR-mammary MCF-7/Adr xenografts, the therapeutic effect of dEpoB was far superior to paclitaxel (89% versus 27% average tumor-size reduction, respectively), although Q2D × 5 doses, i.v infusion over a 6-hr period did not achieve a full cure with either paclitaxel or dEpoB (Fig. 3). For comparison, DX, VBL, and VP-16 were injected i.v. Q2D × 5 to nude mice bearing the refractory MCF-7/Adr xenograft by using the manufacturers’ formulations. In these experiments, DX demonstrated a lack of therapeutic effect even at the near-lethal dose, and both VBL and VP-16 showed little therapeutic effect (Fig. 3). For the cross-resistant CCRF-CEM/paclitaxel human T cell leukemic cells that were 57-fold resistant to paclitaxel, dEpoB achieved a full cure against the nude mice xenografts with dEpoB (Q2D × 5 doses, 6-hr i.v. infusion), whereas paclitaxel given under exactly the same conditions showed no significant therapeutic effects (Fig. 4).
Figure 3.

Therapeutic effect of dEpoB and paclitaxel in nude mice bearing MCF-7/Adr xenografts following Q2D × 5 i.v. treatment. DX-resistant human mammary adenocarcinoma (MCF-7/Adr) tissue (50 mg) was implanted s.c. into nude mice on day 0. Six-hour i.v. infusions for control, dEpoB, and paclitaxel and i.v. injections for VBL, DX, and VP-16 were given on days 8, 10, 12, 14, and 16. ⋄, control with vehicle only; □, VBL 0.8 mg/kg; ▵, Taxol 24 mg/kg; +, DX 3 mg/kg; ○, VP-16 30 mg/kg; and ×, dEpoB 30 mg/kg. The average tumor volumes of the control group on days 14, 16, and 18 were 1,281 ± 145, 1,767 ± 161 and 2,381 ± 203 mm3 (mean ± SEM; n = 5), respectively. The vehicle for 6-hr i.v. infusion was 100 μl of Cremophor/EtOH (1:1) + 3.5 ml of saline solution. The vertical bars are the SEM for the control, dEpoB, and Taxol.
Figure 4.

Therapeutic effect of dEpoB and Taxol in nude mice bearing CCRF-CEM/paclitaxel xenografts after Q2D × 5 6-hr i.v. infusion. Human T cell lymphoblastic leukemia (CCRF-CEM/paclitaxel) cells resistant to Taxol (107 cells) were inoculated s.c. into nude mice on day 0. I.v. infusion was given on days 6, 8, 10, 12, and 14. ⋄, control with vehicle only; □, Taxol 20 mg/kg; and ×, dEpoB 30 mg/kg. The average tumor volumes of the control group on day 12, 14, 16, 18, and 24 were 20 ± 3, 53 ± 6, 77 ± 8, 119 ± 22, and 415 ± 62 mm3, respectively (mean ± SEM; n = 3). The vehicle for 6-hr i.v. infusion was 100 μl of Cremophor/EtOH (1:1) + 3 ml of saline solution.
Structure-Activity Relationships.
Previous research from our laboratory (6, 14, 18), and others (ref. 28, and references therein), has allowed for division of the epothilones into zones with respect to their degrees of tolerance for structural modification. Previous investigations determined that modification of the functionality extending from C-12 was tolerated well. Table 3 depicts the relative effects of modifying functionality at C-12 as it relates to both the sensitive (CCRF-CEM) and resistant (CCRF-CEM/VBL) cell lines.
Table 3.
Structure-activity relationship of C-12 substitutions
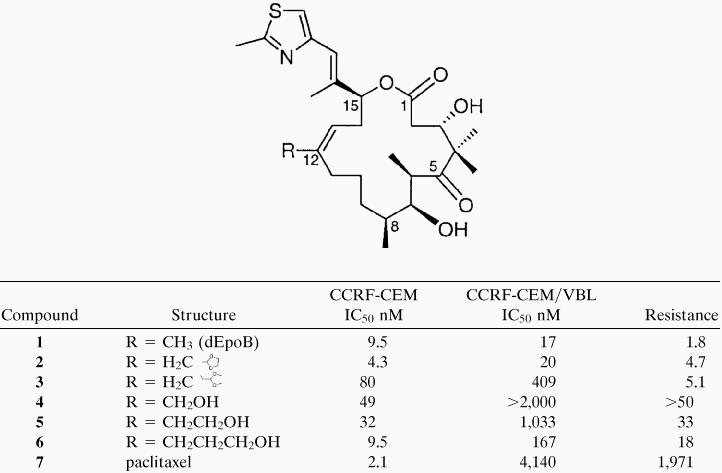 |
Results of in vitro studies performed on both sensitive, CCRF-CEM, and resistant (CCRF-CEM/VBL) cell lines. IC50 values for both the sensitive and the resistant cell lines are given in nM concentrations. Resistance relates the efficacy of the compound relative to sensitive and resistant cell lines for each individual drug. Thus, resistance = [IC50 resistant]/[IC50 sensitive], for a given compound.
In this account, we demonstrate further that the functional groups present on C-12 have a profound consequence on the susceptibility of the compound to succumb to multidrug resistance (Table 3). Thus, the more polar alcohol functionality provided analogues (compounds 4–6) that were MDR substrates and were presumably exported from the cell more efficiently by the drug-efflux transport P-glycoprotein. Interestingly, the length of the alkyl chain in the alcohol series significantly affects the multidrug-resistance susceptibility. By contrast, the analogues that are in the aldehyde oxidation state but protected as the ethylene glycol acetal (compounds 2 and 3) are active and show little resistance to MDR cell lines. The potency of the drug is related to the number of carbons between C-12 and the acetal linkage. Thus, the 3-carbon acetal (compound 3) is less potent that the 2-carbon acetal (compound 2). Interestingly, compound 2 is both resistant to drug efflux and is also 2-fold more potent against the sensitive tumor CCRF-CEM than lead compound dEpoB.
DISCUSSION
Our recent report (21) indicated that when administered i.p., dEpoB showed much better therapeutic results than paclitaxel against MX-1 and MCF-7/Adr xenografts and significantly better therapeutic effects than adriamycin and camptothecin against MCF-7/Adr xenografts in nude mice. However, an i.p. route of administration for dEpoB may lead to an unfair comparison with clinically used paclitaxel because a much higher maximal tolerated dose can be achieved for paclitaxel when it is administered either by i.v. injection or by infusion with a Cremophor/EtOH formulation. This notion is substantiated by our finding (21) that similar antitumor efficacy can be reached when dEpoB (i.p. in DMSO) and paclitaxel (i.v. in Cremophor/EtOH) are deployed against non-MDR SK-OV-3 ovarian tumor, wherein the dose for paclitaxel can be increased at least 3-fold over its i.p. route. Therefore, in the present studies, i.v. infusion with Cremophor/EtOH as a vehicle was used for both dEpoB and paclitaxel. Indeed, both drugs showed remarkable therapeutic results against MX-1 with a complete cure. The onset of action for paclitaxel was faster (Fig. 2). Likewise, both paclitaxel and dEpoB performed similarly against sensitive HT-29 colon xenografts when administered as above by slow i.v. infusion.
The most intriguing conclusions in this study are the indications that for cross-resistant, refractory tumors (such as MCF-7/Adr or CCRF-CEM/paclitaxel), the therapeutic effects of dEpoB are far superior than those of paclitaxel when each drug is administered over a 6-hr period by i.v. infusion using a Cremophor/EtOH formulation (Figs. 3 and 4). In fact, dEpoB actually diminished the established tumor and proved to be curative against CCRF-CEM/paclitaxel, whereas paclitaxel showed no significant efficacy. Another impressive finding is that dEpoB displays remarkable antitumor effects against non-MDR human tumor xenografts as well as refractory human tumor xenografts resistant to widely used antitumor agents such as paclitaxel, adriamycin, vinblastine, and etoposide.
Acknowledgments
This research was supported by the National Institutes of Health Grants CA-28824 (S.J.D.) and Core Grant CA-08748 (T.C.C. and S.K.I.). Postdoctoral Fellowship support is gratefully acknowledged by C.R.H. (American Cancer Society, PF-98-173-001), S.D.K. (US Army Breast Cancer Research Fellowship, DAMD 17-98-1-1854), A.B. (National Institutes of Health Grant CA-GM 72231), and K.A.S. (National Institutes of Health Grant QM-18248). Dr. G. Höfle of the Gesellschaft für Biotechnologische Forschung is gratefully acknowledged for providing natural epothilone A and epothilone B for comparative analysis. We thank Quen-Hui Tang and Luan-Ing Chen for technical assistance.
ABBREVIATIONS
- dEpoB
Z-12,13-desoxyepothilone B (NSC-703147)
- VBL
vinblastine
- DX
doxorubicin (Adriamycin or Adr)
- DMSO
dimethyl sulfoxide
- MDR
multidrug-resistant
Footnotes
Taxol is a trademark of Bristol-Myers Squibb.
Desoxyepothilone B (Epothilone D) has been encountered as a very minor fermentation product in mixtures where epothilone A and epothilone B were the principle products.
References
- 1.Landino L M, MacDonald T L. In: The Chemistry and Pharmacology of Taxol and Its Derivatives. Favin V, editor. New York: Elsevier; 1995. Chap. 7. [Google Scholar]
- 2.Giannakakou P, Sackett D L, Kang Y-K, Zhan Z, Buters J T, Fojo T, Poruchynsky M S. J Biol Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 3.Rose W C. Anti-Cancer Drugs. 1992;3:311–321. [PubMed] [Google Scholar]
- 4.Rowinsky E K, Eisenhauer E A, Chaudhry V, Arbuck S G, Donehower R C. Semin Oncol. 1993;20:1–15. [PubMed] [Google Scholar]
- 5.Ojima I, Bounaud P-Y, Bernacki R J. CHEMTECH. 1998;28:31–36. [Google Scholar]
- 6.Su D-S, Balog A, Meng D, Bertinato P, Danishefsky S J, Zheng Y-H, Chou T-C, He L, Horwitz S B. Angew Chem Int Ed Engl. 1997;36:2093–2096. [Google Scholar]
- 7.Schiff P B, Fant J, Horwitz S B. Nature (London) 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 8.Bollag D M, McQuency P A, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods C M. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 9.Kowalski R J, Giannakakou P, Hamel E. J Biol Chem. 1997;272:2534–2541. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 10.Hoefle G, Bedorf N, Steinmetz H, Schumburg D, Gerth K, Reichenbach H. Angew Chem Int Ed Engl. 1996;35:1567–1569. [Google Scholar]
- 11.Muhlradt P F, Sasse F. Cancer Res. 1997;57:3344–3346. [PubMed] [Google Scholar]
- 12.Service R E. Science. 1996;274:2009. doi: 10.1126/science.274.5295.2009. [DOI] [PubMed] [Google Scholar]
- 13.Balog A, Meng D, Kamenecka T, Bertinato P, Su D-S, Sorensen E J, Danishefsky S J. Angew Chem Int Ed Engl. 1996;35:2801–2803. [Google Scholar]
- 14.Su D-S, Meng D, Bertinato P, Balog A, Sorensen E J, Danishefsky S J, Zheng Y-H, Chou T-C, He L, Horwitz S B. Angew Chem Int Ed Engl. 1997;36:757–759. [Google Scholar]
- 15.Nicolaou K C, Winssinger N, Pastor J A, Ninkovic S, Sarabia F, He Y, Vourloumis D, Yang Z, Li T, Giannakakou P, Hamel E. Nature (London) 1997;387:268–272. doi: 10.1038/387268a0. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z, He Y, Vourloumis D, Vallberg H, Nicolaou K C. Angew Chem Int Ed Engl. 1997;36:166–168. [Google Scholar]
- 17.Schinzer D, Limberg A, Bauer A, Böhm O M, Cordes M. Angew Chem Int Ed Engl. 1997;36:523–524. [Google Scholar]
- 18.Meng D, Su D-S, Balog A, Bertinato P, Sorensen E J, Danishefsky S J, Zheng Y-H, Chou T-C, He L, Horwitz S B. J Am Chem Soc. 1997;119:2733–2734. [Google Scholar]
- 19.Meng D, Bertinato P, Balog A, Su D-S, Kamenecka T, Sorensen E J, Danishefsky S J. J Am Chem Soc. 1997;119:10073–10092. [Google Scholar]
- 20.Balog A, Harris C, Savin K, Zhang X-G, Chou T-C, Danishefsky S J. Angew Chem Int Ed Engl. 1998;37:2675–2678. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2675::AID-ANIE2675>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Chou T-C, Zhang X-G, Balog A, Su D-S, Meng D, Savin K, Bertino J R, Danishefsky S J. Proc Natl Acad Sci USA. 1998;95:9642–9647. doi: 10.1073/pnas.95.16.9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou T-C, Zhang X-G, Danishefsky S J. Proc Am Assoc Cancer Res. 1998;39:163–164. [Google Scholar]
- 23.Goldin A, Venditti J M, MacDonald J S, Muggia F M, Henney J E, DeVita J T., Jr Eur J Cancer. 1981;17:129–142. doi: 10.1016/0014-2964(81)90027-x. [DOI] [PubMed] [Google Scholar]
- 24.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren J T, Bokesch H, Kenny S, Boyd M R. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 25.Scudiero D A, Shoemaker R H, Paull K D, Monks A, Tierney S, Nofziger T H, Currens M J, Seniff D, Boyd M R. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]
- 26.Chou T-C, Talalay P T. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 27.Chou J, Chou T-C. Dose-Effect Analysis with Microcomputers: Quantitation of ED50, Synergism, Antagonism, Low-Dose Risk, Reception-Rigand Binding and Enzyme Kinetics, IBM-PC software and manual. Cambridge, U. K.: Biosoft; 1987. [Google Scholar]
- 28.Nicolaou K C. Angew Chem. Int Ed Engl. 1998;37:2014–2045. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2014::AID-ANIE2014>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]