

Abstract
Two small polypeptides, PorA and PorH, are known to form cell wall channels in Corynebacterium glutamicum and in Corynebacterium efficiens. The genes coding for both polypeptides are localized in close proximity to one another between the genes coding for GroEl2 and a polyphosphate kinase (PKK2). In this study, we investigated the relationship of PorA and PorH to one another. The results suggested that the major cell wall channels of Corynebacterium glutamicum, Corynebacterium efficiens, and Corynebacterium diphtheriae need the obligatory presence of two distinct polypeptides, one of class PorA and one of class PorH, to form an active cell wall channel. Identification of genes coding for homologous proteins in the chromosome of Corynebacterium callunae suggested a similar result for this strain. Contrary to our previous reports on channel-forming proteins in these strains, a heterooligomeric structure composed of PorA and PorH is needed in all of them to form the major cell wall channel. This was concluded from complementation experiments using a porH- and porA-deficient C. glutamicum strain. The stringent necessity of proteins of either class to recover the wild-type channels was demonstrated by black lipid bilayer experiments using detergent or organic solvent extracts of the complemented porH- and porA-deficient C. glutamicum strain. The channel-forming capability of recombinant expressed, affinity-purified PorA and PorH proteins of C. glutamicum revealed that the channels consisted solely of these two components. This agreed with results obtained from a transcript coding for both channel-forming components identified in C. glutamicum by Northern blot analysis and reverse transcription-PCR analysis. The transcription start point of the genes was determined by the rapid amplification of cDNA ends approach, allowing the prediction of the −35 and −10 regions of the promoter. The results demonstrate that the cell wall channels within the genus Corynebacterium may be formed by two-component oligomers.
Micrococcus glutamicus, first described in 1957 by Kinoshita and coworkers, was isolated from a soil sample at Ueno Zoo (Tokyo, Japan) (27). In a screening project, the Gram-positive, rod-shaped, and nonsporulating species turned out to naturally secrete l-glutamate, the substance responsible for the spicy taste of the Asian cuisine called umami. Promoted by this finding, M. glutamicus (later renamed Corynebacterium glutamicum) has made its impact on the fermentation industry and in the following decades became the main producer of the flavor enhancer l-glutamate and the feed additive l-lysine (16, 20, 29, 41). Assisted by the multiple and independent decipherment of the genomes of two C. glutamicum species (23, 25, 67), identification of proteins involved in the synthesis and translocation of amino acids was intensively pursued. In recent years, various amino acid exporters have been suggested, such as LysE, ThrE, and BrnFE, taking part in the flux of lysine, threonine, and methionine, respectively (55, 63, 66). Due to the distinctive features of the cell envelope of the genus Corynebacterium, overall fluxes of these and other substances cannot, however, be explained exclusively on the basis of cytoplasmic-membrane-located transporters (12).
Organisms of the genera Corynebacterium, Mycobacterium, and Nocardia constitute the CMN group of Gram-positive bacteria. Characteristic of these bacteria is the existence of two lipid layers as part of their envelope. The inner layer is the cytoplasmic membrane, which contains glycerophospholipids found in most other biological membranes. In addition to the cytoplasmic membrane, the members of the CMN group have another efficient permeability barrier, the so-called mycolate membrane (7, 33, 34, 39, 48, 56, 60, 69). This membrane consists mainly of mycolic acids (alpha-branched, beta-hydroxylated fatty acids) attached covalently to an arabinogalactan-peptidoglycan complex and free trehalose derivatives (11, 43, 62). Similar to the lipopolysaccharide layer of Gram-negative bacteria, this membrane prevents the passage of hydrophilic and charged compounds (24, 38, 39, 60, 69). In line with the situation in Gram-negative bacteria, in members of the CMN group (the mycolata), facilitation of the uptake of nutrients and other chemical substances is presumably also provided by specialized water-filled channels. Channel-forming proteins in the cell wall of mycobacteria (35, 36, 61), corynebacteria (10, 21, 30), nocardiae (47), and closely related genera (45, 46) support the assumption.
In industry, Corynebacterium species used in the fermentative processing of the amino acid l-glutamate are nevertheless further permeabilized to obtain higher productive yields. Methods commonly applied to increase cell wall permeability include the addition of detergents or chemicals, biotin limitation, and a temperature upshift. Although these rather unspecific methods have different points of contact, they all affect the mycolate layer by changing its lipid composition or fluidity, which indicates that the corynebacterial outer membrane represents a barrier to the flux of solutes (15, 18, 28, 44). A deeper knowledge of the porins of mycolata that establish the natural main passage across the outermost membrane could make such treatments unnecessary and could presumably be a great economic advantage. Similarly, this knowledge could also be of great importance for the design of drugs, which has, in recent years, become a major challenge to meet the fast worldwide emergence of highly antibiotic-resistant pathogens such as Mycobacterium tuberculosis, Corynebacterium jeikeium, or Corynebacterium urealyticum (54, 57, 58). For the development of new antimicrobial compounds precisely targeting essential intracellular components, knowledge of the structure of cell wall channels could be a tremendous help.
Here, we report that the main cell wall channel of C. glutamicum, its close relative Corynebacterium efficiens (14, 49), and a nonpathogenic strain of Corynebacterium diphtheriae (2) is composed of two small proteins designated PorH and PorA. A heterooligomer is thus predicted to form the cell wall channels of these corynebacteria. This was concluded from reconstitution experiments in which the respective major cell wall channels were expressed in a C. glutamicum strain deficient in PorH and PorA. The reconstitution of the channels was studied by the black lipid bilayer technique using cell extracts and purified proteins. This report is of further interest because the PorH and PorA homologues of Corynebacterium callunae, another prominent amino acid producer, were identified.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in this study are summarized in Table 1. C. glutamicum was grown in brain heart infusion (BHI) medium (Difco Laboratories) or CGXII minimal medium [20 g (NH4)2SO4, 5 g urea, 1 g KH2PO4, 1 g K2HPO4, 0.25 g MgSO4 · 7H2O, 42 g morpholinepropanesulfonic acid (MOPS), 10 mg CaCl2, 10 mg FeSO4 · 7H2O, 0.2 mg CuSO4, 0.02 mg NiCl · 6H2O, 10 mg MnSO4 · 2H2O, 1 mg ZnSO2 · 7H2O, 30 mg protocatechuic acid (3,4-dihydroxybenzoic acid), and 0.2 mg biotin/liter of distilled water, adjusted with NaOH to pH 7.0] supplemented with 2.5% glucose at 30°C with shaking at 150 rpm or maintained on BHI agar. If required, agar plates and liquid media were supplemented with 20 or 40 μg/ml chloramphenicol.
TABLE 1.
Strains used in this study for cloning, RNA isolation, and porin complementation
| Strain | Description | Reference |
|---|---|---|
| C. glutamicum | ||
| ATCC 13032 | Wild type | DSMZa |
| CglΔA | ATCC 13032 ΔporA | 9 |
| CglΔHΔA | ATCC 13032 ΔporH ΔporA | 53 |
| C. efficiens AJ 12310 | Wild type | DSMZ |
| C. callunae ATCC 15991 | Wild type | DSMZ |
DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany).
Plasmids and DNA manipulations.
The channel-forming capabilities of PorH- and PorA-like proteins of different Corynebacterium strains were studied by cloning the corresponding genes into the multicloning site of the chloramphenicol resistance-conferring shuttle vector pXMJ19 (51). All plasmids were sequenced (Seqlab, Germany) prior their transformation into the channel-deficient C. glutamicum ΔporH ΔporA mutant according to a slightly modified standard electrotransformation method (64).
Cloning of C. glutamicum porins.
From chromosomal C. glutamicum DNA, the individual genes cgporH (i) and cgporA (ii) and a fragment comprising both genes (iii) were PCR amplified, including small flanking regions (data not shown). The PCR mixtures contained 1× Taq buffer, 0.2 mM deoxynucleoside triphosphates (dNTPs), 3 mM MgCl2, 1 U Taq DNA polymerase (Fermentas, St. Leon-Rot, Germany), and 0.4 μM primers in the following combination: FP Cg_HA_XbaI/RP Cg_H_BamHI (i), FP Cg_A_XbaI/RP Cg_HA_BamHI (ii), and FP Cg_HA_XbaI/RP Cg_HA_BamHI (iii) (Table 2). The PCR products were digested with BamHI and XbaI and ligated into the likewise cut plasmid pXMJ19 using T4 DNA ligase (Fermentas). The resulting plasmids were designated pXCg-H (i), pXCg-A (ii), and pXCg-HA (iii).
TABLE 2.
Oligonucleotides used in this studya
| Oligonucleotide | Sequence 5′ → 3′ | Position |
|---|---|---|
| FP Cg_HA_XbaI | GCAAACTGGCAACCATCTCTAGATTTCTTTGCTGGT | 2888391-2888356b |
| RP Cg_HA_BamHI | GGATCAGAGTTTTTGGATCCTTTGCCCGTGGGC | 2887933-2887901b |
| FP Cg_A_XbaI | CACAGCCTTCCCCTCTAGACCTCATCTCAACTCTT | 2888130-2888096b |
| RP Cg_H_BamHI | GGAAGGCTGTGGATCCTAGCCAATCAGCCAAATCG | 2888154-2888119b |
| RP Cg_H_EcoRI | GGAAGGCTGTGAATTCTAGCCAATCAGCCAAATCG | 2888154-2888119b |
| FP Seq | GTGAGCGGATAACAATTTCAC | |
| RP Insert | CTCTCATCCGCCAAAACAGC | |
| FP A_NHisXa_NheI | AGAATTTAAAATGGCTAGCCGCGGATCCCATCACCATCACCATCACCATCACATCGAAGGCCGCGAAAACGTTTACGAGTTCCTTGGA | |
| RP A_NHisXa_NheI | GATGGGATCCGCGGCTAGCCATTTTAAATTCTCCTATTAAGAGTTGAGATGAG | |
| FP H_CHisXa_NheI | ATCGAAGGCCGCGCTAGCCGCGGATCCCATCACCATCACCATCACCATCACTAAGAGAAATCCGATTTGGCTGATTGGCTA | |
| RP H_CHisXa_NheI | GGATCCGCGGCTAGCGCGGCCTTCGATGGAAGAGAAGTTATCCAGATTCTCGCC | |
| FP Ce_HA_XbaI | CAAACTGGCAACCATCTCTAGATTATTTGCTGGTCC | 2727847-2727882c |
| RP Ce_HA_EcoRI | GGACGGGAGCTGAATTCTACGCCGGAGCG | 2727431-2727403c |
| FP Ce_A_XbaI | CCTAGAAGCTGAGATTCTAGACTTTTTCTTAATCTC | 2727628-2727593c |
| RP Ce_H_BamHI | CAGCTTCTAGGTGGATCCCCTAGCAGCAAGA | 2727648-2727618c |
| FP Cd_WTQC1 | GCTATTCACAATAGGATATTTTTGTAGTGCTCTTGCGGC | |
| RP Cd_WTQC1 | GCCGCAAGAGCACTACAAAAATATCCTATTGTGAATAGC | |
| FP Cd_WTQC2 | CTCAAGGAGGAAGATTAGGACATTCAGTTCA | |
| RP Cd_WTQC2 | TGAACTGAATGTCCTAATCTTCCTCCTTGAG | |
| FP Cd_WTQC3 | GAAAGGGGTTAAATTAGCAGAACATTGAGA | |
| RP Cd_WTQC3 | TCTCAATGTTCTGCTAATTTAACCCCTTTC | |
| FP Cg_KO_HA_CHECK2 | ACATGCACGGCAACCTTCCGTTAAC | 2888780-2888756b |
| RP Cg_OPERON_1_5 | CTTCAGCAGCTCGATCTGGAG | 2887621-2887601b |
| FP Cg_H | ATGGATCTTTCCCTTCTCAAGG | 2888336-2888315b |
| RP Cg_H | TTAGGAAGAGAAGTTATCCAGA | 2888184-2888163b |
| RP Cg_A | TTAGCCAAGCAGACCGATGAG | 2887942-2888308b |
| RP Cg_Extension2 | CACCGAAGGTCTCGTAGTTGCCGA | 2888285-2888308b |
| FP Cc_HA1 | GACCACTGAGGCTGTCGTTGCTGACAAG | 2888961-2888988b |
| RP Cc_HA3 | TTGAAGCGCTTAATGGTGCCACCCTTACC | 2887506-2887534b |
In this study, the PorH and PorA proteins of C. glutamicum were purified by immobilized metal ion affinity chromatography (IMAC). Required histidine residues were factor Xa cleavable and attached either to the 3′ end of cgporH or to the 5′ end of cgporA. This was performed by a nested PCR approach using plasmid pXCg-HA as the template, buffer D, and the Failsafe polymerase of the FailSafe PCR system (Biozym Scientific, Oldendorf, Germany) as described by the manufacturer. The PCR products obtained with primers FP Seq/RP Cg_H_CHisXa_NheI and FP Cg_H_CHisXa_NheI/RP Insert (both used to construct fragment HCHisA), as well as the products of FP Seq/RP Cg_A_NHisXa_NheI and FP Cg_A_ NHisXa_NheI/RP Insert (both used to construct fragment HANHis), were NheI digested. Fragments belonging together were ligated and subjected to a further PCR using the flanking primers FP Seq/RP Insert. Subsequent to HindIII-EcoRI double digestion, amplification products were ligated into the equally opened pXMJ19 vector, resulting in pXCg-HCHisA and pXCg-HANHis.
The genes cgporHCHis and cgporANHis were derived by PCR from pXCg-HCHisA and pXCg-HANHis, respectively. cgporHCHis was cloned with primers FP Seq/RP Cg_H_EcoRI, and the amplicon was inserted into the HindIII-EcoRI-cleaved pXMJ19 vector, leading to pXCg-HCHis. cgporANHis was isolated with primers FP Cg_A_XbaI/RP Insert. The PCR product and plasmid pXMJ19 were XbaI-EcoRI digested and ligated to obtain pXCg-ANHis.
Cloning of C. efficiens porins.
Genomic DNA of C. efficiens was used for PCR amplification of chromosomal stretches that were slightly larger than the individual genes ceporH (I) and ceporA (II) and a fragment embedding both genes (III) (22). The PCR products of FP Ce_A_XbaI/RP Ce_HA_EcoRI (II) and FP Ce_HA_XbaI/RP Ce_HA_EcoRI (III) were double digested with XbaI and EcoRI and ligated into the backbone of the equally cut pXMJ19 vector, resulting in pXCe-A (II) and pXCe-HA (III). Subsequent to XbaI and BamHI cleavage, the amplicon of FP Ce_HA_XbaI/RP Ce_H_BamHI (I) was inserted into the suitably digested pXMJ19 vector, and the resulting plasmid was designated pXCe-H.
Cloning of C. diphtheriae porins.
Plasmid pXCd-WT corresponds to the expression plasmid described by Schiffler et al. (53). It contains a PCR-amplified genomic fragment of C. diphtheriae ATCC 11913. Participation of fragment-encoded polypeptides complementing the channel deficiency of the C. glutamicum ΔporH ΔporA mutant was studied by site-directed mutagenesis. Therefore, the TOPO 2.1 vector with the original PCR amplicon of strain ATCC 11913 (53) was subjected to QuikChange (QC) PCR using Pfu DNA polymerase (Fermentas) and QC primers in Table 2 (e.g., FP Cd_WTQC1/RP Cd_WTQC1) as described by the manufacturer. Amplicons that contained the intended mutations were cut from the TOPO 2.1 plasmid with EcoRI and XbaI and inserted into the backbone of pXMJ19. The resulting vectors were designated pXCd-WTQC1, pXCd-WTQC2, and pXCd-WTQC3 (Fig. 1).
FIG. 1.

ORF analyses of a plasmid-encoded C. diphtheriae fragment. Depicted is the fragment of strain C. diphtheriae ATCC 11913 which is an integral part of plasmid pXCd-WT. The fragment complements the channel deficiency of the C. glutamicum ΔporH ΔporA mutant (53). Gene silencing disclosed that the 59-amino-acid small protein CdPorH, together with CdPorA, accounted for the channel activity. The polypeptide sequence of CdPorH shows 20 and 24% identify to PorH of C. efficiens and C. glutamicum, respectively. The genes cdporH and cdporA of C. diphtheriae are highlighted in black. Dots below the sequence of the cloned C. diphtheriae fragment indicate identical nucleotides, whereas mutations are specified in boldface type. Boldface italics show a putative ribosome binding site (RBS) in front of each porin gene. Underlined italics mark the XbaI and EcoRI sites used for cloning. A transcriptional terminator is specified by underlined letters (53). The specific size of the fragment in the plasmid is displayed by the numbers behind the DNA sequence. The asterisks specify plasmids with a functional C. diphtheriae channel domain.
Cloning of the porin domain of C. callunae.
The region that encodes the PorH and PorA homologues of C. callunae was cloned with the FailSafe PCR system. Its amplification was carried out using chromosomal DNA of C. callunae strain ATCC 15991 as the template, primers FP Cc_HA1/RP Cc_HA3 (Table 2), Failsafe polymerase, and buffer E according to the manufacturer's advice.
Extraction of RNA.
Total bacterial RNA was extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany) as described by the supplier, with minor modifications. The bacterial pellet of 10 ml of a C. glutamicum culture grown to an optical density at 600 nm (OD600) of about 3 was resuspended in 700 μl RLT lysis buffer (Qiagen) supplemented with β-mercaptoethanol and transferred to a 2-ml screw-cap tube containing 300 mg glass beats. The sample was homogenized in a Fastprep120 cell disrupter (ThermoSavant) at maximum speed for 45 s at 4°C. Glass beads and cell wall debris were then removed by centrifugation (13,000 rpm, 1 min, room temperature), and the supernatant was mixed with 500 μl ethanol before it was loaded onto an RNeasy column. RNA isolation was then performed as described in the supplier's protocol. The concentration of the eluted RNA was determined by measuring the absorbance at 260 nm. Purity of the RNA eluate was claimed if the 260/280-nm absorbance ratio was above 1.9. For downstream applications, such as Northern blotting, RNA integrity was checked visually by denaturing formaldehyde agarose gel electrophoresis combined with ethidium bromide staining of 16S and 23S rRNA bands.
DIG-labeled DNA probes.
The DNA probes used for transcript analysis were complementary to the cgporH and cgporA RNA sequences of C. glutamicum. Probe_porH (5′-TCCAGATTCTCGCCGGTGGTGTCAGCGAGA-3′) and probe_porA (5′-CCGATGAGGTCAGCAACTGCGCCAGCCCAC-3′) were designed on basis of nonhomologous gene stretches and received as 3′ digoxigenin (DIG)-labeled oligonucleotides (MWG Biotech, Ebersberg, Germany). Prior to hybridization, the probes were denatured by incubation at 95°C for 10 min.
Northern blot analysis.
For Northern blot analysis, 5 μg of C. glutamicum total RNA isolated from the wild-type strain or the ΔPorA mutant was fractionated on a denaturing agarose gel (2% agarose, 1× MOPS [1× MOPS is 20 mM MOPS, 5 mM sodium acetate, 2 mM EDTA, pH 7], 2% formaldehyde). The size-separated total RNA was then transferred to Nytran N membrane (Schleicher & Schuell, Germany) by overnight capillary transfer (50) with 20× SSC (3 M NaCl, 300 mM sodium citrate [pH 7]) and fixed by UV cross-linking (Stratalinker UV Cross-Linker 2400; Stratagene). Reactive membrane binding sites were blocked by prehybridization in DIG Easy Hyb (Roche Applied Science, Mannheim, Germany) for 45 min at 45°C on a rotary shaker. Hybridization with probes (50 ng/ml) was performed in the same solution at 50°C overnight. Subsequent to the removal of the hybridization solution, membranes were first washed twice for 10 min in 2× SSC-0.1% (wt/vol) sodium dodecyl sulfate (SDS) at room temperature and then twice for 15 min in 0.1× SSC-0.1% (wt/vol) SDS at 50°C. Bound, DIG-labeled probes were detected using the DIG Luminescent Detection kit (Roche Applied Science) with CSPD (provided with the kit) as the alkaline phosphatase substrate according to the instructions of the manufacturer. Developed blots were exposed to autoradiography film (HyperfilmMP; GE Healthcare, United Kingdom) for 30 to 60 min. Size determination of detected signals was performed on the basis of ethidium bromide-stained 16S and 23S rRNA bands (1.5 and 2.9 kbp, respectively).
RT.
Traces of contaminating chromosomal DNA in a total RNA eluate were eliminated by DNase I (Turbo DNA; Ambion, Foster City, CA) digestion. Afterwards, the added enzyme was removed with the Ambion DNase inactivation reagent. Reverse transcription (RT) was performed, with minor variations, according to the supplied two-step protocol of the Enhanced Avian RT-PCR kit (Sigma-Aldrich, Munich, Germany). In the first step, 1.5 μg of total RNA was reverse transcribed into single-stranded cDNA. The 20-μl reaction mixture contained 1× avian myeloblastosis virus (AMV) RT buffer, 250 μM dNTPs, 1.25 μM random nonamers, 20 U RNase inhibitor, and 20 U of AMV reverse transcriptase. The reaction mixture was incubated for 15 min at 25°C and for an additional 50 min at 45°C. In the second step, a target of the cDNA was PCR amplified in a 25-μl reaction mixture containing 3 μl of the cDNA, 1× AccuTaq buffer, 200 μM dNTPs, 1.25 U AccuTaq LA DNA polymerase mix, and 0.4 μM primers (see Results). Identical PCRs were set up with not reverse transcribed total RNA used as the template to exclude false-positive results.
5′ RACE.
The transcription start point (TSP) was determined by the rapid amplification of cDNA ends (RACE) method. Following the protocol supplied with the chosen 5′/3′ RACE kit (Roche Applied Science), 1.3 μg of DNase I-treated total C. glutamicum RNA was reverse transcribed into first-strand cDNA using the gene-specific primer RP Cg_H (Table 2). The reaction mixture was incubated for 60 min at 55°C. The cDNA, purified with the High Pure PCR Product kit (Roche Applied Science), was poly(A) tailed by a terminal transferase at its 3′ end. The modified cDNA was subsequently PCR amplified using Taq DNA polymerase (Fermentas) and primers Oligo dT-Anchor (Roche)/RP Cg_Extension2. The resultant PCR product was gel purified and subjected to a second PCR with the nested primers PCR Anchor (Roche)/RP Cg_Extension2. This amplicon was again gel purified and sequenced with primer RP Cg_Extention2 (Seqlab).
Isolation of channel-forming proteins.
For isolation of the channel-forming proteins, a method was used that has previously been described (30). It is based on the extraction of whole cells with detergents or organic solvents and avoids the substantial loss of material caused by sucrose density centrifugation of the cell envelope to separate the cytoplasmic membrane from the cell wall fraction. Cells of a 100-ml overnight BHI culture of C. glutamicum were grown to an OD600 of about 6 and harvested by centrifugation (5,000 rpm for 10 min in a Heraeus Minifuge RF centrifuge). The bacterial pellet was washed twice with 1/10 culture volume of Tris buffer (10 mM Tris [pH 8]). One part of the cells (0.3 g [wet weight] of the bacterial pellet) was resuspended in 5 parts detergent (1.5 ml 1% lauryl dimethylamine n-oxide [LDAO], 10 mM Tris [pH 8]) or an organic solution (1.5 ml of chloroform-methanol at a ratio of 1:2 [vol/vol]). The cell suspension was then agitated in a closed tube for 3 h at room temperature. As the channel-forming proteins were expected in the supernatant (21, 22, 30), cells were pelleted using a tabletop centrifuge (10,000 rpm for 10 min at 4°C) and discarded. Proteins isolated with organic solvents were precipitated overnight at −20°C by adding to the chloroform-methanol solution a volume of ice-cold diethyl ether that was ninefold greater than the volume of the solution. The resultant (protein) pellet was resolved in detergent (0.4 to 1% LDAO, 10 mM Tris [pH 8]). The cell preparations were investigated for the presence of channel-forming proteins with artificial black lipid membranes.
Affinity purification with immobilized Ni2+ ions.
The proteins CgPorHCHis and CgPorANHis were purified to homogeneity from pXCg-HCHis- or pXCg-ANHis-complemented ΔporH ΔporA mutant C. glutamicum cells. Overnight isopropyl-β-d-thiogalactopyranoside (IPTG)-induced whole cells were extracted with detergent buffer (1% LDAO, 20 mM Tris [pH 8]) as described above. The supernatant of a centrifugation step (10,000 rpm for 10 min at 4°C) was loaded onto Ni-nitrilotriacetic acid (NTA) spin columns (Qiagen, Hilden, Germany) preequilibrated with buffer 1 (20 mM Tris-HCl [pH 8]). Proteins that interacted with the Ni2+ matrix were washed 10 times with 650 μl of buffer 2 (0.4% LDAO, 50 mM NaCl, 20 mM Tris-HCl [pH 8], 50 mM imidazole). Bound protein was eluted twice with 200 μl of buffer 3 (buffer 2 supplemented with 250 mM imidazole). All centrifugation steps were done at room temperature in a tabletop centrifuge at 2,000 rpm.
Digestion with factor Xa protease.
When necessary, the histidine tag of IMAC-purified proteins was removed by factor Xa treatment. For this purpose, the eluate containing the purified protein was dialyzed against Xa buffer (0.4% LDAO, 1 mM CaCl2, 50 mM NaCl, 20 mM Tris-HCl [pH 6.5]) using the Spectra/Por Micro DispoDialyser (Carl-Roth, Karlsruhe, Germany) for at least 3 h. The dialysate was equipped with 4 U of factor Xa, and cleavage was allowed to proceed overnight.
Protein electrophoresis, immunoblotting, and antibodies.
Protein samples were size separated by Tris-Tricine-12% or 16.5% polyacrylamide gel electrophoresis (PAGE) (52). For protein visualization, the gels were stained with either Coomassie brilliant blue or silver stain (6) or immunoblotted (59). In the case of the latter method, proteins were first transferred to a 0.1-μm nitrocellulose membrane (Protran, BA79; Whatman) using a wet tank blot system with Towbin buffer (25 mM Tris, 192 mM glycine, 20% methanol). Because of the low molecular mass of the porin proteins, the blotting time was 4 min at 350 mA of current. Reactive binding sites on the membrane were blocked with 5% skimmed milk in TBS-T buffer (0.1% Tween, 0.01 M NaCl, 20 mM Tris-HCl [pH 7.5]). According to which porin protein of C. glutamicum (i.e., CgPorH, CgPorA, CgPorHCHis, or CgPorANHis) was examined, polyclonal rabbit anti-PorH antibodies (21), polyclonal rabbit anti-PorA antibodies (30), or monoclonal mouse anti-His antibodies (Amersham Biosciences, United Kingdom) were used as the primary immunoglobulins at dilutions of 1:2,500, 1:8,000, and 1:5,000, respectively. As secondary antibodies, we applied peroxidase-conjugated anti-rabbit or anti-mouse antibodies (Dako, Denmark) at a dilution of 1:5,000. For visualization of detected proteins, the enhanced-chemiluminescence Western blot detection system (GE Healthcare, United Kingdom) and autoradiography film (HyperfilmMP; GE Healthcare, United Kingdom) were used according to the manufacturers' instructions. The exposure time that was necessary until an observable signal appeared varied from 20 s to a couple of minutes, depending on the sample.
Black lipid bilayer assay.
The principles used for the black lipid bilayer experiments have been described previously in detail (4, 5). The core of the experiment is a Teflon chamber which is divided into two aqueous compartments. Both aqueous compartments are connected by a small circular hole with a surface area of about 0.2 mm2. Artificial lipid bilayers were formed by painting over the hole a 1% (wt/vol) solution of diphytanoyl phosphatidylcholine (PC; Avanti Polar Lipids, Inc., Alabaster, AL) in n-decane. For electrical measurements, two Ag/AgCl electrodes are submerged in each compartment. One electrode was connected to a voltage source, which allowed the application of defined membrane potentials on the cis side of the membrane. The resulting current through the membrane was detected by the second electrode on the trans side, which was connected to a current-to-voltage converter made from a Burr-Brown operational amplifier. The amplified signal was monitored on a digital oscilloscope and recorded with a strip chart recorder. Analytical-grade KCl was obtained from Merck (Darmstadt, Germany) and used as a 1 M unbuffered solution. During all experiments, the temperature was maintained at 20°C.
Nucleotide sequence accession numbers.
The PorA and/or PorH sequences of C. efficiens YS-314, C. glutamicum R, C. diphtheriae NCTC 13129 and ATCC 11913, and C. callunae ATCC 15991 have been deposited in the DDBJ/EMBL/GenBank databases under accession numbers BN001295, BN001296, BN001297, BN001298, FN257304, FN257302, and FN257303, respectively.
RESULTS
Northern analysis of the cgporH and cgporA genes of C. glutamicum shows a bicistronic transcript.
A previous study suggested that the cgporH and cgporA genes of C. glutamicum could belong to a transcriptional unit consisting of up to 13 coding DNA sequences (CDS) (21). Putative transcriptional terminators within this gene cluster indicated, however, the possibility of different-size transcripts. We thus performed Northern blot experiments to identify the transcripts of cgporH and cgporA of C. glutamicum because the products of both genes were shown to possess channel-forming activity in previous studies (21, 30). C. glutamicum was cultured in BHI and CGXII media. The ΔporA mutant strain of C. glutamicum served to exclude unspecific binding of applied DNA probes where possible. Cells of the wild-type and ΔporA mutant strains were harvested at the mid-exponential growth phase by centrifugation, and the bacterial pellet was used for total RNA isolation.
Figure 2 shows a Northern blot analysis of the transcripts of cgporH and cgporA of C. glutamicum. The same amount of total RNA of BHI-cultured C. glutamicum and ΔporA mutant cells was blotted onto a nylon membrane after size separation. Transcripts of cgporH (probe_porH) and cgporA (probe_porA) were detected with DIG-labeled DNA probes derived from nonhomologous regions of the genes. Anti-DIG Fab fragments conjugated to alkaline phosphatase were applied, and chemiluminescent signals were recorded for probe detection. In lanes with total RNA of the wild-type strain, probe_porH and probe_porA detected single bands of comparable sizes. No band was observed in lanes with total RNA of the ΔporA mutant strain. A similar result was obtained in the case of cells cultured in minimal medium (data not shown). This means that the transcription of both porin genes occurred independently of the culture medium. Lack of the probe_porH signal in the lane of the mutant strain indicated that the cgporH transcript is somehow affected by the deletion of cgporA (see Discussion).
FIG. 2.

(A) Arrangement of cgporA and cgporH in the genome of C. glutamicum and primers used for Northern blot analysis. (B) Transcript analysis of cgporH and cgporA of C. glutamicum. Total RNA was isolated from the wild-type strain and a mutant strain deficient in cgporA (9). Northern blot assays of a single membrane in which 5 μg of total RNA of C. glutamicum cells grown in BHI medium was probed with either a cgporH or a cgporA probe. A cotranscript of approximately 600 bases was detected.
Each band in Fig. 2B had a size of about 600 bases. The open reading frames (ORFs) of cgporH (174 bp) (21) and cgporA (138 bp) (31) are too small to account for the size of these bands. According to the deposited C. glutamicum genome data (GenBank accession number NC_003450), direct neighbors of cgporH are NCgl2621 (ORF of 1,647 bp) (3) and cgporA, followed by NCgl2620 (ORF of 921 bp) (32). Because of the sizes of the four genes, we deduced the absence of NCgl2621 and NCgl2620 as part of the detected transcripts. Only a transcriptional unit of cgporH and cgporA (taking into account additionally 83 bases of the region between the two genes, Fig. 3) could account for the observed Northern blot signals.
FIG. 3.

Comparison of the genotypes of wild-type and ΔporH ΔporA mutant C. glutamicum strains. The sequence of the mutant strain used for the alignment with the wild-type (WT) sequence is deduced from a sequenced PCR product obtained with primers FP Cg_KO_HA_CHECK2/RP Cg_OPERON_1_5 (Table 2) and template DNA of the C. glutamicum ΔporH ΔporA mutant (data not shown). The deleted region comprises 472 bp, including both the cgporH and cgporA genes (black highlighted) coding for the components of the main cell wall channel of C. glutamicum. Boldface type indicates CDS adjacent to the porins. The TSP of the cgporH-cgporA cotranscript is marked by a broken arrow. Putative −10 and −35 boxes are shown by underlined italics. Boldface italics indicate putative ribosome binding sites (RBS), whereas double-underlined letters indicate rho-independent hairpin terminators (26). The numbers to the right of the nucleotide sequence correspond to positions in the genome of C. glutamicum (GenBank accession number NC_003450).
RT-PCR analysis confirms the bicistronic transcript of C. glutamicum.
Random nonamers were used to convert the total RNAs of both the wild-type and ΔporA mutant strains of C. glutamicum into cDNA. Using cgporH- and cgporA-specific primers (FP Cg_H and RP Cg_A, Table 2), a single amplicon with a size of about 400 bp (consistent with the predicted size of a transcript containing cgporH-cgporA) was obtained from cDNA of the wild-type strain. According to the cgporA deficiency, no PCR product was detected with these primers in control experiments in which cDNA of the ΔporA mutant strain was used as the template. Chromosomal DNA of the wild-type strain also resulted in a prominent 400-bp PCR product (besides some minor, unspecific PCR products).
Search for the TSP of the cgporH-cgporA transcript.
C. glutamicum cultured in BHI was used for analysis of the 5′ TSP of the porin cotranscript as described in Materials and Methods. Study of the sequenced PCR product determined guanine as the TSP. This point was located 75 bases upstream of the start methionine of CgPorH (Fig. 3). In comparison to the canonical hexamers TTGACA and TATAAT of Escherichia coli and Bacillus subtilis (17, 19), the promoter sequences of Corynebacterium species are less well conserved. Analysis of the region in front of the TSP suggested a putative promoter corresponding to the transcript. The associated −10 TTGAAT and −35 TTGACG hexamers of the porin promoter (Fig. 3) show homology to the −10 TGNGNTA(C/T)AATGG (residues with a high degree of conservation are underlined) and −35 TTGGCA consensus sequences suggested for vegetative genes (42, 65).
Transcriptional terminators flank the cgporH-cgporA region of C. glutamicum.
With the aid of the TransTermHP tool (26), the noncoding region between NCgl2621 and NCgl2620 was searched for inverted repeat sequences that may form stem-and-loop structures. Two distinctive rho-independent terminators were predicted within the region (Fig. 3). The first structure (5′-GCCCTCCCGCACGCTTTGCGGGAGGGC-3′) is located downstream of NCgl2621, whereas the second structure (5′-AAAAACTCTGATCCATATGGATCAGAGTTTTT-3′) is positioned downstream of cgporA. Both sequences presumably form hairpins with free energy levels of −21.8 and −15.7 kcal/mol, respectively (http://www.genebee.msu.su). It is noteworthy that no terminator was found within the intergenic region between cgporH and cgporA.
Functional analysis of the bicistronic porin transcript of C. glutamicum.
The transcriptional unit of the Northern blot analysis suggested the question of whether the CgPorH and CgPorA proteins form the channel in the cell wall of C. glutamicum alone or together. Therefore, we investigated the channel-forming capability of both proteins by taking advantage of the channel deficiency of a ΔporH ΔporA knockout mutant of C. glutamicum (Fig. 4). Detergent and organic solvent preparations of whole cells of this strain do not contain large ion-conducting channels (53) (Fig. 4A). To answer the above question, plasmids coding for PorA (pXCg-A), PorH (pXCg-H), or both (pXCg-HA) were electrotransformed to restore either one or both gene defects of the ΔporH ΔporA mutant. Expression of the porin proteins was induced in each plasmid-complemented strain overnight by IPTG addition. The expressed porin proteins were extracted with organic solvents, and their channel-forming activity was studied using a 1 M KCl solution bathing black lipid membranes formed from PC/n-decane. The precipitate of cgporH- and cgporA-complemented mutant cells (using plasmid pXCg-HA matching the wild-type genotype) showed high channel-forming activity (Fig. 4E). Addition of the precipitate to the aqueous phase (about 10 ng/ml) bathing a black membrane resulted, with a delay of a few minutes, in a stepwise increase in ion conductance. The recorded single-channel events had a long lifetime and were comparable to the prominent 2.5- and 5.5-nS channels of the C. glutamicum wild-type strain, as described previously (21, 30) (Fig. 4D). Surprisingly, no channels were recorded from precipitates of mutant cells complemented solely for the defect of cgporH (pXCg-H) or cgporA (pXCg-A), respectively (Fig. 4B and C).
FIG. 4.

Single-channel recordings for a PC/n-decane membrane in the presence of channel-forming proteins of C. glutamicum. The aqueous phase contained 1 M KCl. The applied membrane potential was 20 mV, and the temperature was 20°C. Arrows indicate fluctuations caused by sample addition. (A to F) Crude organic solvent extracts of C. glutamicum rporHrporA (A), C. glutamicum rporHrporA expressing CgPorA (B), C. glutamicum rporHrporA expressing CgPorH (C), wild-type C. glutamicum (D), C. glutamicum rporHrporA expressing CgPorH and CgPorA (E), and a mixture of C. glutamicum rporHrporA expressing CgPorH and wild-type C. glutamicum (F). (G) A 1:1 protein mixture of purified CgPorHCHis and factor Xa-treated CgPorANHis (20 ng/ml).
Validation of the expression of the CgPorH and CgPorA proteins.
Expression failure could be the cause of the lack of channel-forming activity in mutant cells transformed with pXCg-H or pXCg-A. To check for this possibility, these and other size-separated organic solvent precipitates were immunoblotted with either anti-PorH or anti-PorA antibodies. The 5-kDa CgPorA protein was expressed in the wild-type strain but not in the ΔporH ΔporA mutant strain of C. glutamicum. An identical CgPorA signal (expected because of channel activity) arose from the precipitate of mutant cells complemented with pXCg-HA. Surprisingly, the non-pore-forming precipitate of cells transformed with pXCg-A resulted in an equally strong signal (data not shown). Expression failure and lack of CgPorA were therefore excluded as the causes of the missing channel activity. Immunoblot assays with anti-PorH antibodies unfortunately showed unspecific binding in the low-molecular-mass region (data not shown). The missing CgPorH channel activity in the precipitate of pXCg-H-complemented cells was not readily explained if this protein formed a homooligomeric channel. Based on the Western blot results, we mixed the precipitates from the mutant cells complemented with either pXCg-H or pXCg-A. Interestingly, the mixture (Fig. 4F) showed the same pore-forming activity in the lipid bilayer assay as the precipitate of the mutant strain complemented with pXCg-HA, which agreed again with the assumption that the major cell wall channel in C. glutamicum is formed by a combination of PorH and PorA.
Purification of recombinant CgPorHCHis and CgPorANHis proteins.
Our reconstitution experiments did not provide any indication that other proteins besides CgPorH and CgPorA are involved in the formation of the C. glutamicum major cell wall channel. In order to exclude such a possibility, CgPorH and CgPorA were expressed in the ΔporH ΔporA mutant of C. glutamicum using plasmids pXCg-HCHis and pXCg-ANHis, respectively. This means that after IPTG induction, the cell wall of the mutant contained either recombinant protein CgPorHCHis or CgPorANHis. These proteins were purified to homogeneity using Ni2+ affinity chromatography. According to an overloaded and silver-stained Tricine-containing SDS-PAGE, both purified proteins were free of contaminants (Fig. 5).
FIG. 5.

Silver staining of Ni-NTA-purified, histidine-tagged PorA and PorH proteins of C. glutamicum separated by 12% Tricine SDS-PAGE. The proteins were expressed independently of each other in a ΔporH ΔporA mutant strain of C. glutamicum. Note that the forms of the bands are caused by some overloading of the gels and the highly hydrophobic nature of PorA and PorH.
The cell wall channel of C. glutamicum is exclusively composed of the CgPorH and CgPorA proteins.
From the study of the channel-forming activity of the C. glutamicum ΔporH ΔporA mutant after electrotransformation with plasmids pXCg-HCHisA and pXCg-HANHis, it was found that porin proteins in the combinations CgPorHCHis/CgPorA and CgPorH/CgPorANHis, respectively, did not show inhibition of channel formation (data not shown). This means that an additional 17 amino acids attached to the C terminus of CgPorH or an additional 18 amino acids attached to the N terminus of CgPorA did not result in loss of channel activity. However, when a combination of the purified CgPorHCHis and CgPorANHis proteins was applied in the reconstitution experiments, it was not possible to observe any channel-forming activity. Thus, CgPorANHis was treated with factor Xa to remove the His tag. The mixture of pure CgPorHCHis and CgPorA again caused a stepwise increase in conductance, similar to the CgPorH/CgPorA wild-type combination, as shown in Fig. 4G. Statistical analysis of 164 recorded single-channel events collected from more than three individual membranes suggested that prominent units had single-channel conductances of 2.5 and 5.5 nS (Fig. 6). As in previous studies, it was not possible to decide if the maximum of 5.5 nS was caused by two channels opening at once or by the different configurations of the PorA/PorH channels (21, 30, 31). This means that channels formed by the purified CgPorHCHis and CgPorA monomers matched units of previously examined whole-cell preparations (30). No pore was recorded in control experiments when only detergent or a single component of the C. glutamicum channel was added to lipid membranes (data not shown). These important results indicated that the main cell wall channel of C. glutamicum can be formed solely from PorH and PorA monomers.
FIG. 6.

Histogram of the probability of the occurrence of ascertained conductivity units observed in the experiment of Fig. 4G. The most frequent single-channel conductances were 2.5 nS (left-side maximum) and 5.5 nS (right-side maximum), for a total of 164 single-channel events. The data were collected from more than three individual membranes.
Heterologous expression of PorH and PorA homologues of C. efficiens and study of their channel-forming capabilities.
The major cell wall channel of C. efficiens was previously isolated and characterized in lipid bilayers (22). The slightly anion-selective channel was proposed to be formed by an oligomer of the 6-kDa CePorH protein (Table 3). Its gene, ceporH, is located in close proximity to ceporA (22), the predicted homologous gene to cgporA of C. glutamicum (53). The two Corynebacterium species are close relatives (14), which means that it is possible that the major cell wall channel of C. efficiens is also a heterooligomer formed by the combination of CePorH and CePorA, similar to the situation in C. glutamicum. This question was addressed by heterologous expression of CePorH and CePorA in the C. glutamicum ΔporH ΔporA mutant using plasmids pXCe-H and pXCe-A, containing the genes ceporH and ceporA, respectively, of C. efficiens. Plasmid pXCe-HA harbored both the ceporH and ceporA genes together.
TABLE 3.
Alignment of the PorH and PorA amino acid sequences of C. glutamicum ATCC 13032, C. glutamicum R, C. efficiens AJ 12310, C. callunae ATCC 15991, C. diphtheriae NCTC 13129, and C. diphtheriae ATCC 11913a
 |
The alignment shown was created by European Bioinformatics Institute Sequence Analysis (http://www.ebi.ac.uk/Tools/clustalw2/index.html). The polarities of the charged residues of the proteins are indicated above the sequences. Residues conserved in the homolog proteins are in boldface (100%) or underlined (>50%).
N, calculated number of amino acids.
MW, molecular mass.
pI, isoelectric point (http://www.expasy.org/tools/pi_tool.html).
The protein preparation of the C. glutamicum mutant complemented with ceporH showed no channel-forming activity. The same result was observed when CePorA was expressed in another experiment. A mixture of these non-channel-forming precipitates (in a 1:1 molar ratio), however, yielded high channel-forming activity (Fig. 7A). Likewise, the expression of both proteins in the C. glutamicum ΔporH ΔporA mutant using plasmid pXCe-HA resulted in a protein sample that formed major cell wall channels of C. efficiens (Fig. 7B). Most of the steps (about 60% of all conductance fluctuations) in this experiment had conductances of 2.0 and 4.0 nS (Fig. 7C). The latter step either represented dimers of the 2.0-nS channel that could not be separated with the time resolution of our experimental setup or represented another state of the PorA/PorH channel of C. efficiens, similar to the case of C. glutamicum (see above). It is noteworthy that these results are in good agreement with the previous study of the major cell wall channel of C. efficiens (purified from strain AJ 12310), where an average single-channel conductance of 2.3 ± 0.3 nS was obtained (22). However, it is clear from this study that the major cell wall channel of C. efficiens is not formed by CePorH alone but also needs the presence of CePorA, indicating that the channel-forming unit is also a heterooligomer formed by CePorH and CePorA (Table 3).
FIG. 7.

Single-channel recordings and single-channel analysis of porin proteins of C. efficiens expressed in the C. glutamicum ΔporH ΔporA mutant. The aqueous phase contained 1 M KCl. The applied membrane potential was 20 mV, and the temperature was 20°C. (A) A combination of organic solvent cell wall preparations which contained either the CePorH protein (using plasmid pXCe-H) or the CePorA protein (using plasmid pXCe-A). Each sample itself was non-channel forming. (B) PorH and PorA of C. efficiens were expressed together using plasmid pXCe-HA. About 20 ng/ml ether-precipitated organic solvent extract was added to the aqueous phase on both sides of the membrane. (C) Statistical analysis of the channel conductance of 143 single-channel events of panel B derived from more than three individual membranes.
Identification of a porH-like ORF in close proximity to the cdporA gene of C. diphtheriae.
C. diphtheriae contains a cell wall channel (43). The channel-forming protein was suggested to be CdPorA (for C. diphtheriae PorA). This was determined from the expression of a cloned chromosomal fragment of strain ATCC 11913 in the C. glutamicum ΔporH ΔporA mutant strain (53). In this study, we became interested in the question of whether the cloned C. diphtheriae segment of our previous study also codes for proteins other than CdPorA. Figure 8 demonstrates that the C. diphtheriae fragment (cloned into vector pXCd-WT) indeed codes for additional putative ORFs. Depending on the ATG start codon used, three possible ORF-encoded proteins of 102, 97, and 59 amino acids could be identified preceding the TAG stop codon at nucleotides 474 to 476 in the plasmid insert (Fig. 8). Their genes were localized near cdporA, similar to the situation in the genomes of C. glutamicum and C. efficiens, where porA and porH are cotranscribed. To study the function of the gene products in more detail, these four genes were inactivated by point mutating the respective translational initiation sites (Fig. 8).
FIG. 8.
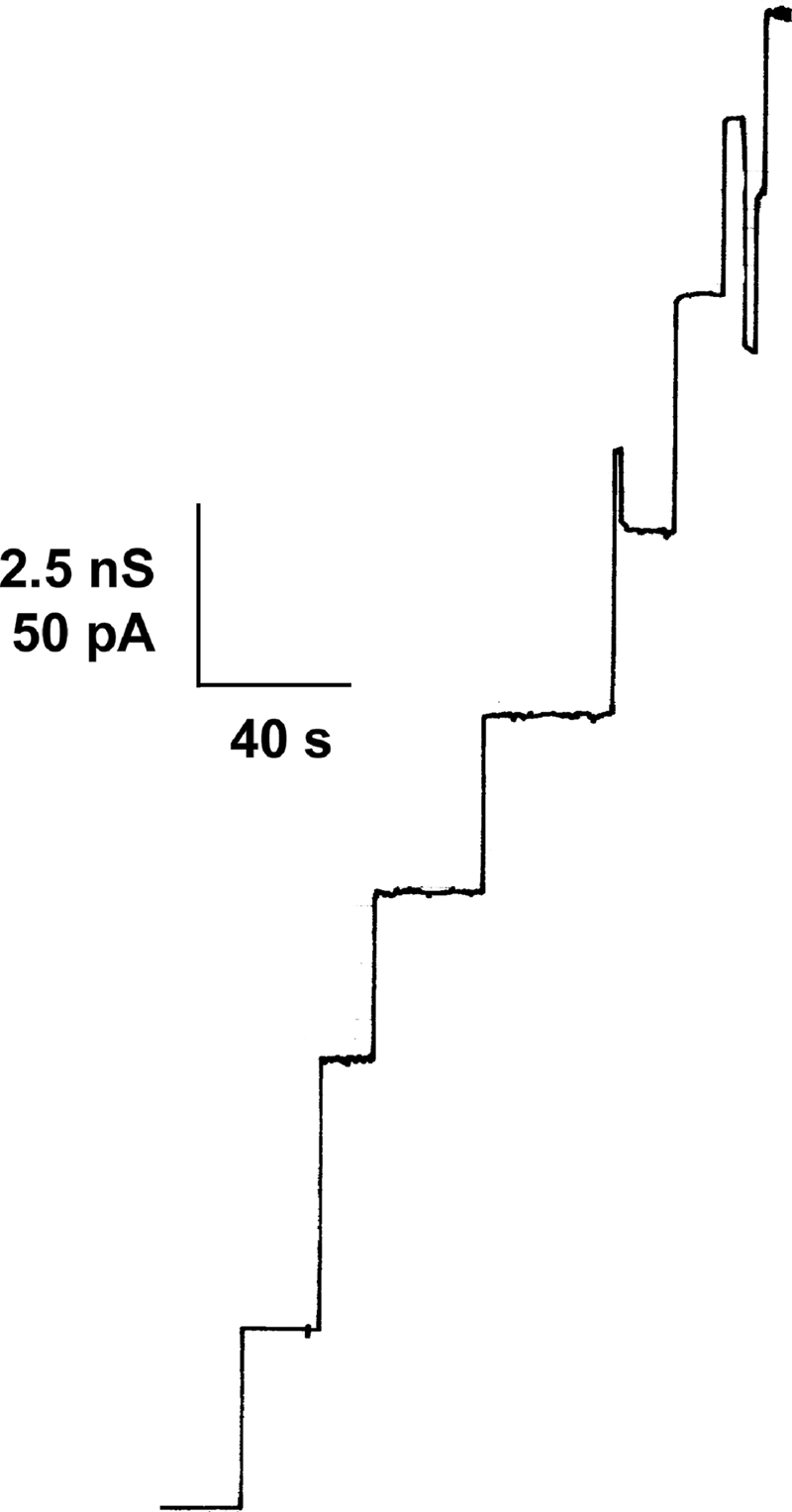
Single-channel recording of a PC/n-decane membrane in the presence of channel-forming proteins of C. diphtheriae expressed in the C. glutamicum ΔporH ΔporA mutant. About 20 ng/ml ether-precipitated organic solvent extract (see text) was added to the aqueous phase on both sides of the membrane. The aqueous phase contained 1 M KCl. The applied membrane potential was 20 mV. The temperature was 20°C.
Reconstitution of the major cell wall channel of C. diphtheriae requires the presence of PorH and PorA homologues of this bacterium.
To identify the proteins involved in forming the major cell wall channel of C. diphtheriae, we transformed the C. glutamicum ΔporH ΔporA mutant strain with plasmids pXCd-WT, pXCd-WTQC1, pXCd-WTQC2, and pXCd-WTQC3 (Fig. 8), respectively. In line with reference 53, the protein preparation of pXCd-WT-transformed mutant cells showed high channel-forming activity with a major conductance step of about 2.3 nS. Results obtained with mutant cells harboring pXCd-WTQC1 were similar (Fig. 9). However, neither pXCd-WTQC2 (expression of CdPorA) nor pXCd-WTQC3 (expression of CdPorH) provided protein samples that were active in the lipid bilayer assay (data not shown). Activity was only observed when CdPorH and CdPorA were expressed together in the C. glutamicum ΔporH ΔporA mutant strain. A BLAST (1) search of the nonredundant GenBank database (6,970,043 sequences, 06/08; National Center for Biotechnology Information) with the CdPorH sequence found only two matches, a significant match to DIP2019 of C. diphtheriae NCTC 13129 (8) and a match to cgR_2618 of C. glutamicum R (67). As Table 3 shows, the protein of the latter CDS is no doubt the homologue of PorH of C. glutamicum ATCC 13032 (21, 23, 25) and also of C. efficiens AJ 12310 (22, 40). DIP2019 (annotated as a putative transposase) corresponds to ORF1 in Fig. 8. The PorH homologue of C. diphtheriae NCTC 13129 thus represents the C-terminal part of DIP2019. As in Fig. 8, cdporH and cdporA (DIP2018) are adjacent to each other in the genome of strain NCTC 13129. It is notable that the two C. diphtheriae PorH sequences exhibit only 69% amino acid sequence identity. cdporH of strain 13129 is 177 bp long and encodes a 58-amino-acid polypeptide containing six aspartates and glutamates, compared to five lysines, i.e., an almost neutral protein. The deduced CdPorH sequence of strain 11913 is 59 amino acids long and contains nine aspartates and glutamates, compared to five lysines and a single histidine, giving the protein an acidic character. Likewise, it is intriguing that the two C. diphtheriae PorA sequences exhibit only 74% amino acid identity (Table 3).
FIG. 9.

Agarose (0.8%) gel electrophoresis of PCR-amplified homologous porin regions of C. glutamicum and C. callunae. Amplicons were obtained with primers FP Cc_HA1 and RP Cc_HA3 using chromosomal DNA of both strains as the template. The sequence of the C. callunae PCR product is shown in Fig. 10.
Cloning of the prospective channel domain of C. callunae.
To date, C. callunae strain ATCC 15991 is the fourth member of the genus Corynebacterium with a well-studied major cell wall channel (22). The cation-selective pore is associated with a low-molecular-mass protein which is clearly a homologue of PorH of C. glutamicum and C. efficiens. Its sequence, however, is only partially known (22). When the results of this study with C. glutamicum, C. efficiens, and C. diphtheriae are considered (see above), it is clear that the major cell wall channel of C. callunae is presumably also formed by two proteins, a PorH homologue and a PorA homologue. Unfortunately, the genome of C. callunae is not yet known well enough to allow a clear judgment. To decide this question, we cloned the presumable porin region of C. callunae using primers FP Cc_HA1/RP Cc_HA3 (Table 2). Both were derived from the CDS NCgl2620/NCgl2621, CE2558/CE2561, and DIP2016/DIP2020 that surround the porH-porA region of C. glutamicum, C. efficiens, and C. diphtheriae, respectively. With C. callunae genomic DNA as the template, these primers yielded a 1,500-bp PCR product (Fig. 10). The C. callunae amplicon was cloned into the TOPO 2.1 vector and sequenced with primers M13 forward and reverse (Fig. 10).
FIG. 10.

Analysis of the C. callunae porin domain. The amplicon of Fig. 9 was cloned into the TOPO 2.1 vector and sequenced. The primers used for cloning of the C. callunae region are marked by underlined italics. The boldface type represents the C. callunae homologues of, e.g., NCgl2620 and NCgl2621 of C. glutamicum ATCC 13032. The identified C. callunae ccporH and ccporA genes (black background) thus have the same chromosomal localization as the porH and porA genes of other corynebacteria investigated in this study. As with C. glutamicum, a putative RBS shown in boldface italics attends each porin gene. Both genes are surrounded by distinct hairpin structures marked as double-underlined letters. Numbers behind the DNA sequence display the specific size of the PCR product inserted into the TOPO 2.1 vector.
Identification of the genes coding for the PorH and PorA homologues of C. callunae.
Two small coding sequences, each with a putative ribosome binding site (5′-AGGAG-3′), were disclosed by a search of the C. callunae amplicon for ORFs (Fig. 10). The protein sequence derived from the first CDS (ccporH) corresponded (with minor variations) to partial amino acid stretches of the directly sequenced CcPorH protein (22). The mature CcPorH protein is 57 amino acids long and has a calculated molecular mass of 5,963 Da. Nine aspartates and glutamates, compared to one lysine, give the polypeptide an overall negative charge (Table 3). Only 80 bp downstream of ccporH is the location of the second CDS. The deduced polypeptide sequence comprises 45 amino acids (with the start methionine) and shows about 24 to 53% homology to PorA of C. diphtheriae, C. efficiens, and C. glutamicum (Table 3). We assumed that the second CDS encodes the PorA protein of C. callunae and named it ccporA (for C. callunae pore-forming gene A). CcPorA has a calculated molecular mass of 4,790 Da and is overall negatively charged (six glutamates and aspartates, compared to two lysines). The results obtained by the investigation of other Corynebacterium species suggest that the major cell wall channel of C. callunae is also a heterooligomer of CcPorH and CcPorA. In this respect, it is interesting that the acidic characters of both proteins agree well with the cation selectivity of the C. callunae cell wall channel (22).
Transcriptional terminators flank the ccporH-ccporA region of C. callunae.
An analysis of inverted repeat sequences that likely form stem-loop structures revealed that there is one transcriptional terminator in the noncoding region downstream of ccgroEL and a second terminator behind the gene of ccporA (Fig. 10); the free-energy levels of the 5′-ATAAGCTCCTCCCGCTTCTTGCGGGAGGAGCTTAT-3′ and the 5′-AAAGCTCCGATCCTTAACAGGATCGGAGCTTT-3′ sequences are −25.7 and −25.9 kcal/mol, respectively (http://www.genebee.msu.su). As no distinct rho-dependent terminator has been spotted between ccporH and ccporA, we believe that these genes form a transcriptional unit of the porin operon responsible for the main cell wall channel of C. callunae, similar to the situation in the genomes of the other Corynebacterium species studied here.
DISCUSSION
The cgporH and cgporA genes constitute a porin operon in C. glutamicum.
In this study, we investigated the transcriptional relationship of the cgporH and cgporA genes of C. glutamicum. The products of these genes have been associated in the past with individual channel-forming units in the cell wall of C. glutamicum; i.e., it was assumed that CgPorH and CgPorA formed homooligomeric channels (21, 31). A 600-bp transcript was found in Northern blot experiments (Fig. 2) that suggested that the two genes were cotranscribed. This cotranscript was observed for C. glutamicum grown under rich and minimal medium conditions, which means that both proteins are expressed under a range of conditions. This cotranscript is responsible for a coordinated expression of CgPorH and CgPorA in equimolecular amounts to form channels of the PorH/PorA type in the cell wall. Other Northern experiments were performed with a C. glutamicum ΔporA mutant strain to study the cgporH transcript. Interestingly, this transcript was not observed in the chromosome of the C. glutamicum ΔporA mutant strain (Fig. 2B). Since cgporH is upstream of cgporA, there was no obvious reason for this observation. Thus, we additionally checked the correctness of the porA deletion on the chromosome of the ΔporA mutant strain (Fig. 11). In RT-PCR experiments using the C. glutamicum ΔporA mutant strain and primers directed against porH (FP Cg_H/RP Cg_H [Table 2]), we became aware of a very faint porH amplimer (data not shown), suggesting that this gene is transcribed. Therefore, we assume that the cgporA deletion affects the stability of the residual cgporH transcript in the ΔporA mutant strain and causes its fast degradation.
FIG. 11.

Ethidium bromide-stained agarose (0.8%) gel electrophoresis of PCR amplification products obtained with primers FP Cg_KO_HA_CHECK2 and RP Cg_OPERON_1_5 (Table 2) binding in the flanking regions of the porH-porA genes of C. glutamicum. Clearly visible are the absence of cgporA in lane 3 and the absence of cgporA and cgporH in lane 4 when the data are compared to PCR in the presence of wild-type (WT) DNA (lane 2).
Consistent with the C. glutamicum genomic data (Fig. 3), RT-PCRs yielded an ∼400-bp PCR product. Compared to the Northern blot analysis results, the smaller size of the RT-PCR amplicon is due to the different experimental setups used. Primers annealing at the 5′ end of cgporH (ATG) and at the 3′ end of cgporA (TAA) yielded only the cgporH-cgporA region. Thus, the noncoded flanking regions which are part of the full-length Northern blot signal (such as the region from the TSP to the start codon of cgporH and the region from the stop codon of cgporA to the putative terminator) are missing in the RT-PCR amplicon (which therefore has a smaller size than the Northern blot signal).
The TSP heading the cgporH-cgporA transcript was determined by RACE analysis. This enabled the prediction of the −10 box. Corynebacterial promoters often show a considerable variation of the nucleotides in this conserved region, which has the extended consensus sequence TGNGNTA(C/T)AATGG (residues with a high degree of conservation are underlined) (42). In seven positions, the postulated −10 box agrees with the tridecamer of vegetative promoters shown (Fig. 3). The −35 box of the promoter of the CgPorH/CgPorA operon has four nucleotides identical to the −35 consensus region for vegetative C. glutamicum promoters (65). Sequence analysis of the flanking regions of the porin operon discovered two inverted repeats, the potential terminator of the bicistronic porin transcript and the potential terminator of NCgl2621, which represents the shock-regulated GroEL chaperon (3). This means that separate cggroEL and cgporH-cgporA transcripts should be formed. It is noteworthy that elements of the C. glutamicum porin operon were also found in the homologous region of the C. callunae (Fig. 10), C. diphtheriae, and C. efficiens chromosomes (22, 53). Inverted-repeat structures flank the proximate porH/porA genes of these species in a similar way, which suggests that these porin genes are also cotranscribed.
The oligomeric major cell wall channels of C. glutamicum and its close relatives consist of homologous PorH and PorA subunits.
The results of the transcriptional analysis suggested that the major cell wall pores of C. glutamicum could be oligomers composed of two proteins, CgPorH and CgPorA. Pore-forming activity in a ΔporH ΔporA mutant C. glutamicum strain was only observed when both proteins were expressed, as in the fully complemented C. glutamicum ΔporAΔporH mutant or the wild-type strain of C. glutamicum. In addition, recombinant expressed and affinity-purified proteins were only active in the lipid bilayer assay when both proteins were present. These data rule out the possibility that another protein is involved in channel formation. It is noteworthy that this observation is consistent with the results of the transcriptional analysis showing a bicistronic porin transcript for C. glutamicum. Consideration of the bilayer results obtained from the CgPorH and CgPorA channel reconstitution experiments also allowed the reconstruction of the PorH/PorA cell wall pore of C. efficiens and C. diphtheriae in C. glutamicum by heterologous expression. As in the case of C. glutamicum, functional channels could only be observed in these cases when both PorH and PorA of these strains were expressed in C. glutamicum. The knowledge of the location of the porA and porH genes in the genomes of C. efficiens, C. diphtheriae, and C. glutamicum also allowed the cloning and sequencing of the complete porin region of C. callunae strain ATCC 15991. Oligomeric channels are not rare among the members of the taxon mycolata. MspA of Mycobacterium smegmatis and PorB of Corynebacterium glutamicum also form oligomeric channel structures; MspA is an octamer, whereas PorB could be a pentamer (13, 68). The PorH and PorA proteins contain, presumably because of their small sizes, only a single membrane-spanning domain. This means that the higher-structure heterooligomer is needed to form the stable, large, ion-conducting channels in the mycolic acid layer and artificial membranes. We do not have a defined picture of the stoichiometry of PorA and PorH in the pore-forming complex. The genetic organization of the different porA and porH genes suggests that the complex contains the same number of PorA and PorH monomers. Lipid bilayer experiments with different combinations of PorA and PorA tend to support this assumption because the highest pore-forming activity was observed when the PorA/PorH mixtures contained approximately equal amounts of the two proteins (data not shown). However, a final answer can only be derived from studies of the three-dimensional structure of the pore-forming complex.
In previous reports, we favored the view that C. glutamicum, C. callunae, and C. efficiens contained two major cell wall channels, each formed by oligomers of PorA and oligomers of PorH (9, 21, 22, 31). This clearly contradicts the finding of this study that the architecture of the major cell wall channels of all of these bacteria, including that of C. diphtheriae, is formed by a heterooligomer of PorH and PorA (Table 3). PorH or PorA alone cannot form a cell wall channel. The previous results were presumably caused by incomplete separation of the PorH and PorA proteins of C. glutamicum, C. callunae, and C. efficiens because of their low molecular mass. Trace amounts of PorH in PorA, or vice versa, that could not be detected by analytical or immunological methods were sufficient for the reconstitution of active channels in the lipid bilayer assay.
For C. glutamicum, previous reconstitution experiments with artificial lipid membranes revealed a prominent single-channel conductance of about 5.5 nS in 1 M KCl (30, 37). A significant but smaller amount of recorded channels had a conductance of 2.5 nS in these experiments. This study revealed a comparable result, but the 2.5-nS channel unit appeared to be more frequent (Fig. 6). This means that it could be that the lower-conductance unit (2.5 nS) corresponded to a single C. glutamicum cell wall channel, whereas the higher-conductance unit (5.5 nS) was most likely caused by the incorporation of two channels at once. This could be concluded from other studies with corynebacterial PorH/PorA cell wall channels. For instance, the single-channel conductance of PorH/PorA channels of C. diphtheriae and C. efficiens was only about 2.3 nS (22, 53). The sequences, as well as other intrinsic properties, of the PorH and PorA proteins aligned in Table 3 suggest that their structures should be very similar, which supports the view that their conductance should also be similar, i.e., close to 2.5 nS in 1 M KCl and not 5.5 nS.
Acknowledgments
We thank Christian Andersen for helpful discussions.
This investigation was supported by grants of the Deutsche Forschungsgemeinschaft (Be865-13/1) and the Fonds der Chemischen Industrie.
Footnotes
Published ahead of print on 4 December 2009.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Barksdale, L. 1959. Lysogenic conversions in bacteria. Bacteriol. Rev. 23:202-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barreiro, C., E. Gonzalez-Lavado, M. Patek, and J. F. Martin. 2004. Transcriptional analysis of the groES-groEL1, groEL2, and dnaK genes in Corynebacterium glutamicum: characterization of heat shock-induced promoters. J. Bacteriol. 186:4813-4817. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Benz, R. 2001. Porins—structure and function, p. 227-246. In G. Winkelmann (ed.), Microbial transport systems. Wiley-VCH, Weinheim, Germany.
- 5.Benz, R., K. Janko, W. Boos, and P. Läuger. 1978. Formation of large, ion-permeable membrane channels by the matrix protein (porin) of Escherichia coli. Biochim. Biophys. Acta 511:305-319. [DOI] [PubMed] [Google Scholar]
- 6.Blum, H., H. Beier, and H. J. Gross. 1987. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8:93-99. [Google Scholar]
- 7.Brennan, P. J., and H. Nikaido. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29-63. [DOI] [PubMed] [Google Scholar]
- 8.Cerdeno-Tárraga, A. M., A. Efstratiou, L. G. Dover, M. T. Holden, M. Pallen, S. D. Bentley, G. S. Besra, C. Churcher, K. D. James, A. De Zoysa, T. Chillingworth, A. Cronin, L. Dowd, T. Feltwell, N. Hamlin, S. Holroyd, K. Jagels, S. Moule, M. A. Quail, E. Rabbinowitsch, K. M. Rutherford, N. R. Thomson, L. Unwin, S. Whitehead, B. G. Barrell, and J. Parkhill. 2003. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 31:6516-6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa-Riu, N., A. Burkovski, R. Kramer, and R. Benz. 2003. PorA represents the major cell wall channel of the Gram-positive bacterium Corynebacterium glutamicum. J. Bacteriol. 185:4779-4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa-Riu, N., E. Maier, A. Burkovski, R. Krämer, F. Lottspeich, and R. Benz. 2003. Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol. Microbiol. 50:1295-1308. [DOI] [PubMed] [Google Scholar]
- 11.Daffé, M., and P. Draper. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131-203. [DOI] [PubMed] [Google Scholar]
- 12.Eggeling, L., and H. Sahm. 2001. The cell wall barrier of Corynebacterium glutamicum and amino acid efflux. J. Biosci. Bioeng. 92:201-213. [Google Scholar]
- 13.Faller, M., M. Niederweis, and G. E. Schulz. 2004. The structure of a mycobacterial outer-membrane channel. Science 303:1189-1192. [DOI] [PubMed] [Google Scholar]
- 14.Fudou, R., Y. Jojima, A. Seto, K. Yamada, E. Kimura, T. Nakamatsu, A. Hiraishi, and S. Yamanaka. 2002. Corynebacterium efficiens sp. nov., a glutamic-acid-producing species from soil and vegetables. Int. J. Syst. Evol. Microbiol. 52:1127-1131. [DOI] [PubMed] [Google Scholar]
- 15.Gebhardt, H., X. Meniche, M. Tropis, R. Krämer, M. Daffé, and S. Morbach. 2007. The key role of the mycolic acid content in the functionality of the cell wall permeability barrier in Corynebacterineae. Microbiology 153:1424-1434. [DOI] [PubMed] [Google Scholar]
- 16.Georgi, T., D. Rittmann, and V. F. Wendisch. 2005. Lysine and glutamate production by Corynebacterium glutamicum on glucose, fructose and sucrose: roles of malic enzyme and fructose-1,6-bisphosphatase. Metab. Eng. 7:291-301. [DOI] [PubMed] [Google Scholar]
- 17.Harley, C. B., and R. P. Reynolds. 1987. Analysis of E. coli promoter sequences. Nucleic Acids Res. 15:2343-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto, K., H. Kawasaki, K. Akazawa, J. Nakamura, Y. Asakura, T. Kudo, E. Sakuradani, S. Shimizu, and T. Nakamatsu. 2006. Changes in composition and content of mycolic acids in glutamate-overproducing Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 70:22-30. [DOI] [PubMed] [Google Scholar]
- 19.Helmann, J. D. 1995. Compilation and analysis of Bacillus subtilis sigma A-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA. Nucleic Acids Res. 23:2351-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermann, T. 2003. Industrial production of amino acids by coryneform bacteria. J. Biotechnol. 104:155-172. [DOI] [PubMed] [Google Scholar]
- 21.Hünten, P., N. Costa-Riu, D. Palm, F. Lottspeich, and R. Benz. 2005. Identification and characterization of PorH, a new cell wall channel of Corynebacterium glutamicum. Biochim. Biophys. Acta 1715:25-36. [DOI] [PubMed] [Google Scholar]
- 22.Hünten, P., B. Schiffler, F. Lottspeich, and R. Benz. 2005. PorH, a new channel-forming protein present in the cell wall of Corynebacterium efficiens and Corynebacterium callunae. Microbiology 151:2429-2438. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda, M., and S. Nakagawa. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99-109. [DOI] [PubMed] [Google Scholar]
- 24.Jarlier, V., and H. Nikaido. 1990. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J. Bacteriol. 172:1418-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalinowski, J., B. Bathe, D. Bartels, N. Bischoff, M. Bott, A. Burkovski, N. Dusch, L. Eggeling, B. J. Eikmanns, L. Gaigalat, A. Goesmann, M. Hartmann, K. Huthmacher, R. Kramer, B. Linke, A. C. McHardy, F. Meyer, B. Mockel, W. Pfefferle, A. Pühler, D. A. Rey, C. Ruckert, O. Rupp, H. Sahm, V. F. Wendisch, I. Wiegrabe, and A. Tauch. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5-25. [DOI] [PubMed] [Google Scholar]
- 26.Kingsford, C. L., K. Ayanbule, and S. L. Salzberg. 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 8:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinoshita, S., S. Udaka, and M. Shimono. 2004. Studies on the amino acid fermentation. Part 1. Production of l-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 50:331-343. [PubMed] [Google Scholar]
- 28.Kumagai, Y., T. Hirasawa, K. Hayakawa, K. Nagai, and M. Wachi. 2005. Fluorescent phospholipid analogs as microscopic probes for detection of the mycolic acid-containing layer in Corynebacterium glutamicum: detecting alterations in the mycolic acid-containing layer following ethambutol treatment. Biosci. Biotechnol. Biochem. 69:2051-2056. [DOI] [PubMed] [Google Scholar]
- 29.Lakshman, M., and M. R. Raghavendra Rao. 1981. Excretion of lysine by Micrococcus glutamicus. J. Biosci. 3:51-55. [Google Scholar]
- 30.Lichtinger, T., A. Burkovski, M. Niederweis, R. Krämer, and R. Benz. 1998. Biochemical and biophysical characterization of the cell wall porin of Corynebacterium glutamicum: the channel is formed by a low molecular mass polypeptide. Biochemistry 37:15024-15032. [DOI] [PubMed] [Google Scholar]
- 31.Lichtinger, T., F. G. Riess, A. Burkovski, F. Engelbrecht, D. Hesse, H. D. Kratzin, R. Krämer, and R. Benz. 2001. The low-molecular-mass subunit of the cell wall channel of the Gram-positive Corynebacterium glutamicum. Immunological localization, cloning and sequencing of its gene porA. Eur. J. Biochem. 268:462-469. [DOI] [PubMed] [Google Scholar]
- 32.Lindner, S. N., D. Vidaurre, S. Willbold, S. M. Schoberth, and V. F. Wendisch. 2007. NCgl2620 encodes a class II polyphosphate kinase in Corynebacterium glutamicum. Appl. Environ. Microbiol. 73:5026-5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marienfeld, S., E. M. Uhlemann, R. Schmid, R. Krämer, and A. Burkovski. 1997. Ultrastructure of the Corynebacterium glutamicum cell wall. Antonie Van Leeuwenhoek 72:291-297. [DOI] [PubMed] [Google Scholar]
- 34.Minnikin, D. E., P. Patel, and M. Goodfellow. 1974. Mycolic acids of representative strains of Nocardia and the ‘rhodochrous’ complex. FEBS Lett. 39:322-324. [DOI] [PubMed] [Google Scholar]
- 35.Molle, V., N. Saint, S. Campagna, L. Kremer, E. Lea, P. Draper, and G. Molle. 2006. pH-dependent pore-forming activity of OmpATb from Mycobacterium tuberculosis and characterization of the channel by peptidic dissection. Mol. Microbiol. 61:826-837. [DOI] [PubMed] [Google Scholar]
- 36.Niederweis, M., S. Ehrt, C. Heinz, U. Klöcker, S. Karosi, K. M. Swiderek, L. W. Riley, and R. Benz. 1999. Cloning of the mspA gene encoding a porin from Mycobacterium smegmatis. Mol. Microbiol. 33:933-945. [DOI] [PubMed] [Google Scholar]
- 37.Niederweis, M., E. Maier, T. Lichtinger, R. Benz, and R. Kramer. 1995. Identification of channel-forming activity in the cell wall of Corynebacterium glutamicum. J. Bacteriol. 177:5716-5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikaido, H., and V. Jarlier. 1991. Permeability of the mycobacterial cell wall. Res. Microbiol. 142:437-443. [DOI] [PubMed] [Google Scholar]
- 40.Nishio, Y., Y. Nakamura, Y. Kawarabayasi, Y. Usuda, E. Kimura, S. Sugimoto, K. Matsui, A. Yamagishi, H. Kikuchi, K. Ikeo, and T. Gojobori. 2003. Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res. 13:1572-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnishi, J., M. Hayashi, S. Mitsuhashi, and M. Ikeda. 2003. Efficient 40 degrees C fermentation of l-lysine by a new Corynebacterium glutamicum mutant developed by genome breeding. Appl. Microbiol. Biotechnol. 62:69-75. [DOI] [PubMed] [Google Scholar]
- 42.Pátek, M., J. Nesvera, A. Guyonvarch, O. Reyes, and G. Leblon. 2003. Promoters of Corynebacterium glutamicum. J. Biotechnol. 104:311-323. [DOI] [PubMed] [Google Scholar]
- 43.Puech, V., M. Chami, A. Lemassu, M. A. Laneelle, B. Schiffler, P. Gounon, N. Bayan, R. Benz, and M. Daffé. 2001. Structure of the cell envelope of corynebacteria: importance of the non-covalently bound lipids in the formation of the cell wall permeability barrier and fracture plane. Microbiology 147:1365-1382. [DOI] [PubMed] [Google Scholar]
- 44.Radmacher, E., K. C. Stansen, G. S. Besra, L. J. Alderwick, W. N. Maughan, G. Hollweg, H. Sahm, V. F. Wendisch, and L. Eggeling. 2005. Ethambutol, a cell wall inhibitor of Mycobacterium tuberculosis, elicits l-glutamate efflux of Corynebacterium glutamicum. Microbiology 151:1359-1368. [DOI] [PubMed] [Google Scholar]
- 45.Riess, F. G., and R. Benz. 2000. Discovery of a novel channel-forming protein in the cell wall of the non-pathogenic Nocardia corynebacteroides. Biochim. Biophys. Acta 1509:485-495. [DOI] [PubMed] [Google Scholar]
- 46.Riess, F. G., M. Elflein, M. Benk, B. Schiffler, R. Benz, N. Garton, and I. Sutcliffe. 2003. The cell wall of the pathogenic bacterium Rhodococcus equi contains two channel-forming proteins of different properties. J. Bacteriol. 185:2952-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riess, F. G., T. Lichtinger, A. F. Yassin, K. P. Schaal, and R. Benz. 1999. The cell wall porin of the gram-positive bacterium Nocardia asteroides forms cation-selective channels that exhibit asymmetric voltage dependence. Arch. Microbiol. 171:173-182. [DOI] [PubMed] [Google Scholar]
- 48.Ruimy, R., P. Boiron, V. Boivin, and R. Christen. 1994. A phylogeny of the genus Nocardia deduced from the analysis of small-subunit ribosomal DNA sequences, including transfer of Nocardia amarae to the genus Gordona as Gordona amarae comb. nov. FEMS Microbiol. Lett. 123:261-267. [DOI] [PubMed] [Google Scholar]
- 49.Ruimy, R., P. Riegel, P. Boiron, H. Monteil, and R. Christen. 1995. Phylogeny of the genus Corynebacterium deduced from analyses of small-subunit ribosomal DNA sequences. Int. J. Syst. Bacteriol. 45:740-746. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 51.Schäfer, A., A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach, and A. Pühler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69-73. [DOI] [PubMed] [Google Scholar]
- 52.Schägger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368-379. [DOI] [PubMed] [Google Scholar]
- 53.Schiffler, B., E. Barth, M. Daffé, and R. Benz. 2007. Corynebacterium diphtheriae: identification and characterization of a channel-forming protein in the cell wall. J. Bacteriol. 189:7709-7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shah, N. S., A. Wright, G. H. Bai, L. Barrera, F. Boulahbal, N. Martin-Casabona, F. Drobniewski, C. Gilpin, M. Havelkova, R. Lepe, R. Lumb, B. Metchock, F. Portaels, M. F. Rodrigues, S. Rusch-Gerdes, A. Van Deun, V. Vincent, K. Laserson, C. Wells, and J. P. Cegielski. 2007. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg. Infect. Dis. 13:380-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simic, P., H. Sahm, and L. Eggeling. 2001. l-Threonine export: use of peptides to identify a new translocator from Corynebacterium glutamicum. J. Bacteriol. 183:5317-5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutcliffe, I. C. 1997. Macroamphiphilic cell envelope components of Rhodococcus equi and closely related bacteria. Vet. Microbiol. 56:287-299. [DOI] [PubMed] [Google Scholar]
- 57.Tauch, A., O. Kaiser, T. Hain, A. Goesmann, B. Weisshaar, A. Albersmeier, T. Bekel, N. Bischoff, I. Brune, T. Chakraborty, J. Kalinowski, F. Meyer, O. Rupp, S. Schneiker, P. Viehoever, and A. Pühler. 2005. Complete genome sequence and analysis of the multiresistant nosocomial pathogen Corynebacterium jeikeium K411, a lipid-requiring bacterium of the human skin flora. J. Bacteriol. 187:4671-4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tauch, A., E. Trost, A. Tilker, U. Ludewig, S. Schneiker, A. Goesmann, W. Arnold, T. Bekel, K. Brinkrolf, I. Brune, S. Gotker, J. Kalinowski, P. B. Kamp, F. P. Lobo, P. Viehoever, B. Weisshaar, F. Soriano, M. Droge, and A. Pühler. 2008. The lifestyle of Corynebacterium urealyticum derived from its complete genome sequence established by pyrosequencing. J. Biotechnol. 136:11-21. [DOI] [PubMed] [Google Scholar]
- 59.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trias, J., and R. Benz. 1994. Permeability of the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 14:283-290. [DOI] [PubMed] [Google Scholar]
- 61.Trias, J., V. Jarlier, and R. Benz. 1992. Porins in the cell wall of mycobacteria. Science 258:1479-1481. [DOI] [PubMed] [Google Scholar]
- 62.Tropis, M., X. Meniche, A. Wolf, H. Gebhardt, S. Strelkov, M. Chami, D. Schomburg, R. Krämer, S. Morbach, and M. Daffé. 2005. The crucial role of trehalose and structurally related oligosaccharides in the biosynthesis and transfer of mycolic acids in Corynebacterineae. J. Biol. Chem. 280:26573-26585. [DOI] [PubMed] [Google Scholar]
- 63.Trötschel, C., D. Deutenberg, B. Bathe, A. Burkovski, and R. Krämer. 2005. Characterization of methionine export in Corynebacterium glutamicum. J. Bacteriol. 187:3786-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van der Rest, M. E., C. Lange, and D. Molenaar. 1999. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl. Microbiol. Biotechnol. 52:541-545. [DOI] [PubMed] [Google Scholar]
- 65.Vasicová, P., M. Patek, J. Nesvera, H. Sahm, and B. Eikmanns. 1999. Analysis of the Corynebacterium glutamicum dapA promoter. J. Bacteriol. 181:6188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vrljic, M., H. Sahm, and L. Eggeling. 1996. A new type of transporter with a new type of cellular function: l-lysine export from Corynebacterium glutamicum. Mol. Microbiol. 22:815-826. [DOI] [PubMed] [Google Scholar]
- 67.Yukawa, H., C. A. Omumasaba, H. Nonaka, P. Kos, N. Okai, N. Suzuki, M. Suda, Y. Tsuge, J. Watanabe, Y. Ikeda, A. A. Vertes, and M. Inui. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042-1058. [DOI] [PubMed] [Google Scholar]
- 68.Ziegler, K., R. Benz, and G. E. Schulz. 2008. A putative alpha-helical porin from Corynebacterium glutamicum. J. Mol. Biol. 379:482-491. [DOI] [PubMed] [Google Scholar]
- 69.Zuber, B., M. Chami, C. Houssin, J. Dubochet, G. Griffiths, and M. Daffé. 2008. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190:5672-5680. [DOI] [PMC free article] [PubMed] [Google Scholar]