

Summary
Asymmetric localization of intracellular proteins and signals directs movement during axon guidance, endothelial cell invasion, and immune cell migration. In these processes, cell movement is guided by external chemical cues in a process known as chemotaxis. In particular, leukocyte migration in the innate immune system has been studied in the human neutrophil-like cell line (HL-60). Here, we describe the maintenance and transfection of HL-60 cells and explain how to analyze their behavior with two standard chemotactic assays. Finally, we demonstrate how to fix and stain the actin cytoskeleton of polarized cells for fluorescent microscopy imaging.
Keywords: Migration, Chemotaxis, Neutrophil, HL-60, Actin cytoskeleton, Amaxa transfection, Micropipette, EZ-TAXIS assay
1. Introduction
Directed cell migration toward chemical cues, or chemotaxis, is critical in eukaryotic cells for immune response, wound healing, axon guidance, and embryogenesis (1). An especially useful model for eukaryotic chemotaxis is the human neutrophil. Neutrophils seek infectious bacteria to engulf at wound sites as part of the innate immune system. They follow gradients of formylated peptides released by the bacteria (1). Yet, many open questions remain in neutrophil and eukaryotic cell migration. How do cells interpret shallow gradients or initially establish polarity? How is their cytoskeleton rearranged during a turn, and what limits this rearrangement to one part of the cell and not another? What signaling components and circuitry are required to accomplish these processes?
The human promyelocytic leukemia (HL-60) cell line was developed as a simple model system to study neutrophil cell migration without the need to derive cells from primary tissue (2). The immortal cell line can be sustained for extended periods of time in culture and may be frozen for longer-term storage. This is impossible with bone marrow or peripheral blood derived neutrophils. When needed, neutrophil-like cells can be derived from HL-60 cells through differentiation with DMSO (3). Differentiated HL-60 cells (dHL-60) can then be used in chemotaxis assays and to visualize the cytoskeleton of a polarized cell (4). Amaxa nucleofection may be used in dHL60 cells to knock genes down (5–8) or transfect cells with a fluorescent tag to analyze protein localization (our unpublished results). Cell behavior can be analyzed either in response to a point source of chemoattractant (9–11), or using the EZ-TAXIS system, which allows simultaneous measurement of directionality and speed for six different assay conditions (12, 13).
2. Materials
2.1. HL-60 Cell Culture and Differentiation
Culture medium. Roswell Park Memorial Institute (RPMI) 1640 plus l-glutamine and 25 mM HEPES (Fisher Scientific) supplemented with antibiotic/antimycotic (Invitrogen) and 15% heat-inactivated fetal bovine serum (FBS, Invitrogen); store at 4°C (see Note 1).
Dimethyl sulfoxide (DMSO), endotoxin, and hybridoma tested (Sigma).
2.2. Amaxa Nucleofection of HL-60 cells
Recovery medium. Monocyte medium (Amaxa) supplemented with 2 mM glutamine (Invitrogen) and 20% FBS.
100 μl of transfection solution containing supplement (Amaxa) mixed with 2 μg DNA per reaction (see Note 2).
2.3. Plating Cells for Microscopy
Fibronectin from bovine plasma (Sigma); store lyophilized protein at −20°C.
Ca2+/Mg2+free phosphate buffered saline (PBS). 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 (Invitrogen).
Gold Seal cover glass 20 × 40 mm No. 1.5 (Fisher Scientific).
Lab-Tek 8 well permanox chamber slide (Nunc).
Chemoattractant. 10 mM formylated Methione–Leucine–Phenylalanine (fMLP, Sigma) in DMSO; store at −20°C. Prepare 100 μM working stocks in RMPI and store at 4°C up to 1 month (see Note 3).
2.4. Micropipette Assay
Glass capillary with filament (World Precision Instruments) (see Note 4).
Alexa594 working stock. 10 mM Alexa Fluor 594 hydrazide (sodium salt, Invitrogen) in DMSO; store at 4°C and protect from light.
Chemoattractant solution. 200 nM fMLP, 10 μM Alexa594 in RPMI culture medium; protect from light.
2.5. EZ-TAXIScan Assay
RPMI culture medium.
Chemoattractant solution. 200 nM fMLP in RMPI culture medium.
2.6. Staining the Actin Cytoskeleton
Intracellular buffer (2×). 280 mM KCl, 2 mM MgCl2, 4 mM EGTA, 40 mM Hepes of pH 7.5, 0.4% low endotoxin albumin from human serum (Sigma) (see Note 5).
Fixation buffer (2×). 640 mM sucrose, 7.4% formaldehyde (Sigma) in 2× intracellular buffer; store at 4°C (see Note 6).
Stain buffer. 0.2% triton, 2 μl/ml rhodamine phalloidin (Invitrogen) in 1× intracellular buffer (see Note 7).
3. Methods
3.1. Maintenance of HL-60 Cell Culture Line
Unless imaging, all cell work is performed under a biological safety cabinet.
HL-60 cells are passaged when the cells reach a density between 1 and 2 million cells/ml in 25-cm2 cell culture flasks with 0.2-μm vent cap. Split cells to 0.15 million cells/ml in a total volume of 10 ml prewarmed culture medium. Cells will need to be passaged again after 2–3 days (Fig. 1). Maintain cells at 37°C and 5% CO2 in standard tissue culture incubator (see Note 8).
Differentiate cells in culture medium containing 1.3% DMSO. Because DMSO is more viscous and denser than culture medium, premix medium with DMSO before adding cells. Cells stop proliferating upon differentiation and typically achieve a density of 1–2 million cells/ml at 7 days postdifferentiation (Fig. 1). Cells are most active 5–6 days postdifferentiation, but can still respond even after 8 days (see Note 9).
To freeze cells, pellet cells by spinning at 100 × g for 10 min. Aspirate medium and resuspend in chilled culture medium plus 10% DMSO at 10 million cells/ml. Aliquot 1.8 ml each into cryovials, place in Nalgene cryofreezing container with isopropanol at −80°C for 2 days, and then transfer to liquid nitrogen storage (see Note 10).
Thaw cells by quickly warming a cryovial at 37°C just until last bit of ice has melted. Dilute thawed cells in 10 ml of prewarmed culture medium and spin at 100 × g for 10 min. Remove supernatant, resuspend pellet in 20 ml culture medium, and recover in a 75-cm2 culture flask.
Fig. 1.
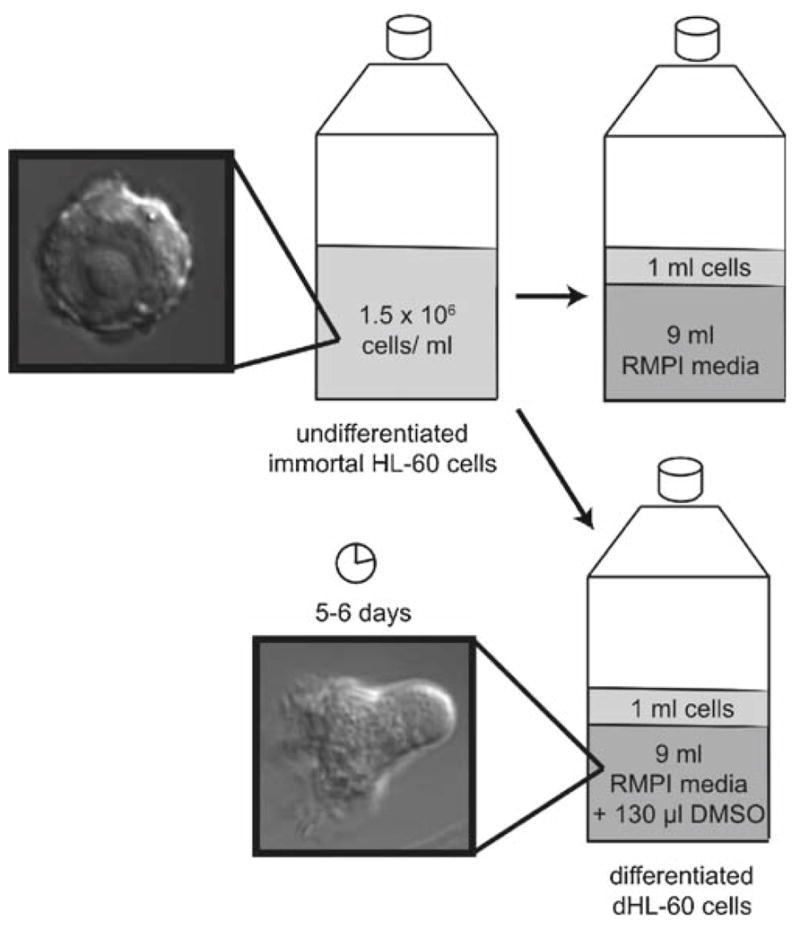
Passaging and differentiating HL-60 cells. When cells reach a density between 1 and 2 million cells/ml, split to 0.15 million cells/ml in a total volume of 10 ml prewarmed culture medium. Differentiate cells in culture medium plus 1.3% DMSO; cells take ~5 days to become migratory.
3.2. Transient Transfection of DNA into HL-60 Cells
Prepare ~2 ml of recovery medium per transfection in a 6-well plate and let equilibrate at 5% CO2 and 37°C for 15 min or more. Add 500 μl of equilibrated recovery medium to an Eppendorf tube per transfection (see Note 11).
Spin 5 million cells in a 10-ml Falcon tube at 100 × g for 10 min. Use a separate Falcon tube for each transfection.
After spin, remove all medium with aspirator and gently resuspend cells in 100 μl transfection solution with an L-1000 pipette (see Note 12).
As quickly as possible, transfer transfection solution to nucleofection cuvette. Electroporate in single-chamber nucleofector on program Y-001.
Immediately remove cuvette, use a plastic pipette to obtain 500 μl recovery medium from Eppendorf tube, flush chamber, and replace medium containing cells in Eppendorf tube.
Incubate for 30 min in Eppendorf tube at 37°C (see Note 13).
Transfer 500 μl recovery medium and cells with L-1000 pipette to 1.5 ml recovery medium in a 6-well plate. Expression in viable cells occurs in ~2 h with best behavior <8 h after transfection (Fig. 2).
Fig. 2.

Transient transfection of HL-60 cells with amaxa nucleofection. Spin ~5 million cells at 100 × g. Aspirate supernatant and resuspend pellet in 100 μl transfection solution per reaction and nucleofect with amaxa program “Y-001.” Flush with prewarmed recovery medium and incubate in an Eppendorf tube for 30 min. Transfer to a 6-well dish with 1.5 ml of recovery medium; expression occurs after 2 h. Shown is an example of HL-60 cells 5 h after transfection with GFP visualized with DIC and fluorescence microscopy.
3.3. Preparing for live Cell Microscopy
Dissolve 1 ml of sterile water in 1 mg fibronectin and let sit at room temperature for 1 h. Avoid shaking or physically mixing because this may denature the fibronectin.
Add 4 ml of Ca2+/Mg2+ free PBS to fibronectin solution to obtain 200 μg/ml solution (Fig. 3a): store fibronectin solution at 4°C for up to 2 weeks.
Remove exterior gaskets from permanox chamber slide and the interior gasket with a razor blade. Discard 8-well plastic walls to expose underlying epoxy mold. With razor, cut single-well epoxy chambers and adhere to glass coverslips (see Note 14).
Add 125 μl fibronectin solution to epoxy chamber on coverslip. Incubate at room temperature for 1 h.
Aspirate fibronectin solution and rinse once in RPMI culture medium. Add 250 μl RPMI culture medium until ready to plate cells. Fibronectin-coated slides may be stored up to 2 days at 4°C (Fig. 3b).
For cell plating, remove culture medium on slide and replace with 250 μl differentiated cells (in RPMI culture medium) or transfected cells (in monocyte medium). Incubate at 37°C for 5–15 min (see Note 15).
Aspirate cells at the corner of the epoxy chamber (avoid touching the coverslip), rinse in RPMI, and replace with 125 μl RPMI. Cells are most migratory at 37°C, but also function at room temperature.
During image acquisition, 125 μl of 200 nM fMLP may be added to coverslip (final concentration is 100 nM) to show random migration in response to uniform chemoattractant.
Fig. 3.

Preparing a coverslip for live cell microscopy. (a) Dissolve 1 mg of bovine fibronectin in sterile water. After 1 h, add 4 ml of PBS and store 200 μg/ml fibronectin solution at 4°C. (b) Remove gaskets from plastic permanox 8-well chamber. Cut epoxy mold squares and stick to No. 1.5 gold seal cover glass. Add 125 μl of fibronectin, let sit for 1 h, rinse once with RPMI culture medium, and store in RPMI medium until ready to image.
3.4. Micropipette-Stimulated Cell Migration
Pull glass filaments on Sutter Model P-87 (Program: heat = 750, pull = 0, velocity = 20, time = 250, pressure = 100, loops 2 or 3 times) to achieve ~2–3 μm needle diameter.
Backfill needles with chemoattractant solution and connect to a needle holder. Flick to remove air bubbles at the tip. The needle holder is held by a micromanipulator and agonist flow controlled by adjusting balance pressure between 0 and 3 psi on a Narishige IM-300 injection system (see Note 16).
Place cells seeded on a coverslip on microscope and find focus of cells in brightfield.
Orient needle in light path and switch to the back focal plane of the objective (usually labeled “B” for Bertrand lens). Find the needle and lower it until just above the cells; then switch back to the normal focal plane for viewing.
Image dye in fluorescence or Total Internal Reflection Fluorescence microscopy using appropriate filter sets with ~20 ms exposures and near-maximal multiplication on an EM-CCD camera (see Note 17). An example of a cell migrating toward a micropipette filled with chemoattractant is shown in Fig. 4.
Fig. 4.
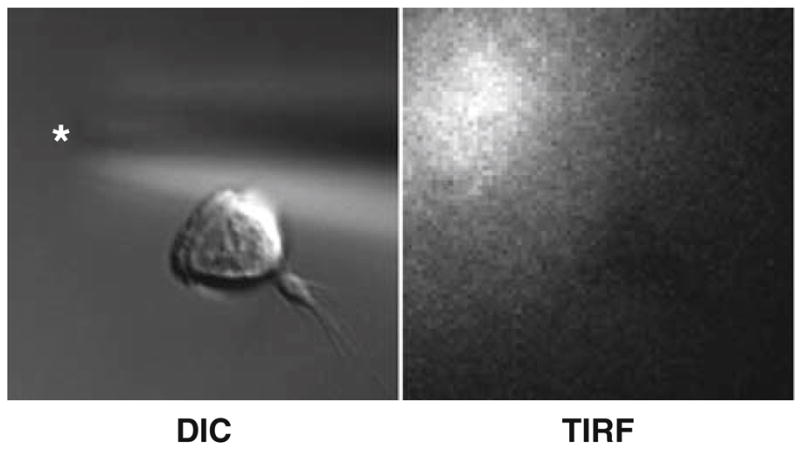
An example of an HL-60 cell crawling toward a micropipette visualized with DIC and TIRF microscopy. Asterisk indicates micropipette tip.
3.5. Ascertaining Directionality and Velocity of Migrating Cells with the “EZ-TAXIS” Assay System
Place “40 Glass” (41 Glass is used in conjunction with a #1.5 coverslip) in holder base. Fill with 3 ml of culture medium, place small “o” ring beneath wafer housing, and add wafer housing into holder base. Place large “o” ring on top of wafer housing. Gently close the inner lever.
Place EZ-TAXIScan chip gently into wafer housing with forceps. Chip should fit snuggly on top of glass and between wafer holder (see Note 18). Place rubber gasket (to protect chip) beneath the wafer clamp and place wafer clamp on wafer housing. Gently close outer lever.
Place assembled device on top of preheated EZ-TAXIS microscope (Fig. 5a,b).
Using syringe guide, add 1 μl of cells to lower chamber using microsyringe. Use a plastic pipette to draw cells into imaging surface.
Add 1 μl of 100 nM–1 μM chemoattractant to upper chamber. Begin image acquisition immediately (Fig. 5c).
Fig. 5.

The components of the EZ-TAXIS system are shown in (a) with the individual components in their order of assembly from top to bottom shown in (b). (c) An example of HL-60 cells migrating toward chemoattractant in the EZ-TAXIS assay visualized with brightfield microscopy.
3.6. Staining the actin Cytoskeletons
Stimulate adherent cells with micropipette or uniform chemoattractant. Alternatively, you can stimulate cells in suspension.
Add one volume of 2× fixation buffer to cells to give a final concentration of 320 mM sucrose and 3.7% formaldehyde. Fix for 20 min at 4°C (see Note 19).
Aspirate supernatant (adherent cells) or spin down cells at 400 × g for 1 min and then aspirate supernatant (suspended cells).
Permeabilize cells with 125 μl stain buffer and protect from light for 20 min (Fig. 6).
Remove supernatant, replace with intracellular buffer, and image (see Note 20).
Fig. 6.
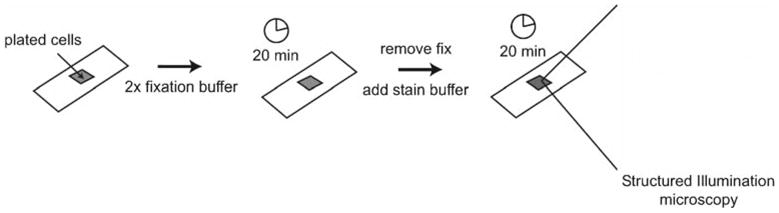
Staining the actin cytoskeleton. Add 2× fixation buffer to plated cells and fix for 20 min at 4°C. Remove fixation buffer and replace with stain buffer for 20 min; protect from light. Shown is an example of an HL-60 cell stained with rhodamine phalloidin visualized with structured illumination microscopy.
Acknowledgments
We would like to thank the NIH R01GM084040 and a National Defense Science and Engineering Graduate Fellowship to A. Millius.
Footnotes
HL-60 cells acidify their medium quite rapidly, so HEPES is essential to counterbalance the pH.
Poor expression occurs in retroviral LTR-containing vectors.
If an exact concentration of fMLP is required, make stocks fresh weekly because formyl peptide will slowly oxide in aqueous solutions over time.
Capillaries with filaments are easier to load than capillaries without filaments.
HL-60 cells may be spuriously stimulated with albumin containing endotoxins. It is important to add the albumin fresh to the intracellular buffer and let sit at room temperature for 30 min before mixing because the albumin will oxidize in aqueous solution to products that may also activate HL-60 cells.
Sucrose is important to maintain osmolarity.
Unlike Alexa Fluor 594, rhodamine phalloidin increases its fluorescence upon binding to actin filaments. This makes it preferable to Alexa Fluor 594. However, for imaging in the green channel, we would use Alexa Fluor 488 or similar fluorophore over the rapidly bleaching FITC.
Keep a basin of water on the bottom shelf of the incubator to maintain humidity.
Cells may also be differentiated with retinoic acid (14).
As an alternative to nalgene freezing chambers, two styrofoam 15-ml Falcon tube holders may be sandwiched around tubes to provide insulation during slow freezing process.
Our protocol differs significantly from the standard Kit V protocol from amaxa, because we have optimized for cells to retain chemotactic function (not necessarily highest transfection efficiency).
It is important to remove every drop of medium from the Falcon tube because FBS will interfere with transfection. Large-tip pipettes minimize shearing of cells and increases the number of healthy cells post-transfection.
Cells will adhere to each other and form a thin, white film during the 30-min incubation in Eppendorf tube. This step increases recovery efficiency. Additionally, cells are fragile after transfection and they behavior poorly if centrifuged.
An 8-well chamber will yield four molds. Only bottom surface (side touching permanox chamber slide) of epoxy mold is sticky. Additionally, Lab-Tek 8 well cover glass #1.5 chamber slide (Nunc) may be used for epifluorescence and brightfield microscopy. However, the epoxy that hold the chamber walls to the coverslip is autofluorescent and thus less suitable for TIRF.
Cells may be plated in culture medium containing chemoattractant for a higher proportion of adherent cells.
We use a universal needle holder (MINJ4, Tritech) connected to a micromanipulator (MM-89, Narishige). Additionally, it is difficult to handle capillaries with gloves on. Once the solution is loaded into the needle, flick the needle vigorously to remove microscopic bubbles. Do not try to force air bubbles through the tip by increasing air pressure. Invariably, the bubbles will become stuck and the needle rendered useless.
For quantification of gradient, TIRF imaging is superior to epifluorescence microscopy because TIRF reveals the agonist concentrations in the same plane as the cell. The signal obtained from epifluorescence is a combination of the agonist the cell experiences plus out of focus blur from fluorescent dye above the cell. Confocal imaging can also be used to quantitate the gradient.
Chips come in four different sizes (4 μm, 5 μm, 6 μm, and 8 μm) depending on the gap space between the chip and glass. We have used the 4–6 μm chips with success.
Formaldehyde can partially permeabilize cells, allowing water influx due to high internal protein concentration.
Can repeat wash if background too high. If pressed for time, can immediately image after adding stain buffer. However, this will give high background staining. You can also mount cells in Vectashield (Vector Laboratories) for decreased spherical aberrations and photobleaching.
Contributor Information
Arthur Millius, Cardiovascular Research Institute and Department of Biochemistry, University of California, San Francisco, CA, USA.
Orion D. Weiner, Cardiovascular Research Institute and Department of Biochemistry, University of California, San Francisco, CA, USA
References
- 1.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 2.Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 1977;270:347–349. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- 3.Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci USA. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao KM, Currie MS, Ruff JC, Cohen HJ. Lack of correlation between induction of chemotactic peptide receptors and stimulus-induced actin polymerization in HL-60 cells treated with dibutyryl cyclic adenosine monophosphate or retinoic acid. Cancer Res. 1988;48:6721–6726. [PubMed] [Google Scholar]
- 5.Carter BZ, Milella M, Tsao T, McQueen T, Schober WD, Hu W, et al. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia. 2003;17:2081–2089. doi: 10.1038/sj.leu.2403113. [DOI] [PubMed] [Google Scholar]
- 6.Nakata Y, Tomkowicz B, Gewirtz AM, Ptasznik A. Integrin inhibition through Lyn-dependent cross talk from CXCR4 chemokine receptors in normal human CD34+ marrow cells. Blood. 2006;107:4234–4239. doi: 10.1182/blood-2005-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Fuente J, Manzano-Roman R, Blouin EF, Naranjo V, Kocan KM. Sp110 transcription is induced and required by Anaplasma phagocytophilum for infection of human promyelocytic cells. BMC Infect Dis. 2007;7:110. doi: 10.1186/1471-2334-7-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gattenloehner S, Chuvpilo S, Langebrake C, Reinhardt D, Muller-Hermelink HK, Serfling E, et al. Novel RUNX1 isoforms determine the fate of acute myeloid leukemia cells by controlling CD56 expression. Blood. 2007;110:2027–2033. doi: 10.1182/blood-2007-02-074203. [DOI] [PubMed] [Google Scholar]
- 9.Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, Bourne HR. Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol. 1999;1:75–81. doi: 10.1038/10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, et al. PI(3)Kγ has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9:86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- 13.Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- 14.Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]