

Abstract
Nucleotide excision repair (NER) is universally used to recognize and remove many types of DNA damage. In eubacteria, the NER system typically consists of UvrA, UvrB, UvrC, the UvrD helicase, DNA polymerase I, and ligase. In addition, when DNA damage blocks transcription, transcription-repair coupling factor (TRCF), the product of the mfd gene, recruits the Uvr complex to repair the damage. Previous work using selected mutants and assays have indicated that pathogenic Neisseria spp. carry a functional NER system. In order to comprehensively examine the role of NER in Neisseria gonorrhoeae DNA recombination and repair processes, the predicted NER genes (uvrA, uvrB, uvrC, uvrD, and mfd) were each disrupted by a transposon insertion, and the uvrB and uvrD mutants were complemented with a copy of each gene in an ectopic locus. Each uvr mutant strain was highly sensitive to UV irradiation and also showed sensitivity to hydrogen peroxide killing, confirming that all of the NER genes in N. gonorrhoeae are functional. The effect of RecA expression on UV survival was minor in uvr mutants but much larger in the mfd mutant. All of the NER mutants demonstrated wild-type levels of pilin antigenic variation and DNA transformation. However, the uvrD mutant exhibited higher frequencies of PilC-mediated pilus phase variation and spontaneous mutation, a finding consistent with a role for UvrD in mismatch repair. We conclude that NER functions are conserved in N. gonorrhoeae and are important for the DNA repair capabilities of this strict human pathogen.
Neisseria gonorrhoeae (the gonococcus [Gc]) is a strict human pathogen and is the sole causative agent of the human sexually transmitted disease gonorrhea. Gc and its closest relatives are only found naturally within the human population with no known animal, insect, or environmental reservoirs. Disease symptoms consistent with gonorrhea have been described throughout history (25), leading to the hypothesis that most of the genus Neisseria have evolved within humans for >50,000 years. This adaptation to life within a single organism has produced a streamlined genome and genetic and physiology focused on life within humans. Although these organisms have not been regularly exposed to UV light, extremes of temperature, desiccation, or a wide range of chemicals during their exclusive residence in the human population, the Gc genome encodes a variety of genes involved, or predicted to be involved in DNA repair, including photoreactivation, base excision repair, nucleotide excision repair (NER), recombination repair, and replication restart (10, 18). Among the predicted DNA repair pathways is a full NER pathway.
NER is the most ubiquitous DNA repair pathway and is found in the sequenced genomes from almost every kingdom, genus, and species that has been examined. The core set of genes involved in the Escherichia coli NER system are uvrA, uvrB, uvrC, uvrD, and mfd (43). All of these gene products have been purified, and their roles in NER have been defined. The UvrAB complex recognizes and binds to the distortion in the DNA duplex caused by DNA damage. UvrB recruits UvrC to the lesion, where it acts as a single-stranded DNA endonuclease cleaving the DNA 5′ and 3′ from the lesion. UvrD (helicase II) then removes the single stranded segment carrying the lesion. DNA polymerase I resynthesizes the DNA copying the undamaged template, and ligase completes the repair (43). The mfd gene product TRCF recognizes stalled transcription machinery and targets the NER components to correct lesions that block transcription (36). The NER systems of all organisms are used to recognize and remove chemical lesions in DNA. Although uvrB and uvrC are induced by the damage-inducible SOS system in E. coli and this system is associated with the removal of pyrimidine dimers resulting from UV irradiation, many types of chemical damage can be recognized and repaired by NER, including psoralen monoadducts, cisplatin cross-links, thymine glycol products, O6-methyl guanine, and others, as well as abasic sites (43). In a subset of organisms including E. coli, a paralog of UvrC has been identified that allows the removal of larger chemical substitutions on DNA, like cholesterol (28).
Although NER has not been extensively studied in the Neisseria, several reports have concluded that a functional NER system exists in Neisseria spp. In 1984, Campbell and Yasbin demonstrated the excision of nucleotides carrying pyrimidine dimers in UV-irradiated Gc by using an alkaline agarose gel system (7). In 1997, Black et al. demonstrated that the Gc uvrA gene could complement an E. coli uvrA mutant for UV survival (3), and in 1995 the same group demonstrated that a uvrB mutant was deficient in UV survival (4). Finally, Davidsen et al. demonstrated that a Neisseria meningitidis uvrA mutant was also deficient for UV survival but not altered for survival to alkylating agents (11). The involvement of NER in other DNA recombination and repair pathways in pathogenic Neisseria, such as natural competence, repair of spontaneous mutations, and pilus variations, has not been defined.
In order to fully address the role of NER in Gc, each gene involved in the repair system was cloned and mutated by a transposon insertion and then transformed into laboratory strain FA1090. As expected, NER mutants had substantial sensitivity to UV irradiation, and further studies revealed a complex relationship between the NER system and RecA in repairing UV-mediated DNA damage. NER mutants were also more sensitive to other types of DNA damage but had no defect in the recombination-dependent events of DNA transformation and pilin antigenic variation. Finally, the participation of NER mutants in the control of spontaneous mutation was assayed, and only uvrD was found to influence mutation frequencies, presumably due to the role for this helicase in mismatch correction.
MATERIALS AND METHODS
Strains and growth conditions.
Gc strain FA1090, pilin variant 1-81-S2, is a piliated (P+) Gc strain carrying a defined pilE sequence (35). The FA1090 1-81-S2 recA6 derivative carries a recA gene under the control of lac regulatory elements (34). The recA9 mutant is recA deficient due to an insertion of an erythromycin resistance cassette into recA (34). Where indicated, spontaneous nonreverting nonpiliated (P−) variants of FA1090 1-81-S2 were isolated (ΔpilE), and deletion of the pilE gene confirmed by PCR. Gc strains were grown on GC Medium Base (Difco) plus select agar (Invitrogen) with Kellogg's supplements (17) and, when appropriate, 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Fisher Scientific) was added. Gonococci were grown at 37°C in 5% CO2 unless otherwise indicated. E. coli TOP10 (Invitrogen) were grown on solid Luria-Bertani (LB) agar at 37°C or in LB liquid medium at 37°C with rotation. Antibiotics were used as follows: kanamycin at 50 μg/ml, chloramphenicol at 1 μg/ml, and erythromycin at 0.5 μg/ml for Gc or 250 μg/ml for E. coli.
Identification and cloning of the Gc NER genes.
The annotated but unpublished Gc genome (http://stdgen.northwestern.edu) was used to identify the gene sequences of uvrA, uvrB, uvrC, uvrD, and mfd in the chromosome of Gc strain FA1090. Primers were designed to amplify an internal region of each gene and PCR was performed by using Taq polymerase (Promega). Primers to uvrB, uvrC, and uvrD were designed to incorporate the Gc uptake sequence (GCCGTCTGAA, underlined in the primer sequences below [13]) into PCR-amplified DNA for efficient transformation of Gc. Gc uvrA was amplified by using the primers UVRA-1 (CGAAAAAATCCTCAAAGAAATC) and UVRA-2 (GAGTTCCAAGGCGAGTTTG). The primers used to PCR amplify uvrB were UVRB-1 (TTGCCGTCTGAACGAGTATCAACAAATGGTGCTG) and UVRB-2 (GTACGTCAAACAGGCCGAGC). The primers for uvrC were UVRC-1 (TTGCCGTCTGAACGAAGAAGACTACTGCGACAGC) and UVRC-2 (GCAAGGCCGGGCTATTAGGAG). The primers for amplification of uvrD were UVRD-1 (TTGCCGTCTGAACATGATTGGGTGCTACGCCG) and UVRD-2 (CATGGTGATGTAGAGGCGTTTG). For mfd, primers were MFD-1 (CGAAGATGCGCTGTTTGTCTC) and MFD-2 (GGTCTTCGGTTTCCTCGTAGC). The individual PCR products were cloned into pCR2.1 (Invitrogen) and transformed into TOP10 chemically competent E. coli (Invitrogen) according to the manufacturer's instructions. Plasmids were isolated from kanamycin-resistant E. coli colonies and a diagnostic enzyme digest was performed with EcoRI to confirm the insert was the correct size. The presence of the correct DNA sequence was confirmed by sequencing with M13 forward and reverse primers flanking the insert.
In vitro transposition and transformation of Gc.
In vitro transposition of the plasmid clones, pCR2.1::uvrA, uvrB, uvrC, uvrD, and mfd was performed by using the EZ::Tn5<KAN-2> transposition system (Epicentre Biotechnologies). A 1:1 molar ratio of each plasmid to transposon was used for the in vitro transposition reaction (33). The gaps were repaired by using T4 DNA polymerase (New England Bio Labs) and T4 DNA ligase (New England Biolabs). The DNA was introduced into the chromosome of strain FA1090 1-81-S2 recA6 by spot transformation (14) in the presence of 1 mM IPTG to allow RecA-dependent homologous recombination to take place (34). Kanamycin-resistant Gc transformants were screened by PCR using gene-specific primers to confirm that the transposon had inserted into the appropriate genes. The PCR products were then sequenced by using KAN-2_FP-1 and KAN-2_RP-1 to map the position of the transposon insertion. The primers used read out of the transposon, KAN-2_FP-1 (ACCTACAACAAAGCTCTCATCAACC) and KAN-2_RP-1 (GCAATGTAACATCAGAGATTTTGAG). Southern blotting was performed on all of the strains with gene-specific DIG-labeled probes (Roche) according to the manufacturer's instructions to confirm proper insertion into the correct chromosomal locus. Mutations in the NER genes were reintroduced into FA1090 1-81-S2 (wild-type recA) by spot transformation with genomic DNA from the recA6 NER mutants, followed by selection for kanamycin resistance. Retention of the 1-81-S2 pilE was confirmed by sequencing with PILRBS and SP3A primers (45).
Complementation of uvrB and uvrD.
uvrB and uvrD were complemented by using the neisserial intergenic complementation system, where full-length coding sequences of each gene are inserted between the lctP and aspC genes on the Gc chromosome, a site that has no known effect on Gc recombination or repair processes (26). The primers for complementation of uvrB were UVRB_PacI-1 (GCTTAATTAAGCAAACGTATCACAGGCAAG) and UVRB_PacI-2 (GCTTAATTAACAAGCTGCGTACATGATTTC), and the primers used for amplification of uvrD were UVRD_PacI-1 (CATTAATTAAGCGCATTCCATTTTTCAGAC) and UVRD_PacI-2 (TCTTAATTAAGCCTTGTCCTTGCTACGGT). The genes were amplified by using KOD Hot Start DNA polymerase (Novagen) to lessen the likelihood of any mutation occurring during amplification and cloned into PacI-digested plasmid pGCC4. The ligations were transformed into TOP10 chemically competent E. coli cells and plated on LB-erythromycin. Erythromycin-resistant colonies were screened for insertion of uvrB and uvrD using PacI and for correct orientation of the open reading frames using PacI, MluI, and PflMI. Complementation constructs were introduced into the chromosome of strain FA1090 1-81-S2 by transformation. Proper transformation of all strains was confirmed by PCR and Southern blotting (data not shown), and retention of the 1-81-S2 pilE sequence was confirmed by DNA sequence analysis as described above.
Gc phenotypic assays.
(i) UV sensitivity assay. Serial dilutions of FA1090 1-81-S2 recA6 Gc and isogenic NER mutants were plated on GCB agar containing 1 mM IPTG and exposed to UV at fluences of 0 to 80 J/m2 by using a Stratalinker 1800 (Stratagene). To test the effect of recA on the NER UV phenotype, dilutions of Gc were plated on GCB agar ± 1 mM IPTG and exposed to UV fluences of 0 to 20 J/m2. Survival is expressed relative to unexposed Gc of the same strain. Assays were performed four times.
(ii) H2O2 sensitivity assay.
Survival of FA1090 ΔpilE Gc and isogenic uvrA, uvrB, and uvrD mutants after exposure to 0 to 10 mM H2O2 was assessed as previously described (39), except the first plate growth was limited to 10 h. Assays were performed four times.
(iii) Kinetic PDCMC assay.
The kinetic assay for pilus-dependent colony morphology changes (PDCMC) was performed as previously described (33). Assays were performed four times per strain.
(iv) PilC expression analysis.
FA1090 1-81-S2 recA6 uvrD Gc were grown on GCB without IPTG. P− colonies emerging from the culture were propagated, and the resulting lysates were separated on a 10% polyacrylamide gel and transferred to 0.45-μm-pore-size polyvinylidene difluoride Immobilon-P membrane (Millipore). PilC expression was probed by using a polyclonal antibody (a gift from A. Jonsson), followed by horseradish peroxidase-conjugated anti-rabbit IgG (Chemicon). ECL Plus chemiluminescence (Amersham) was used to develop the blot after transfer. FA1090 Gc that are P− due to an antigenic variation event that gives rise to a truncated pilE sequence (8) served as a positive control for PilC expression.
(v) Sequencing assay for pilin Av.
The DNA sequencing-based assay for pilin Av was performed for FA1090 1-81-S2 recA6 and the isogenic uvrD mutant as previously described (8). Only P+ colonies arising from the P+ parent were passaged and examined for sequence changes at pilE. The assay was performed three times.
(vi) Transformation efficiency assay.
Quantitative transformation was performed as previously described (23). FA1090 1-81-S2 recA6 and isogenic NER mutants were exposed to 1 μg of chloramphenicol-resistant Gc genomic DNA carrying a 1.1-kb cloned cat insertion in the recX locus (FA1090ΔrecX/recX [41]) for 30 min. The transformation frequency was measured as the number of chloramphenicol-resistant CFU arising after transformation divided by the total CFU present in the transformation mix. The assay was conducted four times.
(vii) Spontaneous mutation assay.
FA1090 ΔpilE (wild-type recA) and isogenic mutants and complements in the NER system were resuspended to a concentration of 107 to 108 CFU/ml in GCBL containing 1 mM IPTG for induction of the complement constructs. Gonococci were plated on GCB containing 50 ng of rifampin (Sigma)/ml or 0.8 μg of nalidixic acid (Sigma)/ml, as well as on GCB to enumerate total CFU. Antibiotic-resistant CFU were enumerated after incubation for ∼40 h at 37°C with 5% CO2 and expressed as a fraction of the CFU plated. The median frequency of spontaneous mutation to antibiotic resistance was determined from 10 independent experiments.
RESULTS
Mutation of NER genes affect DNA repair in N. gonorrhoeae.
Gene products that mediate the NER and TRCF pathways are conserved throughout evolution and play key roles in the viability of prokaryotes and eukaryotes. Homologs of the E. coli uvrA, uvrB, uvrC, uvrD, and mfd genes were identified in the chromosome of Gc strain FA1090 (Fig. 1). Each of these genes is found at discrete sites in the chromosome (Fig. 1). Therefore, Gc appears to encode the full complement of NER and TRCF genes. Each of these genes was cloned, and a mini-Tn5 transposon containing a kanamycin resistance cassette was isolated within the coding region of each gene after in vitro transposition (see Materials and Methods). The mutated genes were reintroduced into the piliated Gc strain FA1090, pilin variant 1-81-S2, by DNA transformation, and antibiotic-resistant clones were isolated and verified by PCR and Southern blot analysis (data not shown).
FIG. 1.

Location of NER genes in the chromosome of Gc strain FA1090. The locations and orientations of uvrA, uvrB, uvrC, uvrD, and mfd orthologs identified in the chromosome of Gc strain FA1090 are shown along with flanking open reading frames. The figure is drawn to scale. Bar, 1 kb. Gray triangles indicate sites of insertion of the EZ::Tn5<KAN-2> minitransposon to create insertions in each gene.
In E. coli, the uvr gene products protect the organism from various types of DNA damage, including UV irradiation (43). To assess the involvement of the putative NER genes found in the Gc chromosome in DNA repair, we measured the sensitivity of the Gc strains lacking the NER and TRCF genes to DNA damage caused by UV light B (UV) irradiation (Fig. 2). Parental FA1090 Gc exhibited dose-dependent sensitivity to UV irradiation, with a 90% reduction in survival after exposure to 20 J/m2 UV and a 3-log reduction in survival after exposure to 80 J/m2 UV. As expected, inactivation of the uvr genes rendered Gc exquisitely sensitive to UV irradiation. Survival of the uvr mutants was reduced by ∼4 logs relative to the parental strain after exposure to 20 J/m2 UV, a statistically significant decrease (P < 0.05 [Student t test]), and at higher dosages of UV there was no recovery of uvr mutant Gc. The decrease in survival after UV irradiation was much lower than that of the recA-null strain, which exhibited a 2- to 3-log decrease in survival relative to the RecA+ parent at all UV fluences tested. Complementation of the uvrB and uvrD mutants, achieved by supplying a wild-type copy of each gene at a second site in the Gc chromosome, restored the survival of the mutants to near-parental levels. In contrast to the uvr mutants, the mfd mutant was not as sensitive to UV as the other strains. The mfd mutant exhibited a 15-fold decrease in survival after exposure to 20 J/m2 UV, ∼10-fold higher than for the recA mutant, but at higher dosages of UV the mfd mutant showed sensitivity to UV similar to the recA mutant. We conclude that the uvrABCD genes are critical to Gc survival after UV irradiation but that the mfd gene product plays a minor role in protecting the bacteria from damage caused by UV light. Although complementation was only performed with the uvrB and uvrD mutants, the expected and identical UV sensitivity phenotypes of the uvrABCD mutants strongly suggests that none of these phenotypes are the result of polar effects on downstream genes or second site mutations.
FIG. 2.

Sensitivity of Gc NER mutants to UV irradiation. The FA1090 1-81-S2 parental strain and recA, uvrA, uvrB, uvrC, uvrD, mfd isogenic mutants, as well as uvrB and uvrD complements in which wild-type copies of each gene were inserted elsewhere in the Gc chromosome (uvrB/uvrB+ and uvrD/uvrD+), were irradiated with the indicated doses of UV light. Survival was calculated relative to nonirradiated bacteria of the same strain. The curves for the uvrA, uvrB, uvrC, and uvrD mutants are nearly identical. Error bars represent the standard deviation of the means of four independent experiments. No CFU were recovered from the uvrA, uvrB, uvrC, and uvrD mutants irradiated with >20 J/m2 UV. The differences in survival between all NER mutants and the parent at each UV dose were significant at P < 0.05 (Student t test).
NER is the predominant mechanism for repairing UV damage to DNA in Gc.
recA plays an important role in recombinational DNA repair in Gc and protects the organism from damage caused by UV light (21). The extreme sensitivity of the Gc NER mutants to UV irradiation led us to examine the effect of recA inactivation on this sensitivity phenotype. In order to address this question, the NER genes were inactivated in a strain where the recA gene is under the control of lac regulatory elements; thus, recA expression can be ectopically stimulated or repressed by the presence or absence of IPTG, respectively (34). The resulting Gc strains were subjected to UV fluences ranging from 0 to 20 J/m2 in the presence or absence of recA expression, dosages that reduced the survival of the NER mutants compared to the parental strain (Fig. 3 and 4).
FIG. 3.

RecA dependence of the sensitivity of Gc uvrA, uvrB, and uvrC mutants to UV irradiation. uvrA (A), uvrB (B), and uvrC (C) mutants were constructed in the FA1090 recA6 genetic background, in which recA expression is under the control of lac regulatory sequences (34). The parental recA6 strain and isogenic mutants in each of the NER genes were grown in the presence (+) or absence (−) of IPTG to induce or repress RecA expression, respectively. Gonococci were then irradiated with the indicated fluences of UV light, and survival for each strain with or without recA expression was calculated as in Fig. 2. Error bars represent the standard deviation of the mean of four independent experiments.
FIG. 4.

RecA dependence of the sensitivity of Gc uvrD and mfd mutants to UV irradiation. uvrD (A) and mfd (B) mutants were constructed in the FA1090 1-81-S2 recA6 genetic background, and their survival after exposure to the indicated UV fluences was assessed in the presence (+) or absence (−) of RecA expression as described in Fig. 3. Error bars represent the standard deviation of the mean of six independent experiments.
We observed a bipartite effect of recA on the survival of the uvrA, uvrB, and uvrC mutants after UV irradiation (Fig. 3). At the lowest dose of UV tested (2 J/m2), removal of recA expression from these three mutants decreased their survival by ∼4 logs. However, at doses of 5 to 20 J/m2, no significant difference in survival was observed for the uvrA, uvrB, and uvrC mutants whether or not recA was also expressed. These results show that NER is the predominant repair pathway for UV-induced DNA lesions in Gc and that under conditions where DNA damage is minimal, RecA can aid in repairing damaged DNA by a pathway independent of the NER gene products.
In contrast to the uvrABC mutants, the uvrD and mfd mutants exhibited a less pronounced response to the lowered dosages of UV irradiation (Fig. 4). In the presence of recA expression, the uvrD mutant survived significantly better after exposure to 2 to 10 J/m2 UV than when recA was not induced, but this difference was minimized at 20 J/m2 UV (Fig. 4A). Thus, the effect of uvrD inactivation on repair of UV-damaged DNA is not as extreme as inactivation of the uvrABC genes. At doses of 2 to 20 J/m2, inactivation of mfd had minimal effects on Gc survival after UV irradiation, indicating that TRCF participates in repairing a small subset of all NER lesions in Gc (Fig. 4B). The combined effect of inactivation of mfd and recA on Gc survival was significantly greater than inactivating either gene alone. These observations lend further credence to the hypothesis that recombinational repair and nucleotide excision repair independently correct DNA lesions induced by UV irradiation.
NER genes help protect from oxidative DNA damage.
As a strict human pathogen primarily infecting the urogenital tract, Gc has not been regularly exposed to UV light for tens of thousands of years (25). We therefore reasoned that there must be other types of DNA damage encountered by Gc that are repaired by the NER system. One DNA-damaging agent Gc is likely to encounter during infection is reactive oxygen species (ROS), which can be produced by lactobacilli, neutrophils, and epithelial cells. To test whether one role for the Gc NER system is to protect against ROS-mediated damage, the sensitivity of FA1090 uvrA, uvrB, and uvrD mutants to 0 to 20 mM H2O2 was determined (Fig. 5 and data not shown). The NER mutants were more sensitive to 2.5 and 5 mM H2O2, exhibiting ∼10-fold reduction in survival relative to the FA1090 parent. At higher concentrations of H2O2, there was no difference in survival between the NER mutants and the parental strain; surprisingly, the bacteria survived slightly better at higher H2O2 concentrations, for reasons that are unclear (data not shown). These results suggest that the NER system plays a role in repairing low levels of oxidative damage to DNA.
FIG. 5.

Sensitivity of Gc NER mutants to oxidative damage. FA1090 ΔpilE and isogenic mutants in uvrA, uvrB, and uvrD were exposed to 5 mM H2O2 for 15 min. Survival was calculated relative to untreated bacteria of each strain. Experiments were repeated four times.
NER genes are not required for DNA transformation.
Gc are naturally competent for DNA transformation (37), which can occur at frequencies as high as 10−1 to 10−2 transformants per CFU (5). We examined whether the NER mutants participated in the recombination events underlying DNA transformation by measuring the efficiency with which Gc incorporated a chloramphenicol resistance cassette into their chromosome. Parental Gc exhibited a frequency of transformation to chloramphenicol resistance of (4.6 ± 2.0) × 10−3 transformants/CFU, which was not significantly different in the NER mutants (data not shown). We conclude that the NER gene products are not important for the natural competence of Gc. Due to the high frequency of pilus phase variation of the uvrD mutant this mutant was not tested for transformation competence (see next section).
NER genes do not participate in pilin antigenic variation, but the loss of function of uvrD results in a higher frequency of PilC phase variation.
Gc possesses a type IV pilus that is important for host infection (27). Changes in piliation state arise rapidly during in vitro passage to yield colonies with characteristic morphologies. Piliated Gc give rise to “P+” colonies showing a compact, domed morphology under the stereomicroscope, while underpiliated and nonpiliated Gc yield “P−” colonies with flattened morphology (17, 42). The parental strain used in the present study, FA1090 1-81-S2, exhibits a P+ colony morphology that was retained in the uvrA, uvrB, uvrC, and mfd mutants. In contrast, we observed a high frequency of kanamycin-resistant colonies with a P− morphology arising after Gc transformation with the uvrD mutant, and this phenotype was retained after backcross (data not shown). We validated this observation by using the PDCMC assay, which quantifies the number of P− outgrowths arising from uniformly P+ colonies over time (33). FA1090 uvrA, uvrB, uvrC, and mfd colonies grown on IPTG to induce recA expression produced P− outgrowths at the same rate as in the IPTG-induced parental strain, whereas the number of P− outgrowths was consistently higher in the uvrD mutant than in any of the other strains tested (Fig. 6A).
FIG. 6.
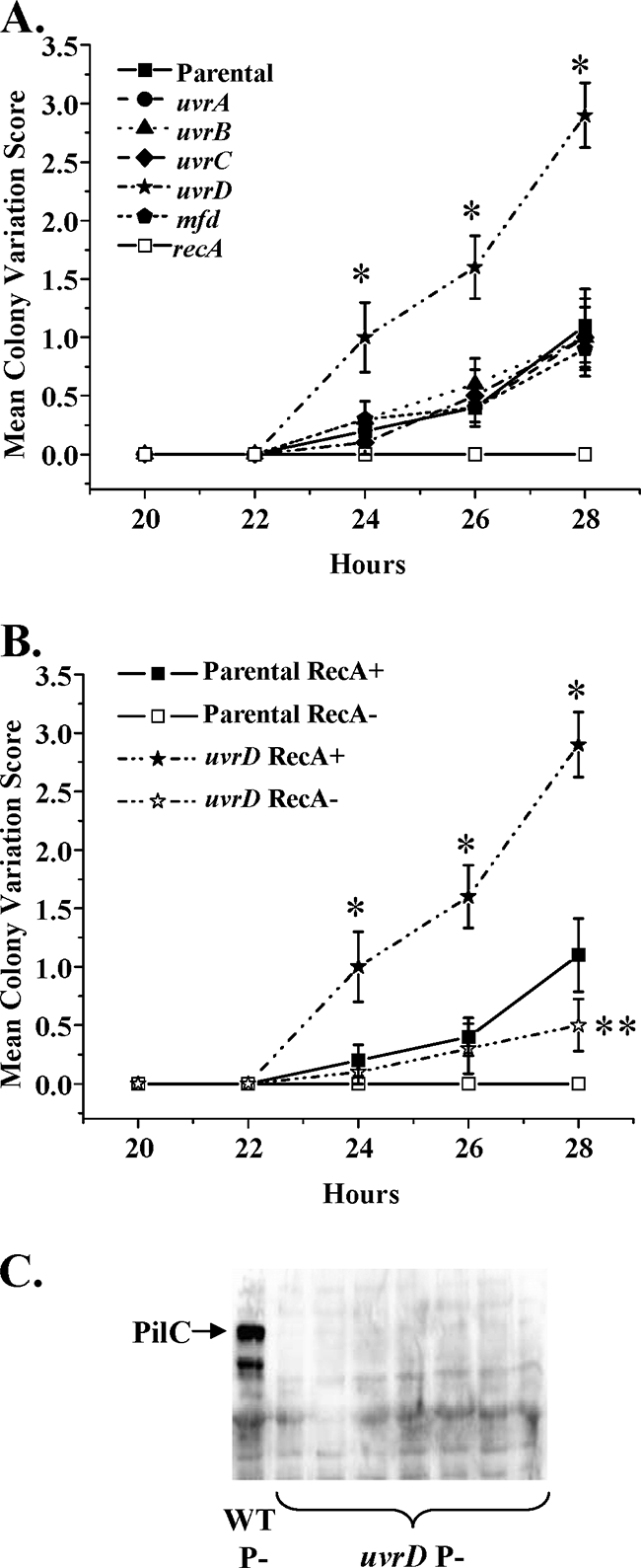
uvrD inactivation leads to a higher frequency of PilC pilus-dependent phase variation events. (A) The parental strain FA1090 recA6 and isogenic mutants in uvrA, uvrB, uvrC, uvrD, and mfd were passaged on solid medium containing 1 mM IPTG to induce RecA expression. The appearance of P− outgrowths from 10 independent P+ colonies was measured from 20 to 28 h as reported by Sechman et al. (33). The mean PDCMC index ± the standard deviation from 10 independent colonies is reported from four experiments. Asterisks represent significant (P < 0.025 [Student two-tailed t test]) increases over the parental strain grown on IPTG at the indicated time points. FA1090 recA6 that were maintained on medium lacking IPTG, which are phenotypically RecA−, served as a negative control for PDCMC (recA). (B) The parental strain FA1090 recA6 and the isogenic uvrD mutant were passaged on solid medium containing 1 mM IPTG to induce RecA expression (+) or without IPTG to repress RecA expression (−). The mean PDCMC index is reported as in panel A. Asterisks represent significant (*, P < 0.025; **, P < 0.05 [Student two-tailed t test]) increases in PDCMC over the parental strain when maintained under the same growth conditions at the indicated time points. (C) FA1090 recA6 uvrD was grown on medium lacking IPTG, and lysates from seven P− CFU arising after passage were immunoblotted for PilC expression as described in Materials and Methods (lanes 2 to 8). A lysate from FA1090 Gc that is P− due to a pilin antigenic variation event (WT P−) served as a positive control for PilC expression (lane 1).
PDCMCs can be produced from both antigenic and phase variation events. Changes in the sequence of the pilE gene that encodes pilin, the major subunit of the Gc pilus, arise from a DNA recombination-based process called pilin antigenic variation (Av), while the loss of expression of the PilC protein due to slipped-strand mispairing events in the pilC promoter prevents presentation of the pilus on the bacterial surface (16). Pilin Av absolutely requires RecA expression, whereas PilC phase variation is RecA independent. To begin to determine whether one or both of these processes were affected in the uvrD mutant, we first examined whether PDCMCs occurred in the absence of RecA expression. In the absence of IPTG to induce RecA expression, FA1090 parental colonies did not give rise to any detectable P− outgrowths, in keeping with the observation that the frequency of the RecA-dependent process of pilin Av is >100-fold higher than that of PilC phase variation (unpublished observations). In contrast, P− outgrowths were still detected in the uvrD mutant even when recA was not expressed (Fig. 6B). This result suggested that the increased PDCMC in the uvrD mutant was attributable to increased PilC phase variation. To directly test this possibility, uvrD mutant Gc were grown in the absence of IPTG, and lysates from seven P− colonies arising after passage were assayed for PilC expression by immunoblotting. All of the uvrD P− colonies had lost PilC expression (Fig. 6C). We conclude that the uvrD mutant exhibits a higher frequency of phase variation of PilC, leading to an increase in the emergence of P− colonies. We attribute this increased frequency of phase variation to reports in other bacteria that in addition to NER, UvrD also participates in mismatch repair, which normally corrects slipped-strand mispairing events, as seen in PilC phase variation (24).
Since the possibility remained that the uvrD mutant might also exhibit a higher frequency of pilin Av, we directly measured the frequency of recombination at pilE using a DNA sequencing-based assay (8). The frequency of pilE recombination was calculated as the number of recombination events at pilE detected in P+ FA1090 and uvrD colonies arising from the parental 1-81-S2 pilE variant divided by the total number of pilE sequences analyzed. In three independent experiments, the average frequency of pilin Av in the FA1090 parent was 0.11 recombination events per CFU, as previously reported for this pilin variant (8), while the average frequency detected in the uvrD mutant was 0.09 events per CFU (Table 1). There were no statistically significant differences in the frequency of pilin Av between parental and uvrD mutant Gc in any experiment. We conclude that uvrD is dispensable for pilin Av, and effects of uvrD inactivation on pilus-dependent colony morphology in Gc are solely due to increased PilC-dependent phase variation.
TABLE 1.
Pilin antigenic variation frequencies
| Strain | Avg frequency of pilin Av ± SEM |
Overall frequency | ||
|---|---|---|---|---|
| Expt 1 | Expt 2 | Expt 3 | ||
| FA1090 | 0.11 ± 0.02 | 0.10 ± 0.03 | 0.10 ± 0.02 | 0.11 |
| uvrD mutant | 0.14 ± 0.06 | 0.05 ± 0.03 | 0.08 ± 0.02 | 0.09 |
Mutations in uvrD result in a statistically higher frequency of spontaneous mutations.
Given the effect of a uvrD mutant on phase variation events in Gc, we examined whether the uvrD or other NER mutants yielded a higher frequency of spontaneous antibiotic resistance mutations. The antibiotics chosen for these experiments were rifampin and nalidixic acid. Rifampin inhibits the initiation of RNA synthesis by binding to the β subunit of RNA polymerase (rpoB), while nalidixic acid inhibits the activity of DNA gyrase encoded by gyrB. Point mutations in rpoB or gyrB render the bacteria resistant to rifampin or nalidixic acid, respectively (1). FA1090 ΔpilE Gc and each of the NER mutants were plated on solid medium containing either of these antibiotics, and the frequency of mutation was calculated as the number of antibiotic-resistant CFU divided by the total CFU arising on antibiotic-free medium. The median frequency of rifampin resistance in FA1090 Gc was 5 × 10−8 mutants per CFU and for nalidixic acid resistance was 5.6 × 10−6 mutants per CFU (Table 2). Deletion of uvrA, uvrB, uvrC, or mfd had no effect on the median frequency with which Rifr or Nalr colonies arose (Table 2). However, the uvrD-inactivated strain exhibited a 22-fold increase in spontaneous rifampin resistance and a 10-fold increase in spontaneous nalidixic acid resistance (Table 2), increases that were statistically significant relative to the FA1090 parent. Complementation of uvrD reduced the frequency of spontaneous mutations below what was observed for the FA1090 parent, which may be attributed to higher levels of UvrD expression driven by lac regulatory sequences (Table 2). The differential effect of the uvrD and uvrABC mutants on spontaneous mutation frequency indicates that NER is not important for correcting mutations that give rise to spontaneous antibiotic resistance and instead implies that the effect of the uvrD mutation on this process is due to an additional role in Gc mismatch repair.
TABLE 2.
Spontaneous mutation frequencies
| Antibiotic resistance and strain | Spontaneous mutation frequencya |
||
|---|---|---|---|
| Median | 25th/75th percentile | Range (low/high) | |
| Rifr | |||
| Parental strain | 4.99E−08 | 0/1.44E−07 | 0/2.86E−06 |
| uvrA mutant | 0 | 0/0 | 0/2.13E−07 |
| uvrB mutant | 0 | 0/0 | 0/1.67E−07 |
| uvrC mutant | 1.18E−08 | 0/2.04E−07 | 0/4.00E−07 |
| uvrD mutant | 1.09E−06 | 3.89E−07/2.19E−06 | 0/1.09E−03 |
| uvrD/uvrD+ mutant | 0 | 0/0 | 0/6.25E−08 |
| mfd mutant | 5.30E−08 | 0/1.06E−07 | 0/1.67E−06 |
| Nalr | |||
| Parental strain | 5.62E−06 | 9.65E−07/8.61E−06 | 4.21E−07/2.33E−05 |
| uvrA mutant | 1.28E−06 | 8.11E−07/1.73E−05 | 5.09E−07/5.00E−05 |
| uvrB mutant | 3.14E−06 | 7.56E−07/9.98E−06 | 3.08E−07/2.10E−05 |
| uvrC mutant | 1.28E−06 | 3.83E−07/7.24E−06 | 4.55E−08/2.61E−05 |
| uvrD mutant | 5.63E−05 | 1.35E−05/1.41E−04 | 4.94E−06/5.81E−04 |
| uvrD/uvrD+ mutant | 4.15E−07 | 1.69E−07/9.51E−07 | 0/7.58E−06 |
| mfd mutant | 1.92E−06 | 6.07E−07/3.76E−06 | 2.00E−07/1.16E−05 |
The frequency of spontaneous mutation of FA1090 ΔpilE and isogenic NER mutants and complements to Rifr and Nalr was calculated from 10 independent experiments. The differences between FA1090 and the uvrD mutant are significant (P≤ 0.005 [Student t test]).
DISCUSSION
Using a genetic approach, we have demonstrated that Gc possesses a complete NER pathway that participates in the repair of many types of DNA damage, including damage produced by UV light and hydrogen peroxide treatment. However, the phenotypes of the mutants suggest that the NER system has a more significant role in repairing lesions produced by UV irradiation than the damage produced by hydrogen peroxide treatment. Not surprisingly, the Gc NER system has no role in pilin antigenic variation or DNA transformation, which rely on the Rep and PriA helicases, respectively (19, 20). However, inactivation of uvrD demonstrated that the UvrD helicase participates not only in NER but also in mismatch correction, given its effects on PilC phase variation and spontaneous mutation (9a).
The UvrABC excinuclease from E. coli is the prototypical enzyme for NER systems and cooperates with the UvrD helicase and polymerase I to repair a variety of bulky lesions. NER gene expression through the SOS system is well described (6). Although Gc, like many bacteria, operates in the absence of a complete SOS system, the UV sensitivity phenotypes of the uvrABC mutants demonstrate that even without a classical SOS system (2), Gc NER is very efficient at repairing bulky DNA lesions such as pyrimidine dimers, but expression of these genes is not upregulated by hydrogen peroxide treatment (39). Not surprisingly, inactivation of the mfd gene produces a less severe UV sensitivity phenotype than the uvrABC mutants. In E. coli, TRCF (the product of the mfd gene) has been shown to recruit the UvrAB complex to stalled transcription complexes to first release RNA polymerase and, second, to direct NER to the damaged DNA to initiate repair of the damage (36). The original E coli mfd mutant also showed a mild UV sensitivity phenotype (44) but was originally isolated as a mutant which demonstrated “a mutation frequency decline” when irradiated cells are inhibited for protein synthesis (36). We did not test for a diminished mutation frequency after irradiation of mfd Gc since the fastidious nature of this organism makes this analysis difficult to interpret. Since mfd only has a role when a pyrimidine dimer blocks transcription and the uvrABC gene products are important both when transcription and when replication is inhibited, the relative phenotypes fit the present models.
The effect of recombination repair on the NER mutants was tested by using the IPTG regulatable recA6 gene, which provides near wild-type levels of RecA when induced and a phenotypically RecA− cell when no IPTG is in the growth medium (34, 40). RecA expression is necessary for both RecBCD and RecF pathways of recombination, which both contribute to UV survival, but without a classical SOS system does not affect gene regulation in response to DNA damage. Thus, the effect of lowering RecA expression in wild-type cells or uvrABC mutants has a relatively minor effect on UV survival. This is similar to the effect reported by Davidsen et al. in a uvrA recA6 double mutant from the related human pathogen Neisseria meningitidis (11). Intriguingly, we observed a large increase in UV sensitivity in the mfd mutant in the absence of RecA expression. The exacerbated phenotype in the mfd recA6 double mutant grown without IPTG suggests that recombinational repair in Gc may be linked to transcriptional stalls, but the mechanism underlying this linkage is unknown at this time.
The UvrD gene product of E. coli is a general helicase that participates in both NER and mismatch correction (15, 22). The UV sensitivity profile of the Gc uvrD mutant suggests that it contributes to NER in Gc. However, the Gc uvrD mutant also showed a RecA-independent increase in pilus-dependent colony phase variation, which was due to an increased frequency of pilC phase variation. PilC phase variation occurs by slipped-strand mispairing in a mononucleotide tract in the pilC promoter, a mispairing event correctable by the mismatch correction system. We postulate that the increased level of pilC phase variation in the uvrD mutant is due to the additional participation of the UvrD helicase in mismatch correction, which is further supported by the increased frequency of spontaneous mutation to antibiotic resistance in the uvrD mutant but not the uvrABC mutants. We did not observe any increased frequency of opaque colonies that result from the slipped-strand mispairing of a pentameric repeat in the opa gene signal sequence (29). This is not a surprising finding, since we did not observe increased opa gene phase variation in mismatch correction mutants (9a) and since mismatch correction does not influence slipped-strand mispairing of polynucleotide repeats with unit sizes over 4 bp (30).
One unique observation made in the present study is that the uvr mutants show sensitivity to oxidative killing by H2O2 exposure. Oxidative damage to DNA can be manifested as the presence of 8-oxoguanine, as well as a variety of other modified bases (43). E. coli uvr mutants have not been reported to have a strong H2O2 sensitivity phenotype, but human cells that are deficient in NER, obtained from patients with xeroderma pigmentosum, cannot remove 8-oxo-guaniine from DNA (31). Oxidative sensitivity of the Gc NER mutants was only observed at lower levels of H2O2 (2.5 to 5 mM) and not at higher H2O2 concentrations. These results suggest that at high levels of ROS, either a different type of lesion is formed that cannot be repaired by the NER system, or the lethality is due to damage to molecules in addition to DNA such that NER-mediated repair does not alter the survival of the cells. We favor the latter model due to the fact that the NER system is very effective at repairing high levels of UV-induced damage and that H2O2 can damage other molecules, as well as DNA (12).
As an obligate human pathogen whose close relatives are also human specific, most members of the genus Neisseria have not been regularly exposed to UV light for tens to hundreds of thousands of years. Examination of the NER system in Gc allows us to explore why continued expression of this repair system has been selected for throughout evolution, even among organisms such as Gc that are not expected to encounter substantial UV-induced DNA damage. The present study conclusively shows that Gc possesses a complete NER system that is functional in its ability to protect the organism from DNA damage, including that caused by UV irradiation, but does not substantially contribute to natural transformation, pilin Av, or repair of spontaneous mutations. Thus, we speculate that Gc encounters other DNA-damaging agents within humans that are repaired, at least in part, by the NER system. Gc are hypothesized to encounter ROS, such as H2O2, during human infection, and here we have shown that the NER system in Gc can protect the organism from oxidative damage. One potential source of damage-producing oxidants are neutrophils, which migrate abundantly into the genitourinary tract during acute infection, but results from our lab and others suggest that neutrophils do not produce large quantities of ROS upon infection with Gc, at least when actively growing (9). ROS is also produced by cervical epithelial cells and commensal lactobacilli, and we speculate that the Gc NER system may have been retained to counter these insults (32, 38). In addition, Gc grown with aeration generate ROS, and the NER system may protect Gc from their own by-products of oxidative metabolism. Together, these results indicate that there are broader roles for NER than simply protecting organisms from UV irradiation, underscoring the importance of retaining this ancient, conserved repair system in prokaryotes and eukaryotes alike.
Acknowledgments
We thank the Seifert laboratory members for helpful comments on this study.
This study was supported by NIH grants R37AI033493, R01AI055977, and R01AI044239 to H.S.S. A.K.C. was partially supported by grant K99 TW008042.
Footnotes
Published ahead of print on 20 November 2009.
REFERENCES
- 1.Biswas, G., S. Comer, and P. F. Sparling. 1976. Chromosomal location of antibiotic resistance genes in Neisseria gonorrhoeae. J. Bacteriol. 125:1207-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black, C. G., J. A. Fyfe, and J. K. Davies. 1998. Absence of an SOS-like system in Neisseria gonorrhoeae. Gene 208:61-66. [DOI] [PubMed] [Google Scholar]
- 3.Black, C. G., J. A. Fyfe, and J. K. Davies. 1997. Cloning, nucleotide sequence, and transcriptional analysis of the uvrA gene from Neisseria gonorrhoeae. Mol. Gen. Genet. 254:479-485. [DOI] [PubMed] [Google Scholar]
- 4.Black, C. G., J. A. Fyfe, and J. K. Davies. 1995. A promoter associated with the neisserial repeat can be used to transcribe the uvrB gene from Neisseria gonorrhoeae. J. Bacteriol. 177:1952-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle-Vavra, S., and H. S. Seifert. 1996. Uptake-sequence-independent DNA transformation exists in Neisseria gonorrhoeae. Microbiol. 142:2839-2845. [DOI] [PubMed] [Google Scholar]
- 6.Bridges, B. A., and G. M. Brown. 1992. Mutagenic DNA repair in Escherichia coli. XXI. A stable SOS-inducing signal persisting after excision repair of ultraviolet damage. Mutat. Res. 270:135-144. [DOI] [PubMed] [Google Scholar]
- 7.Campbell, L. A., and R. E. Yasbin. 1984. A DNA excision repair system for Neisseria gonorrhoeae. Mol. Gen. Genet. 193:561-563. [DOI] [PubMed] [Google Scholar]
- 8.Criss, A. K., K. A. Kline, and H. S. Seifert. 2005. The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 58:510-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Criss, A. K., and H. S. Seifert. 2008. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol. 10:2257-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Criss, A. K., K. M. Bonney, R. A. Chang, P. M. Duffin, B. E. LeCuyer, and H. S. Seifert. 2010. Mismatch correction modulates mutation frequency and pilus phase and antigenic variation in Neisseria gonorrhoeae. J. Bacteriol. 192:316-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidsen, T., and T. Tonjum. 2006. Meningococcal genome dynamics. Nat. Rev. Microbiol. 4:11-22. [DOI] [PubMed] [Google Scholar]
- 11.Davidsen, T., H. K. Tuven, M. Bjoras, E. A. Rodland, and T. Tonjum. 2007. Genetic interactions of DNA repair pathways in the pathogen Neisseria meningitidis. J. Bacteriol. 189:5728-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang, F. C. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2:820-832. [DOI] [PubMed] [Google Scholar]
- 13.Goodman, S. D., and J. J. Scocca. 1988. Identification and arrangement of the DNA sequence recognized in specific transformation of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 85:6982-6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunn, J. S., and D. C. Stein. 1996. Use of a non-selective transformation technique to construct a multiply restriction/modification-deficient mutant of Neisseria gonorrhoeae. Mol. Gen. Genet. 251:509-517. [DOI] [PubMed] [Google Scholar]
- 15.Hall, M. C., J. R. Jordan, and S. W. Matson. 1998. Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J. 17:1535-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson, A. B., G. Nyberg, and S. Normark. 1991. Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J. 10:477-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kellogg, D. S., Jr., W. L. Peacock, W. E. Deacon, L. Brown, and C. I. Pirkle. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonial variation. J. Bacteriol. 85:1274-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kline, K. A., E. V. Sechman, E. P. Skaar, and H. S. Seifert. 2003. Recombination, repair and replication in the pathogenic neisseriae: the 3 R's of molecular genetics of two human-specific bacterial pathogens. Mol. Microbiol. 50:3-13. [DOI] [PubMed] [Google Scholar]
- 19.Kline, K. A., and H. S. Seifert. 2005. Mutation of the priA gene of Neisseria gonorrhoeae affects DNA transformation and DNA repair. J. Bacteriol. 187:5347-5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kline, K. A., and H. S. Seifert. 2005. Role of the Rep. helicase gene in homologous recombination in Neisseria gonorrhoeae. J. Bacteriol. 187:2903-2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koomey, J. M., and S. Falkow. 1987. Cloning of the recA gene of Neisseria gonorrhoeae and construction of gonococcal recA mutants. J. Bacteriol. 169:790-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumura, K., M. Sekiguchi, A. L. Steinum, and E. Seeberg. 1985. Stimulation of the UvrABC enzyme-catalyzed repair reactions by the UvrD protein (DNA helicase II). Nucleic Acids Res. 13:1483-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long, C. D., D. M. Tobiason, M. P. Lazio, K. A. Kline, and H. S. Seifert. 2003. Low-level pilin expression allows for substantial DNA transformation competence in Neisseria gonorrhoeae. Infect. Immun. 71:6279-6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matson, S. W., D. W. Bean, and J. W. George. 1994. DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. Bioessays 16:13-22. [DOI] [PubMed] [Google Scholar]
- 25.McGrew, R. E. 1985. Gonorrhea, p. 115-116. In Encyclopedia of medical history. McGraw-Hill Book Co., New York, NY.
- 26.Mehr, I. J., and H. S. Seifert. 1998. Differential roles of homologous recombination pathways in Neisseria gonorrhoeae pilin antigenic variation, DNA transformation, and DNA repair. Mol. Microbiol. 30:697-710. [DOI] [PubMed] [Google Scholar]
- 27.Merz, A. J., and M. So. 2000. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16:423-457. [DOI] [PubMed] [Google Scholar]
- 28.Moolenaar, G. F., S. van Rossum-Fikkert, M. van Kesteren, and N. Goosen. 2002. Cho, a second endonuclease involved in Escherichia coli nucleotide excision repair. Proc. Natl. Acad. Sci. U. S. A. 99:1467-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy, G. L., T. D. Connell, D. S. Barritt, M. Koomey, and J. G. Cannon. 1989. Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell 56:539-547. [DOI] [PubMed] [Google Scholar]
- 30.Parker, B. O., and M. G. Marinus. 1992. Repair of DNA heteroduplexes containing small heterologous sequences in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 89:1730-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reardon, J. T., T. Bessho, H. C. Kung, P. H. Bolton, and A. Sancar. 1997. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients Proc. Natl. Acad. Sci. U. S. A. 94:9463-9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saigh, J. H., C. C. Sanders, and W. E. Sanders, Jr. 1978. Inhibition of Neisseria gonorrhoeae by aerobic and facultatively anaerobic components of the endocervical flora: evidence for a protective effect against infection. Infect. Immun. 19:704-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sechman, E. V., M. S. Rohrer, and H. S. Seifert. 2005. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 57:468-483. [DOI] [PubMed] [Google Scholar]
- 34.Seifert, H. S. 1997. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene 188:215-220. [DOI] [PubMed] [Google Scholar]
- 35.Seifert, H. S., C. J. Wright, A. E. Jerse, M. S. Cohen, and J. G. Cannon. 1994. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J. Clin. Investig. 93:2744-2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selby, C. P., and A. Sancar. 1993. Molecular mechanism of transcription-repair coupling. Science 93:53-58. [DOI] [PubMed] [Google Scholar]
- 37.Sparling, P. F. 1966. Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J. Bacteriol. 92:1364-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spurbeck, R. R., and C. G. Arvidson. 2008. Inhibition of Neisseria gonorrhoeae epithelial cell interactions by vaginal Lactobacillus species. Infect. Immun. 76:3124-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stohl, E. A., A. K. Criss, and H. S. Seifert. 2005. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 58:520-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stohl, E. A., and H. S. Seifert. 2006. Neisseria gonorrhoeae DNA recombination and repair enzymes protect against oxidative damage caused by hydrogen peroxide. J. Bacteriol. 188:7645-7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stohl, E. A., and H. S. Seifert. 2001. The recX gene potentiates homologous recombination in Neisseria gonorrhoeae. Mol. Microbiol. 40:1301-1310. [DOI] [PubMed] [Google Scholar]
- 42.Swanson, J. 1978. Studies on gonococcus infection. XII. Colony color and opacity variants of gonococci. Infect. Immun. 19:320-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Houten, B., D. L. Croteau, M. J. DellaVecchia, H. Wang, and C. Kisker. 2005. “Close-fitting sleeves”: DNA damage recognition by the UvrABC nuclease system. Mutat. Res. 577:92-117. [DOI] [PubMed] [Google Scholar]
- 44.Witkin, E. M. 1961. Modification of mutagenesis initiated by ultraviolet light through posttreatment of bacteria with basic dyes. J. Cell Comp. Physiol. 58:135-144. [DOI] [PubMed] [Google Scholar]
- 45.Wright, C. J., A. E. Jerse, M. S. Cohen, J. G. Cannon, and H. S. Seifert. 1994. Nonrepresentative PCR amplification of variable gene sequences in clinical specimens containing dilute, complex mixtures of microorganisms. J. Clin. Microbiol. 32:464-468. [DOI] [PMC free article] [PubMed] [Google Scholar]