

Abstract
Iron is an essential nutrient not freely available to microorganisms infecting mammals. To overcome iron deficiency, bacteria have evolved various strategies including the synthesis and secretion of high-affinity iron chelators known as siderophores. The siderophores produced and secreted by Mycobacterium tuberculosis, exomycobactins, compete for iron with host iron-binding proteins and, together with the iron-regulated ABC transporter IrtAB, are required for the survival of M. tuberculosis in iron deficient conditions and for normal replication in macrophages and in mice. This study further characterizes the role of IrtAB in M. tuberculosis iron acquisition. Our results demonstrate a role for IrtAB in iron import and show that the amino terminus domain of IrtA is a flavin-adenine dinucleotide-binding domain essential for iron acquisition. These results suggest a model in which the amino terminus of IrtA functions to couple iron transport and assimilation.
′
Mycobacterium tuberculosis, the causative agent of human tuberculosis, like most organisms, requires iron to sustain essential cellular functions. Due to the poor aqueous solubility of the ferric ion (Fe3+) in aerobic and neutral pH conditions, free ferric iron is not found in the mammalian host but is bound to iron-binding proteins such as transferrin, lactoferrin, and ferritin (30). A common mechanism by which bacteria acquire iron is the synthesis and secretion of siderophores (high-affinity iron chelators) that can solubilize iron in the environment or remove it from iron-binding proteins of the mammalian host. Fe3+-siderophore complexes are recognized by specific surface receptors and translocated through the plasma membrane by ABC-type transporters, using the energy generated by ATP hydrolysis (13). Dissociation of iron from the incorporated siderophore complex can occur via cleavage of the siderophore or by the action of a ferric reductase (13). Reduction of Fe3+ results in a weaker binding of Fe2+ to the siderophore, allowing release of iron that can then be utilized (21).
To overcome iron limitation, M. tuberculosis synthesizes siderophores named mycobactin and exomycobactin. Mycobactin is very hydrophobic and remains cell associated, whereas exomycobactin (ExMB, also known as carboxymycobactin) is more hydrophilic and is secreted to the medium (8, 16). Fe3+-ExMB complexes can deliver iron to the cell by transfer of iron to mycobactin (7) or by a pathway that is mycobactin independent (17). Previously, we showed that inactivation of M. tuberculosis irtA (Rv1348) or irtB (Rv1349) genes, which encode membrane proteins of the ABC transporter family (2), results in decreased ability of M. tuberculosis to replicate in low-iron medium and to utilize Fe3+-ExMb as the sole iron source. Because IrtA and IrtB each encode a membrane protein with one permease domain fused to an ATPase domain, and irtA and irtB are organized in an operon, we postulated that these two proteins associate to form one ABC transporter necessary for iron acquisition in vitro and also for normal replication of M. tuberculosis in human macrophages and in infected mice lungs (18). We provide here evidence that supports a role for IrtAB as an iron importer and unveils essential properties of the amino-terminal domain (NTD) of IrtA. We propose a model by which IrtA-NTD couples iron transport to assimilation.
MATERIALS AND METHODS
Bacteria, plasmids, media, and growth conditions.
Escherichia coli XL1-Blue was routinely grown in Luria-Bertani broth or agar medium at 37°C and used in DNA cloning procedures. M. smegmatis mc2155 was grown in Middlebrook 7H9 broth or on 7H10 agar (Difco), supplemented with 0.2% glycerol and 0.05% Tween 80. M. tuberculosis strains were grown in the same medium with the addition of albumin-dextrose-NaCl complex (ADN) (9). Antibiotics, when required, were included at the following concentrations: kanamycin (Kan), 10 μg/ml; and hygromycin (Hyg), 100 μg/ml. The plasmids and strains generated and used in the present study are described in Table 1.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | 20 | |
| E. coli | supE44 hsdR17 recA1 gyrA | |
| XL1-Blue | thi relA1 lac F′ | |
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm (DE3) | |
| M. smegmatis mc2155 | High-transformation mutant of M. smegmatis ATCC 607 | 23 |
| M. tuberculosis H37Rv | M. tuberculosis ATCC 25618 | |
| ST73 | irtA::Hyg | 18 |
| ST96 | ST73 complemented with pSM546 wild-type irtA and irtB | 18 |
| ST185 | ST73 complemented with pSM773 (IrtA-R70A and IrtB) | This study |
| ST186 | ST73 complemented with pSM774 (IrtA-Y72A and IrtB) | This study |
| ST187 | ST73 complemented with pSM775 (IrtA-T73A and IrtB) | This study |
| Plasmids | ||
| pET28TEV | Derivative of pET28a (Novagene) with a TEV cleavage site that removes the His tag | 29 |
| pET29 | Expression vector encoding the S-tag peptide sequence to expressed amino-terminal S-tagged fusion proteins | Novagen |
| pSR113 | pMV261 derivative containing the inducible acetamidase promoter of M. smegmatis | 15, 24 |
| pSM430 | pMV261 expressing irtB | This study |
| pSM546 | Integrative vector containing irtA and irtB | 18 |
| pSM547 | Integrative vector containing irtA | This study |
| pSM756 | S-irtA-irtB in pSR113 | This study |
| pSM773 | pSM546 derivative with IrtA-NTD-R70A | This study |
| pSM774 | pSM546 derivative with IrtA-NTD-Y72A | This study |
| pSM775 | pSM546 derivative with IrtA-NTD-T73A | This study |
| pSM776 | pET28TEV harboring IrtA-NTD fused to a His tag at the amino terminus | This study |
| pSM777 | pET28TEV harboring IrtA-NTD-R70A fused to a His tag | This study |
| pSM778 | pET28TEV harboring IrtA-NTD-Y72A fused to a His tag at the amino terminus | This study |
| pSM779 | pET28TEV harboring IrtA-NTD-T73A fused to a His tag at the amino terminus | This study |
| pSM780 | S-irtA-Y72A-irtB in pSR113 | This study |
| pSM784 | S-irtA-T73A-irtB in pSR113 | This study |
| pJEM12 | E. coli-mycobacterium shuttle vector to create PhoA protein fusions | 26 |
| pJEM11 | E. coli-mycobacterium shuttle vector to create LacZ protein fusions | 11 |
For mycobacterial growth in low-iron minimal medium (LIMM), a defined medium was prepared containing 0.5% (wt/vol) asparagine, 0.5% (wt/vol) KH2PO4, 2% glycerol, 0.05% Tween 80, and 10% ADN. The pH was adjusted to 6.8. To lower the trace metal contamination, the medium was treated with Chelex-100 (Bio-Rad), according to the manufacturer's instructions. Chelex was removed by filtration, and the medium was supplemented with 0.5 mg of ZnCl2, 0.1 mg of MnSO4, and 40 mg of MgSO4 per liter. When required, iron-sufficient conditions were obtained by supplementing this medium with 50 μM FeCl3.
Preparation of 55Fe-exomycobactin and iron uptake assays.
Ferri-exomycobactins (Fe+3-ExMB) were extracted from the culture filtrate of M. tuberculosis H37Rv according to published protocols (7) and purified by flash silica gel chromatography as previously described (17). Fe+3-ExMB was deferrated by incubation with 1 volume of 50 mM EDTA (pH 4.0) for 18 h or until the absorbance at 450 nm decreased to <10% of its initial value. The solution was then extracted into chloroform, the chloroform was evaporated, and the residue was resuspended in 50% ethanol. Desferri-ExMB was then mixed with 55FeCl3 (Perkin-Elmer) in a ratio of 2:1 ExMB to iron, and the mixture was incubated for 10 min at room temperature. 55Fe3+-ExMB was then extracted into chloroform and washed twice with water to remove free unbound 55Fe3+. The chloroform was evaporated, and 55Fe3+-ExMB was suspended in 50% ethanol and kept in small aliquots at −20°C.
For iron uptake experiments mycobacterial strains were iron depleted by pregrowing them in LIMM. Cultures were inoculated to give an OD540 nm of 0.05. When they reached an OD540 nm of 0.4, the bacteria were collected by centrifugation, diluted to 2 × 108 bacilli per ml in LIMM, and incubated with 55Fe+3-ExMB (106 cpm) at 0 or 37°C. Samples (0.5 ml) were removed at the indicated time points and immediately placed on ice. Bacteria were collected by centrifugation and washed once with 0.1 M LiCl and twice with cold LIMM. The bacterial pellet was resuspended in scintillation fluid, and radioactivity was determined by using a Beckman LS6500 scintillation counter set at the wide-open window setting. The data are expressed as counts per minute (cpm) incorporated by 108 cells at each time point after subtraction of the radioactivity incorporated in cells incubated on ice, which was usually <10% of total incorporation.
Purification of IrtA-NTDs.
The DNA sequence encoding the first 280 amino acids at the NTD of IrtA were PCR amplified by using the forward primer 5′-GACTCATATGGCACGCGGGTTG-3′, the reverse primer 5′-CCGCAAGCTTTTCGGTTGCTCG-3′, and plasmid pSM546 (Table 1), which includes irtA and irtB (18), as a template. The PCR product was purified from agarose gels and inserted at the NdeI-HindIII sites of pET28TEV (29), a vector modified from pET28a (Novagen) to create an in-frame gene fusion of irtA-NTD with a His tag at the amino terminus that is cleavable by the TEV protease. The resulting plasmid pSM776 was transformed into the E. coli strain BL21(DE3). E. coli harboring pSM776 was grown overnight in 50 ml of LB containing 50 μg of Kan/ml (LB-Kan) at 37°C. Then, 1 liter of LB-Kan was inoculated with 25 ml of this culture, and cells were grown at 37°C with 230 rpm shaking until reaching an optical density at 600 nm (OD600) of 0.6. The cells were then incubated at room temperature at 230 rpm for 1 h. A final concentration of 50 μM IPTG (isopropyl-β-d-thiogalactopyranoside) was added to the culture, and cells were allowed to continue growing for another 3 h under the same conditions. The cultures were centrifuged at 3,500 × g, and the cell pellet was resuspended in 50 mM phosphate buffer (pH 7.4)-0.5 M NaCl. The cells were lysed in the presence of 2 mM phenylmethylsulfonyl fluoride in a French press apparatus at 4°C. The cell lysate was centrifuged at 30,000 × g for 30 min at 4o, and the supernatant and the pellet were collected. The supernatant was loaded onto a nickel column (His-Trap) (Amersham Biosciences). Unbound material was removed by washing the column with a buffer containing 50 mM sodium phosphate (pH 7.4), 0.5 M NaCl, and 20 mM imidazole. His-IrtA-NTD was eluted from the column with a linear gradient of 20 to 300 mM imidazole. Fractions were analyzed by SDS-PAGE and Coomassie blue staining. Those containing His-IrtA-NTD, which migrates as a band of ∼32 kDa (the calculated molecular mass of His-IrtA-NTD is 31.6 kDa), were pooled and digested with TEV protease to cleave the His tag. The His tag-free IrtA-NTD was separated from noncleaved His-IrtA-NTD in a second His-Trap column. Alanine replacement mutants of amino acids R70, Y72, and T73 in IrtA-NTD were generated by QuikChange mutagenesis (Stratagene) and cloned into pET28TEV. The resulting plasmids pSM777, pSM778, and pSM779 were used to transform E. coli BL21(DE3). IrtA-NTD mutant polypeptides were expressed and purified from E. coli as described for the wild-type IrtA-NTD.
Determination of FAD content.
UV/Visible absorption spectra of purified IrtA-NTD was recorded at 25°C by using a NanoDrop 1000 spectrophotometer (Thermo Scientific). Purified IrtA-NTD was also analyzed by mass spectrometry. The FAD content of IrtA-NTD wild type and Ala mutants was determined in a solution containing 50 μg of purified protein after denaturation with 6 M guanidine hydrochloride (Gnd-HCl). Denatured protein was centrifuged and the supernatant filtered through a Microcon YM-10 (Millipore) to remove the protein. The UV/visible absorbance spectrum of the filtrate was recorded between 300 and 550 nm, and the absorbance at 450 nm was used to calculate the FAD concentration by using the value previously determined for the extinction coefficient of free FAD in the presence of Gnd-HCl (ɛ450 = 11,900 M−1 cm−1) (28).
Construction of LacZ and PhoA translational fusions.
A 400-bp DNA fragment containing the promoter region of irtA plus the sequence encoding the first 129, 150, 274, or 320 amino acids of IrtA-NTD were amplified by PCR using irtApApaI (5′-AGCGGATGTGGGTTTGGTC-3′) as a forward primer; 129-B (5′-GGCGGGCTGCTCCTCGGGCACGTC-3′), 150-B (5′-GACCG TTTCGATGATCCCGTTCATCCC-3′), 274-B (5′-ACGCGCCGGGGCAGGCACCGCCGA-3′), and 320-B (5′-TGACAGCTCGACCAACAGCACG-3′), respectively, as reverse primers; and pSM546 (18) as a template. ApaI and BamHI sites were added to the primers at the 5′ and 3′ ends, respectively, to clone each PCR fragment into the same sites in pJM12 (26) and pJM11 (11) in frame with the lacZ and phoA genes, respectively. The resulting plasmids were electroporated into M. smegmatis and transformants selected on 7H10-Kan. Three independent Kan-resistant transformants from each transformation were grown and assayed for β-galactosidase and alkaline phosphatase activity.
Enzymatic activities.
β-Galactosidase and alkaline phosphatase activities were assayed in M. smegmatis strains grown to early stationary phase in LIMM to induce the irtA promoter. The β-galactosidase activity was measured in bead beater lysates of M. smegmatis according to the protocol of Parde et al. (14). Units of β-galactosidase were calculated by the following formula: 1,000 × the OD420 per mg of protein per min. The protein concentration in the bacterial extracts was measured by the Bradford assay (Bio-Rad). M. smegmatis mc2155 transformed with pJM12 was used as negative control.
Alkaline phosphatase activity was measured in a 2.5-ml culture of mycobacteria grown in LIMM to early stationary phase. The culture was centrifuged, and the pellet of cells resuspended in 200 μl of 1 M Tris-HCl (pH 8.0). A 1-ml portion of 20 mM p-nitrophenyl-phosphate (Sigma) dissolved in 1 M Tris-HCl (pH 8.0) was added. The reactions were incubated at 37°C in the dark until a yellow reaction product was visible. The reactions were stopped with the addition of 100 μl of 1 M K2HPO4 (pH 8.0). Bacteria were removed by centrifugation, and the OD420 of the supernatant was measured. The alkaline phosphatase activity was calculated by the following formula: 1,000 × the OD420/(reaction time in minutes) (OD600 of the culture) (volume of suspended cells used in reaction in ml). M. smegmatis mc2155 transformed with pJM11 was used as a negative control.
Complementation assays.
ST73 was transformed with plasmids pSM547, pSM430, and pSM546 (Table 1) expressing irtA, irtB, or irtA-B, respectively, to generate strains ST97, ST193, and ST96. Plasmids were sequenced before electroporation to assure that there were no mutations in the cloned genes. Selected transformants were tested for growth in low iron and compared to ST73 and the wild-type strain H37RV.
Nucleotide changes that replace R70, Y72, or T73 in IrtA with Ala were made in pSM546 by using QuikChange site-directed mutagenesis (Stratagene). Mutated plasmids were sequenced to confirm the intended mutations and to assure that no additional mutations were introduced in irtA or in irtB. The pSM546-derived plasmids were electroporated into the mutant strain ST73, and the resulting strains ST185, ST186, and ST187, as well as the complemented strain ST96, were examined for growth in low iron by cultivating the strains in LIMM with agitation, at 37°C, and monitoring the increase in absorbance at 540 nm.
Protein stability.
S-tagged forms of IrtA, IrtA-Y72A, and IrtA-T73A were generated, expressed in M. smegmatis, and detected by Western blotting of the mycobacterial membranes using an anti-S antibody. First, the irtAB operon was inserted into the E. coli expression vector, pET29a, to generate an S-tagged IrtA. The resulting plasmid, pSM756, was used as a template for site-directed mutagenesis of S-irtA, and the desired mutations were confirmed by DNA sequencing. The tagged wild-type and mutated operons were then subcloned into the acetamide-inducible mycobacterial expression vector, pSR113. The resulting plasmids pSM780, pSM784, and empty pSR113 were electroporated into M. smegmatis mc2155. Expression of the tagged proteins was induced in mid-log-phase cultures by addition of 0.2% acetamide for 2 h. Culture pellets were suspended in 1.5 ml of phosphate-buffered saline (PBS) containing a protease inhibitor cocktail (Roche), and the bacterial cells were lysed by sonication (three pulses of 30 s) on ice. Membranes were isolated as described previously (31), and the protein concentration was determined by a DC protein assay (Bio-Rad).
SDS-PAGE and immunoblotting.
Proteins extracts were resolved on 10 or 7.5% gels by SDS-PAGE (10). The gels were electrophoretically transferred to prewetted PVDF membranes (Millipore). PVDF membranes were incubated with anti-PhoA antibody (Sigma) or with S-tag antibody (Novagen), as indicated, and then with a peroxidase-conjugated secondary antibody (Amersham Biosciences) which was detected by using ECL Western blotting detection kit (GE Healthcare) as recommended by the supplier.
RESULTS
Role of IrtAB in iron uptake.
Previously, we showed that the M. tuberculosis mutant strain ST73, in which irtA was inactivated by insertion of a Hyg resistance cassette, has decreased ability to replicate under iron-deficient conditions and to obtain iron, even when provided as Fe3+-ExMB (18), despite normal siderophore synthesis and secretion (18). ST73 is also less sensitive to the iron-activated antibiotic streptonigrin than the wild-type strain (see Fig. S1 in the supplemental material), which suggests lower levels of intracellular iron in this strain than in the wild type.
There is no intergenic sequence between irtA and irtB, and these two genes are cotranscribed (5, 19), suggesting that the irtA mutation will be polar on irtB. However, quantification of irtB transcript in ST73 and H37Rv by reverse transcription-PCR (RT-PCR) showed 40 to 50% reduction in the levels of irtB transcript in ST73 compared to the wild-type strain (data not shown), suggesting that irtB might be transcribed by an internal promoter on irtA. Since irtB inactivation also results in iron deficiency (18), in order to determine whether ST73's iron deficiency is due to inactivation of irtA, a reduction in the levels of irtB, or both, we complemented ST73 with irtA or irtB separately and together (Fig. 1). irtA partially restored growth in low iron, suggesting that IrtB expressed in ST73, in combination with IrtA, can reconstitute functional transporters. irtB alone did not complement, indicating that iron deficiency in ST73 is not only due to a reduction in IrtB levels but also to the absence of IrtA. As expected, reconstitution of irtA and irtB expression restored normal growth of ST73 in low iron, as observed previously (18). These results indicate that both irtA and irtB are required for normal iron acquisition and support our hypothesis that, by reconstituting the ABC transporter IrtAB, these genes mediate iron uptake in M. tuberculosis.
FIG. 1.

Complementation of ST73. H37Rv (⧫), ST73 (•), and ST73 transformed with a plasmid expressing irtA (▴), irtB (▪), or irtAB (×) were inoculated in LIMM. Growth was monitored each day by measuring the OD540. The results of a representative experiment are shown. The experiment was repeated three times. All strains grew normally in high-iron medium (data not shown).
In order to determine whether the iron deficiency phenotype exhibited by ST73 is the result of decreased iron import, we measured the uptake of 55Fe-ExMB by ST73 and H37Rv pregrown in low-iron conditions. Iron incorporation was considerably reduced in ST73 compared to the wild-type strain (Fig. 2), supporting a role for IrtAB as a high-affinity iron importer in M. tuberculosis. Iron incorporation, however, was not completely eliminated in ST73. The possible reasons for this are discussed bellow.
FIG. 2.

55Fe3+-ExMB uptake. H37Rv and ST73 were cultured in LIMM and incubated with 55Fe3+-ExMB at 0 or 37°C. At the indicated time points, an aliquot of cells was collected, washed as described in Materials and Methods, and resuspended in scintillation liquid. The radioactivity incorporated was measured in a Beckman scintillation counter. Symbols: □, H73Rv; ○, ST73. The data show the radioactivity incorporated at 37°C after subtraction of the radioactivity incorporated at 0°C. The average and standard deviation are derived from three experiments.
Sequence-structure analysis of the amino-terminal domain of IrtA.
Amino acid sequence analysis of IrtA and IrtB show 34% identity between these two proteins. This similarity spans the transmembrane and carboxy-terminal domains. However, IrtA has an N-terminal extension of 272 amino acids that is not present in IrtB. This N-terminal extension (IrtA-NTD) is also absent in the close homolog YbtP, a component of the YbtPQ iron importer of Yersinia pestis (6). ProDom analysis (22) indicated a similarity between IrtA-NTD and several proteins annotated as putative siderophore interacting proteins (SIPs) present in various bacteria. However, since the interaction of these proteins with siderophores has not been demonstrated, we prefer to refer to this family as siderophore utilization proteins (SUPs). The prototype of this family is ViuB, a cytoplasmic protein from Vibrio cholerae that is necessary for ferric-vibriobactin utilization (3). Sequence alignment of IrtA-NTD with ViuB and other annotated SUPs revealed high homology and a total of 16 amino acids perfectly conserved (Fig. 3). The motif RXYS(T) (positions 70 to 73 in IrtA) involved in flavin adenine dinucleotide (FAD) binding by proteins of the ferrodoxin reductase family (4) was found in all SUPs. The crystal structure of the Shewanella putrefaciens SUP was recently determined (PDB entry 2GPJ). This protein cocrystallized with a tightly bound FAD molecule, suggesting that SUPs may represent a group of FAD-binding oxidoreductases. Comparison of the predicted secondary structures of the SUPs included in Fig. 3 and the structure of the S. putrefaciens SUP shows that most of the secondary structural elements are at well-conserved sequence segments, indicating that these proteins are likely to have similar structures. The residues interacting with the bound FAD in the S. putrefaciens SUP structure are identical in IrtA-NTD and the other proteins analyzed (Fig. 3). A variation of the classical mix of alpha and beta sheets known as the Rossmann fold (4), commonly found in mono- and dinucleotide binding proteins (4), is also found in these proteins (Fig. 3).
FIG. 3.

Sequence analysis of IrtA-NTD. A sequence alignment of the M. tuberculosis IrtA-NTD with other putative iron-siderophore utilization proteins is shown. The sequences were aligned by using CLUSTAL W (25), and the figure was generated with ALSCRIPT (1). Terms: SIP-S.Putr, siderophore-interacting protein from Shewanella putrefaciens; SID-IrtAMtb, IrtA-NTD from M. tuberculosis; MxcB-S.aura, myxochelin iron-binding protein from Stigmatella aurantiaca; PSUP-P.put, putative iron-chelator utilization protein from Pseudomonas putida; ViuB-V.chol, vibriobactin utilization protein from V. cholerae; ViuB-R.bal, vulnibactin utilization protein from Rhodopirellula baltica. The secondary structural elements at the top of the sequences are from the crystal structure of SIP-S.putr (PDB ID 2GPJ). Cylinders represent α-helices, and arrows denote β-strands. Residues labeled with an asterisk at the bottom of the sequences are those in the SIP-S.putr structure whose side chains interact with the bound FAD. These residues are identical among the aligned sequences. Residues marked with a triangle are those having direct hydrogen bonds from main chain atoms to the bound FAD. A vertical arrow marks the domain boundary between the N- and C-terminal domains. The C-terminal domain of these proteins has a β1α1β2α2β3α3 Rossmann fold, α4 connecting β3 and β4, and a second Rossmann fold lacking the sixth strand as is observed in other FAD-binding proteins (4).
IrtA-NTD purification.
To characterize IrtA-NTD, we overexpressed and purified this protein segment in E. coli as described in Materials and Methods (Fig. 4). Purified IrtA-NTD exhibited a yellow pigment that was not eliminated after dialysis in the presence of EDTA. UV/visible spectral analysis of IrtA-NTD showed a characteristic absorbance of an oxidized flavoprotein maxima at 275, 370, and 450 nm (Fig. 5A). In addition, liquid chromatography-mass spectrometry analysis of a pure preparation of IrtA-NTD indicated that the molecule associated with IrtA-NTD had a mass corresponding to FAD (data not shown). Denaturation of IrtA-NTD with Gnd-HCl completely dissociated FAD from the protein, indicating that the cofactor was noncovalently bound to IrtA-NTD (Fig. 5B). Quantification of the flavin content in the filtrate of a Gnd-HCl denatured sample of IrtA-NTD of known concentration resulted in a ratio of 1.1 mol of FAD/mol of protein. This ratio was obtained for two independent preparations of purified IrtA-NTD.
FIG. 4.
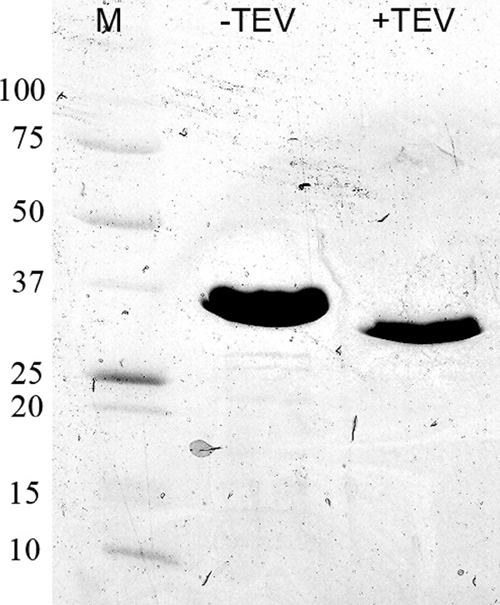
IrtA-NTD purification. IrtA-NTD was expressed and purified as described in Materials and Methods. The protein was expressed with an N-terminal His6 tag and migrated as a band of ∼32 kDa on an SDS-polyacrylamide gel (8 to 25%). The calculated molecular mass is 31.6 kDa. After the His tag was cleaved by TEV protease, the calculated molecular mass is 28.5 kDa. Lane M, molecular markers with molecular masses labeled on the left side in kilodaltons.
FIG. 5.

Spectral analysis of IrtA-NTD. (A) UV/visible spectrum of purified IrtA-NTD. An inset shows an expanded-scale spectrum of characteristic flavin absorbance between 300 and 500 nm. (B) UV/visible spectrum of the filtrate recovered after Gnd-HCl denaturation of IrtA-NTD and filtration through a Microcon YM-10 column.
IrtA-NTD FAD binding is required for iron acquisition.
Having determined that purified IrtA-NTD has bound FAD, we next investigated whether FAD binding was necessary for IrtAB function. We generated mutant IrtA proteins in which the conserved R70, Y72, and T73 in the FAD binding motif were replaced with Ala. These mutations were made in irtA cloned with irtB into an integrative plasmid. Each mutated construct was electroporated into ST73, and the ability of the expressed proteins to complement the low-iron growth defect in this strain was examined. As a positive control, ST73 was transformed with a plasmid carrying wild-type irtAB (18). All strains had equivalent growth rates in iron-sufficient medium (data not shown), whereas in low-iron conditions IrtA-R70A fully complemented the mutant, but IrtA-Y72A and IrtA-T73A did not (Fig. 6A). To rule out the possibility that the failure of IrtA-Y72A and IrtA-T73A to complement ST73 was due to decreased stability of these proteins, they were fused with an S tag at the amino terminus, and their expression and localization on the membrane of M. smegmatis were compared to that of the wild-type S-tag-IrtA. As shown in Fig. 6B, comparable amounts of the mutant and wild-type IrtA proteins were detected in M. smegmatis membrane extracts, indicating that the mutations do not affect the stability of the protein or localization to the membrane. Therefore, we conclude that replacement of Y72 and T73 by Ala in IrtA prevents the normal function of this protein. To determine the ability of the Ala replacement mutants to bind FAD, the same mutations were constructed in the IrtA-NTD isolated domain, and mutated polypeptides were expressed in E. coli and purified as was done for the wild-type IrtA-NTD. The FAD content of each mutant protein was determined. FAD/protein molar ratios of 1.1, 0.29, and 0.17 were obtained for R70A, Y72A, and T73A, respectively. Thus, the ability to bind FAD directly correlated with the complementing capacity of each mutant protein, indicating that IrtA-NTD and its ability to bind FAD is necessary for IrtA function.
FIG. 6.

Growth of mycobacterial strains in low iron and stability of mutated IrtAs. (A) M. tuberculosis strains were grown in LIMM. Growth was monitored each day by measuring the OD540. H37Rv (⧫), ST73 (□), and ST73 complemented with wild-type irtAB (▵), irtAR70A-irtB (▪), irtAY72A-irtB (▴), and irtAT73A-irtB (•) were examined. The results of one representative experiment are shown. The experiment was performed three times. (B) Stability of noncomplementing mutated IrtAs. S-tagged IrtA wild type (lane 1), S-IrtAY72A (lane3) and S-IrtAT73A (lane 4) were expressed in M. smegmatis under acetamide induction. M. smegmatis harboring the acetamide-inducible empty vector was used as a negative control (lane 2). Membrane fractions were isolated, the protein concentration was determined, and an equivalent amount of protein from each extract was loaded on a SDS-7.5% PAGE gel. S-IrtAs were detected by Western analysis with an anti-S monoclonal antibody.
IrtA-NTD topology.
To understand the function of IrtA-NTD, it was important to determine whether IrtA-NTD is in the cytoplasm or faces the external environment. There is no signal sequence at the amino-terminal end of IrtA and prediction of the transmembrane helices and topology of IrtA by HMMTOP (27) indicates six transmembrane helices, the first one encompassing amino acids 291 to 315 and both the NTD and the carboxy-terminal domain (CTD) in the cytoplasm. In contrast, TMHMM and MEMSAT predictions suggest that IrtA-NTD is exposed to the outside. Furthermore, other algorithms predict, with low probability, an additional membrane-spanning segment from amino acids 103 to 123. Given these contradicting predictions suggested by in silico approaches, we decided to determine IrtA-NTD's topology experimentally. For this purpose, IrtA-NTD was fused at increasing lengths to the reporter genes lacZ or phoA. PhoA is assembled into functional dimers only if it is exported across the membrane, whereas LacZ fusions are only active if the fusion site is located in the cytoplasm (12). IrtA-NTD PhoA and LacZ translational fusions were expressed from the native irtA promoter in M. smegmatis grown in low-iron conditions. Alkaline phosphatase and β-galactosidase activities of three independent recombinants were measured. All of the LacZ fusions were positive, and all PhoA fusions were negative except for the longest one at amino acid 320. In this case, the LacZ fusion was negative, and the PhoA fusion was positive (Fig. 7A and B). All PhoA fusion proteins were detected in protein extracts of M. smegmatis (Fig. 7C), indicating that the lack of PhoA activity was not due to instability of the protein fusions. These results are consistent with the topology predicted by HMMTOP in which the first 290 amino acids, i.e., the IrtA-NTD, are cytoplasmic, and IrtA inserts into the membrane via the predicted transmembrane domain that includes amino acids 291 to 315 (Fig. 7D).
FIG. 7.

Determination of NTD-IrtA membrane topology. The β-galactosidase (A) and alkaline phosphatase (B) activities of M. smegmatis expressing IrtA-NTD fused at various points to LacZ and PhoA were determined. The activities were measured in M. smegmatis, cultured in LIMM as described in Materials and Methods. The averages and standard deviations of three independent experiments are shown. M. smegmatis transformed with the vectors pJM12 or pJM11 was used as negative controls. (C) Anti-PhoA Western analysis of M. smegmatis lysates expressing each IrtA-NTD-PhoA fusion. Lanes: 1, pJM11 control; 2, A129-phoA; 3, V150-phoA; 4, R274-phoA; 5, S320-phoA. (D) Predicted topology of IrtA-NTD based on the LacZ and PhoA activities of translational fusions constructed in the present study (indicated by filled circles) and the position of the first transmembrane domain predicted by HMMTOP (pointed by arrows). Additional transmembrane domains illustrated are based on the prediction by HMMTOP.
DISCUSSION
In this study, we have further characterized the role of irtA and irtB in M. tuberculosis. Complementation analysis of the strain ST73, in which irtA is inactivated and irtB expression is reduced, allowed us to confirm that both irtA and irtB are required for normal growth of M. tuberculosis in low-iron conditions. We showed that disruption of the irtAB operon results in decreased iron uptake as demonstrated by reduced incorporation of Fe55 in the ST73 mutant strain compared to the wild-type H37Rv. It is important to note that for the iron uptake assay, the two strains were precultured in low iron; however, uptake was measured before iron depletion affected the viability of the ST73 cells. The extent of this iron depletion is not sufficient to fully induce iron uptake genes in the wild-type strain (G. M. Rodriguez et al., unpublished results). We believe that an even bigger difference in iron uptake would be observed if the wild-type strain is further iron depleted so that stored iron is consumed and uptake is completely derepressed.
Iron incorporation by the ST73 mutant was significantly reduced; however, some iron was still incorporated. ST73 accumulates derepressed mycobactin (18) on the surface, so it is possible that some iron is bound to mycobactin which can receive it from Fe+3-exomycobactin. Another possibility is that in the absence of IrtA, IrtB, which is expressed at reduced levels in ST73, may form functional homodimers and transport iron at lower levels. Alternatively, in addition to IrtAB, M. tuberculosis may possess additional iron transporters that are still functional in ST73. Examining the iron uptake capacity of a double mycobactin-irtAB-deficient strain, determining optimal interactions between the IrtA and IrtB, and analysis of additional iron transporters in M. tuberculosis may help distinguish those possibilities.
The deficiency in iron uptake exhibited by strain ST73, taken together with normal siderophore synthesis and secretion, supports the role of IrtAB in iron transport in M. tuberculosis, which we previously postulated (18). In addition, complementation of ST73 by irtA supports our hypothesis that IrtA and IrtB work together. Recently, Farhana et al. (5) reconstituted recombinant IrtA and IrtB into proteoliposomes and tested IrtA proteoliposomes for siderophore export and IrtB proteoliposomes for siderophore import. Although, the converse, control experiment was not reported and the topology of the proteins in the liposomes was not clearly established, the authors concluded that IrtA acts as an ExMB exporter and IrtB as a Fe+3-ExMB importer, presumably as homodimers. This conclusion is difficult to reconcile with our in vivo experiments in M. tuberculosis, which show that irtA is not required for ExMB export as normal levels of ExMB are detected in the culture filtrate of ST73 (18). Farhana et al. argued that the siderophore detected in the culture filtrate of ST73 resulted from cell lysis after accumulation of intracellular siderophore. Only massive lysis could account for normal siderophore levels in the culture filtrate of ST73 and, in fact, we examined the release of intracellular content from ST73 and H37Rv in the low-iron culture filtrate. IdeR, a cytoplasmic protein, was not detected in the siderophore containing culture filtrate of ST73, ruling out membrane leakage as the source of siderophore (see Fig. S2 in the supplemental material). Interestingly, in the same study, Farhana et al. obtained a mutant of the irtA homolog in M. smegmatis which shows deficient replication in low iron and a negative siderophore assay (CAS), phenotypes that were complemented by the M. tuberculosis irtA. However, not mentioned by these authors is the fact that although M. smegmatis synthesizes very low levels of exomycobactin, the major siderophore secreted by M. smegmatis and responsible for a positive CAS assay is not exomycobactin, but exochelin, a nonrelated peptidic siderophore not made by M. tuberculosis. Thus, their results indicate that in M. smegmatis irtA is necessary for normal exochelin synthesis or secretion (something that was not examined). Whether irtA in M. smegmatis is necessary for exomycobactin secretion was not tested by Farhana et al. The effect of the irtA mutation in the exochelin pathway in M. smegmatis, together with the fact that irtA inactivation does not affect exomycobactin secretion in M. tuberculosis, raises the possibility of functional differences of IrtA in the molecular context of M. smegmatis and M. tuberculosis.
The N-terminal domain of IrtA.
Sequence analysis of the amino-terminal domain of IrtA revealed a primary and secondary structure similarity to ViuB and related proteins. A remarkable conservation in the sequence of these proteins included a typical consensus motif for FAD binding, and we showed that purified IrtA-NTD contains bound FAD. Furthermore, single amino acid replacements in the FAD ligands, which prevented binding of this cofactor, also abrogated IrtAB-mediated iron acquisition, as demonstrated in complementation analysis of ST73. These results indicated an essential role for IrtA-NTD and FAD in M. tuberculosis iron acquisition. On the basis of sequence alignments of IrtA-NTD with other SUPs (Fig. 3) and the crystal structure of the SUP from S. putrefaciens (PDB entry 2GPJ), we constructed a model for the M. tuberculosis IrtA-NTD structure (Fig. 8). Several residues are predicted to interact with the bound FAD molecule and are likely to play important roles in binding FAD. Side chains of residues in the conserved sequence motif, RXYT, are near the bound FAD. The Arg70 side chain binds to the phosphate groups. However, Arg70 probably makes only a small contribution to FAD binding. The guanidinium group is at the surface of the protein, partially exposed to bulk solvent, which weakens its charge-charge interactions with the phosphates. Both phosphate groups are near the N termini of two helices (Fig. 8) and have H bonds to the amino groups of the protein main chain, which hold the phosphates in their position. The FAD molecule is bound mainly through the flavin and adenine groups. Slightly more flexibility at the phosphate groups is unlikely to affect the binding of FAD and the function of the protein. As described in Results, our mutagenesis experiments indicated that mutating R70 to Ala has no effect on FAD binding and IrtA function. Side chains of Y72 and T73, however, are interacting with the flavin group, which is buried deep in the binding pocket. Mutations of these two residues drastically affected FAD binding and iron acquisition. Interestingly, replacement of T73 with Ala further decreased growth of ST73 in low iron, suggesting a dominant-negative effect of this mutation, which we are currently investigating. The structural model also suggests a few other residues that are likely to be important for FAD binding (Fig. 8). The Tyr237 side chain interacts with one side of the flavin rings. Side chains of H91 and W238 sandwich both sides of the adenine group. Experiments to mutate these residues and confirm the model of FAD-binding interactions are in progress.
FIG. 8.

Model structure showing the FAD-binding pocket IrtA-NTD. The model was constructed based on sequence alignments and the structure of the SUP from S. putrefaciens (PDB entry 2GPJ). Only side chains that are directly involved in the binding interactions are shown. These side chains are conserved in most of the SUP sequences. There are also many H-bonds and pi-electron interactions with the protein main chain, which are not shown in the figure for clarity. The residue numbers are from the IrtA protein sequence. The figure was generated by using the program PYMOL (http://www.pymol.org).
Analysis of the topology of IrtA-NTD by construction of protein fusions with β-galactosidase and alkaline phosphatase indicated that IrtA-NTD (amino acid residues 1 to 272) is in the cytoplasm, and the first transmembrane domain mediates insertion of IrtA in the membrane. The cytoplasmic localization suggests that IrtA-NTD functions after translocation of Fe3+-ExMB through the plasma membrane. Ferric iron reduction is the most common way to release iron from a siderophore complex since siderophores have low affinity for ferrous iron. Bacterial ferric reductases are usually flavin reductases. They are found as apoenzymes and function by forming a transient ternary complex with the reductant [usually NAD(P)H] and free flavin. After reduction by NAD(P)H the reduced flavin dissociates from the enzyme and can serve as a chemical reductant of siderophore complexed Fe+3 (21). The ferric reductase from the archaeon Archaeoglobus fulgidus is a flavoprotein, and it has been suggested to work by a “ping-pong” mechanism by which the bound flavin accepts electrons from NAD(P)H or other electron donor and can then directly reduce complexed Fe3+. The discovery that IrtA-NTD is a cytoplasmic, FAD containing domain, and the fact that IrtA-NTD FAD binding is required for the full ability of M. tuberculosis to acquire iron, leads us to propose a model in which IrtA-NTD functions as a flavin/ferric reductase necessary to release iron from the siderophore complex, making it available for utilization (Fig. 9). This model is also supported by the observations regarding the role of the homologous protein ViuB in V. cholerae. ViuB is a cytoplasmic protein required for utilization of iron chelated by vibriobactin (3). Interestingly, viuB can complement an E. coli fes mutant lacking the enterobactin esterase required for the intracellular release of iron from ferric-enterobactin. Because there is no sequence homology to Fes, ViuB presumably allows iron release from enterobactin by a mechanism other than siderophore cleavage. It will be interesting to examine whether ViuB is also a flavoprotein and can function as a ferric reductase. We have not yet detected ferric reductase activity in recombinant IrtA-NTD using NAD(P)H as electron donor. It is likely that it functions not as an independent domain but in association with the whole IrtA protein or even the IrtAB heterodimer. The reductase may also not be active outside the membrane environment and perhaps requires a membrane associated electron donor. We are currently investigating these various possibilities. To the best of our knowledge, if our model were correct, IrtA would be the first transporter with a reductase domain. If this is the case, it is intriguing to ask why would M. tuberculosis couple transport and reduction of iron. It may be that is important to release iron from the siderophore complex as soon as it enters the cell, and perhaps this is necessary for delivery of iron to the appropriate molecules in the cell.
FIG. 9.

Model of IrtA-NTD function. IrtA-NTD is proposed to function as a flavin/ferric reductase that reduces iron in the imported Fe3+-ExMB complex for its assimilation.
From our results it is clear that IrtAB mediates translocation of iron into the cell; however, it remains unknown how ExMB reaches IrtAB in the plasma membrane. Do their amphiphilic properties allow these molecules to penetrate the cell wall, or is there a receptor protein as in most systems? It is evident that there is a lot yet to be learned before we clearly understand how M. tuberculosis acquires iron. Perhaps then we can envision ways to target this essential process.
Supplementary Material
Acknowledgments
This study was supported by NIH grants AI044856 (I.S.) and GM079185 (S.W.).
We thank Luc Gaucreau for providing pSR113.
Footnotes
Published ahead of print on 30 November 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Barton, G. J. 1993. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 6:37-40. [DOI] [PubMed] [Google Scholar]
- 2.Braibant, M., P. Gilot, and J. Content. 2000. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 24:449-467. [DOI] [PubMed] [Google Scholar]
- 3.Butterton, J. R., and S. B. Calderwood. 1994. Identification, cloning, and sequencing of a gene required for ferric vibriobactin utilization by Vibrio cholerae. J. Bacteriol. 176:5631-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dym, O., and D. Eisenberg. 2001. Sequence-structure analysis of FAD-containing proteins. Protein Sci. 10:1712-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farhana, A., S. Kumar, S. S. Rathore, P. C. Ghosh, N. Z. Ehtesham, A. K. Tyagi, and S. E. Hasnain. 2008. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS One 3:e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fetherson, J. D., V. J. Bertolino, and R. D. Perry. 1999. YbtP and YbtQ two ABC transporters required for iron uptake in Yersinia pestis. Mol. Microbiol. 32:289-299. [DOI] [PubMed] [Google Scholar]
- 7.Gobin, J., and M. Horwitz. 1996. Exochelins of Mycobacterium tuberculosis remove iron from human iron-binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J. Exp. Med. 183:1527-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gobin, J., C. H. Moore, J. R. J. Reeve, D. K. Wong, B. W. Gibson, and M. A. Horwitz. 1995. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. U. S. A. 92:5189-5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs, Jr., W. R., G. V. Kalpana, J. D. Cirillo, L. Pascopella, S. B. Snapper, R. A. Udani, et al. 1991. Genetic systems for mycobacteria. Methods Enzymol. 204:537-555. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli, U. K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 11.Lim, E. M., J. Rauzier, J. Timm, G. Torrea, A. Murray, B. Gicquel, and D. Portnoi. 1995. Identification of Mycobacterium tuberculosis DNA sequences encoding exported proteins by using phoA gene fusions. J. Bacteriol. 177:59-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manoil, C. 1991. Analysis of membrane protein topology using alkaline phosphatase and β-galactosidase gene fusions. Methods Cell Biol. 34:61-75. [DOI] [PubMed] [Google Scholar]
- 13.Miethke, M., and M. A. Marahiel. 2007. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71:413-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardee, A. P., J. F. Jacob, and J. Monod. 1959. The genetic control and cytoplasmic expression of “inductibility” in the synthesis of β-galactosidase and tryptophanase induction in Escherichia coli. J. Mol. Biol. 1:3331-3342. [Google Scholar]
- 15.Parish, T., E. Mahenthiralingam, P. Draper, E. O. Davis, and M. J. Colston. 1997. Regulation of the inducible acetamidase gene of Mycobacterium smegmatis. Microbiology 143(Pt. 7):2267-2276. [DOI] [PubMed] [Google Scholar]
- 16.Ratledge, C. 1999. Iron metabolism, p. 260-286. In C. Ratledge and J. Dale (ed.), Mycobacteria: molecular biology and virulence. Blackwell Science Publishers, New York, NY.
- 17.Rodriguez, G. M., R. A. Gardner, N. Kaur, and I. O. Phanstiel. 2008. Utilization of Fe+3-acinetoferrin analogues as an iron source by Mycobacterium tuberculosis. Biometals 21:93-103. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez, G. M., and I. Smith. 2006. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol. 188:424-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez, G. M., M. I. Voskuil, B. Gold, G. K. Schoolnik, and I. Smith. 2002. ideR, an essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70:3371-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook, J., E. K. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Schroder, I., E. Johnson, and S. de Vries. 2003. Microbial ferric iron reductases. FEMS Microbiol. Rev. 27:427-447. [DOI] [PubMed] [Google Scholar]
- 22.Servant, F., C. Bru, E. Courcelle, J. Gouzy, D. Peyruc, and D. Kahn. 2002. ProDom: automated clustering of homologous domains. Brief. Bioinform. 3:246-251. [DOI] [PubMed] [Google Scholar]
- 23.Snapper, S. B., R. E. Melton, S. Mustafa, T. Kieser, and J. W. R. Jacobs. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911-1919. [DOI] [PubMed] [Google Scholar]
- 24.Stover, C. K., V. F. de la Cruz, T. R. Fuerst, J. E. Burlein, L. A. Benson, L. T. Bennett, G. P. Bansal, J. F. Young, M. H. Lee, G. F. Hatfull, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456-460. [DOI] [PubMed] [Google Scholar]
- 25.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timm, J., E. M. Lim, and B. Giquel. 1994. Escherichia coli-mycobacterium shuttle vectors for operon and gene fusions to lacZ: the pJEM series. J. Bacteriol. 176:6749-6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tusnady, G. E., and I. Simon. 1998. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283:489-506. [DOI] [PubMed] [Google Scholar]
- 28.Wagner, M. A., P. Khanna, and M. S. Jorns. 1999. Structure of the flavocoenzyme of two homologous amine oxidases: monomeric sarcosine oxidase and N-methyltryptophan oxidase. Biochemistry 38:5588-5595. [DOI] [PubMed] [Google Scholar]
- 29.Wang, S., J. Engohang-Ndong, and I. Smith. 2007. Structure of the DNA-binding domain of the response regulator PhoP from Mycobacterium tuberculosis. Biochemistry 46:14751-14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinberg, E. D. 1984. Iron withholding: a defense against infection and neoplasia. Physiol. Rev. 64:65-102. [DOI] [PubMed] [Google Scholar]
- 31.Yano, T., L. S. Li, E. Weinstein, J. S. Teh, and H. Rubin. 2006. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J. Biol. Chem. 281:11456-11463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.