

Abstract
Intracellular α-synuclein (α-syn) aggregates are the pathological hallmark in several neurodegenerative diseases including Parkinson's disease, dementia with Lewy bodies and multiple system atrophy. Recent evidence suggests that small oligomeric aggregates rather than large amyloid fibrils represent the main toxic particle species in these diseases. We recently characterized iron-dependent toxic α-syn oligomer species by confocal single molecule fluorescence techniques and used this aggregation model to identify several N′-benzylidene-benzohydrazide (NBB) derivatives inhibiting oligomer formation in vitro. In our current work, we used the bioluminescent protein-fragment complementation assay (BPCA) to directly analyze the formation of toxic α-syn oligomers in cell culture and to investigate the effect of iron and potential drug-like compounds in living cells. Similar to our previous findings in vitro, we found a converse modulation of toxic α-syn oligomers by NBB derivates and ferric iron, which was characterized by an increase in aggregate formation by iron and an inhibitory effect of certain NBB compounds. Inhibition of α-syn oligomer formation by the NBB compound 293G02 was paralleled by a reduction in cytotoxicity indicating that toxic α-syn oligomers are present in the BPCA cell culture model and that pharmacological inhibition of oligomer formation can reduce toxicity. Thus, this approach provides a suitable model system for the development of new disease-modifying drugs targeting toxic oligomer species. Moreover, NBB compounds such as 293G02 may provide useful tool compounds to dissect the functional role of toxic oligomer species in cell culture models and in vivo.
Keywords: Parkinson's disease, Alpha-synuclein, Drug development, Oligomer, Amyloid, Protein-fragment complementation assay
Introduction
The hallmark feature of all common neurodegenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD) is the ability of particular proteins to fold into stable alternative conformations and to form intra- and/or extracellular protein aggregates that accumulate in the brain [11]. PD is characterized neuropathologically by degeneration of dopaminergic neurons in the substantia nigra, which leads to disruption of motor functions. Initial evidence for a central role of α-synuclein (α-syn) in the pathogenesis of PD came from the discovery of point mutations in the α-syn gene in families with familial PD [6]. Subsequently, α-syn has been identified as the major component of Lewy bodies and in Lewy neurites, which are characteristic deposits of aggregated protein in PD, dementia with Lewy bodies, and Lewy body variant of Alzheimer's disease, and the major component of the glial cytoplasmic inclusions that characterize multiple system atrophy [22]. Additional evidence for a fundamental role of α-syn in the pathogenesis of PD came from the observation that an increased gene dose in PD patients, caused by a duplication or triplication of the α-syn gene, is sufficient to trigger disease [21]. α-syn is a presynaptic, natively unfolded cytoplasmic 14 kDa protein that is abundant in brain tissue [24]. Its physiological function has not been understood completely. However, it has been suggested that α-syn plays a role in synaptic transmission, in the stabilization of lipid membranes, and in the interaction with a variety of protein complexes [23].
The transformation of amyloidogenic proteins from the monomeric state into fibrillar aggregates seems to progress via intermediates that have been termed protofibrils, protofilaments or oligomers [5, 9]. Although the cause of neurodegeneration in PD is not fully understood, recent findings suggests that oligomers rather than the fibrillar amyloid deposits of α-syn represent the principal toxic species [7].
Using confocal single particle fluorescence we recently developed a robust in vitro multistep aggregation model in which we characterized two different oligomeric species. Organic solvents (such as dimethyl sulfoxide (DMSO)) [10, 17], were used to trigger aggregation, which resulted in small oligomers (intermediate I). Addition of ferric iron at low micromolar concentrations increased aggregation of α-syn and induced formation of a specific toxic oligomer species (intermediate II) that was relatively SDS-resistant and could form ion-permeable pores in lipid bilayers. We used the aggregation model to identify several N′-benzylidene-benzohydrazide derivatives inhibiting oligomer formation in vitro [12].
In our current work, we used a bioluminescent protein-fragment complementation assay (BPCA) to directly analyze the formation of α-syn oligomers in cell culture and to investigate the effect of potential drug-like compounds affecting oligomer formation in living cells. This approach is based on the co-transfection and co-expression of signal-protein fragments. These fragments are cloned linked to the protein of interest (e.g. α-syn). If oligomers of the protein of interest are formed, the fragments of the signal-proteins are reconstituted and a fluorescent or bioluminescent signal can be detected [15]. Successful application of this approach for the detection of α-syn oligomers has recently been described [18].
Using this approach, we compared the effect of ferric iron and NBB compounds on α-syn aggregation in vitro and in living cells. In both model systems, we found a converse modulation of toxic α-synuclein oligomers by NBB derivates and ferric iron, which was characterized by an increase in aggregate formation by iron and an inhibitory effect of certain NBB compounds.
Material and methods
Confocal Single Particle Analysis
Fluorescence correlation spectroscopy (FCS), fluorescence intensity distribution assay (FIDA), and scanning for intensely fluorescent targets (SIFT) measurements were carried out on an Insight Reader (Evotec-Technologies, Hamburg, Germany) with dual color excitation at 488 and 633 nm, using a 40 × 1.2 numerical aperture microscope objective (Olympus, Japan) and a pinhole diameter of 70 μm at FIDA setting. Excitation power was 200 μW at 488 nm and 300 μW at 633 nm. Measurement time was 10 s. Scanning parameters were set to 100 μm scan path length, 50 Hz beam scanner frequency, and 2000 μm positioning table movement. This is equivalent to ∼10 mm/s scanning speed. All measurements were performed at room temperature.
The fluorescence data were analyzed by autocorrelation analysis using the FCSPP evaluation software version 2.0 (Evotec-Technologies). Two-color cross-correlation amplitudes G(0) and FIDA data were evaluated using the same software. For FIDA [8] and SIFT [2] analysis, fluorescence from the two different fluorophores was recorded simultaneously with two single-photon detectors. Photons were summed over time intervals of constant length (bins) using a bin length of 40 μs. The frequency of specific combinations of “green” and “red” photon counts was recorded in a two-dimensional intensity distribution histogram.
Aggregation Assay
A 5-fold stock solution of fluorescently labeled α-syn was prepared by mixing α-syn labeled with Alexa-488 and α-syn labeled with Alexa-647. The concentrations of α-syn-Alexa-488 and α-syn-Alexa-647 were adjusted to ∼10 molecules/focal volume and 15 molecules/focal volume, respectively. Quality control SIFT measurements were used to confirm that the stock solution was free of α-syn aggregates. Aggregation was induced by the addition of 1 % DMSO [17] to a mixture of α-syn monomers labeled with Alexa-488 or Alexa-647 at a final protein concentration of approximately 10 nM in 50 mM Tris-HCl buffer at pH 7.0 in a total assay volume of 20 μL Compounds were added together with DMSO. N′-benzylidene-benzohydrazide (NBB) derivates (ChemBridge Corp., San Diego, CA, USA) were prepared as stock solutions in DMSO and used at a final concentration of 10 μM. Values for cLogP and topological polar surface area (TPSA) were calculated using web-based software (http://www.molinspiration.com/services). All experiments were performed in 96-well plates with a cover slide bottom (Evotec-Technologies). To reduce evaporation, plates were sealed with adhesive film. Typically, aggregation was monitored for at least five hours in two to four independent samples for each experimental group. In some experiments, FeCl3 (Merck, Germany) was added to a final concentration of 10 μM.
Cell culture and luciferase assay
Human H4-neuroglioma cells (HTB-148 - ATCC, Manassas, USA) were maintained in OPTI-MEM medium plus 10 % fetal bovine serum (FBS, both Invitrogen, Carlsbad, CA, USA)on 96 well plates (toxicity assay), 24 well plates (luciferase assay) and 60 mm dishes (western blot) respectively. Prior to transfection, cells were incubated at 37 °C, 5 % CO2 for 24 h, growing to a confluence of 80-90 %. α-Syn was subcloned into the NotI/ClaI sites of constructs containing optimized fragments of hGLuc (1-93; 94-185). Cells were co-transfected with the gaussia luciferase constructs Syn-hGLuc(1) and Syn-hGLuc(2). In some experiments, control cells were transfected with full length luciferase. Transfection was performed using Superfect (Qiagen, Chatsworth, CA, USA). NBB compounds dissolved in DMSO were added two hours after transfection in a final concentration of 10 μM resulting in a concentration of 0.1 % DMSO in the cell culture media. FeCl3 was added 24 h after transfection at a concentration of 1 mM and 100 μM, respectively. After 24 h of incubation in the presence of compound/ferric iron, cells were washed with phosphate buffered saline (PBS) at room temperature and covered with phenol red- and FBS-free OPTI-MEM. Coelenterazine (20 μM, Prolume Ltd.) was used as substrate for gaussia luciferase. Luminescene was measured in a Wallac Victor2 1420 Multilabel Counter (PerkinElmer, USA) at 480 nm and a signal integration time of two seconds.
Toxicity assay
For toxicity assay, the release of adenylate kinase (AK) was measured by ToxyLight BioAssay Kit (Lonza, Switzerland). For a late treatment paradigm, cells were split into 96 well plates 24 hours prior to transfection. Transfections were done according to Superfect product instructions. Two hours after transfection, the media (OPTI-MEM medium plus 10 % fetal bovine serum) was replaced with media containing 10 μM of compound (diluted 1:1000 from 10 mM stock solution in DMSO), which results in the presence of 0.1 % DMSO in the culture media. Toxicity assays were performed 24 hours after transfection.
For an early treatment paradigm, cells were split into 96 well plates and treated with 10 μM of compound, five to eight hours later. Cells were transfected 24 hours later as described above. No compound or vehicle was present during the two hours of transfection. Post transfection, the compounds/vehicle were replaced in the media. Toxicity assays were performed 24 h after transfection.
Western Blot
H4 cells were washed with room temperature PBS and harvested 24 hours after transfection and treatment similar to the luciferase assay paradigm. For denaturing conditions, samples were lysed using lysis buffer containing Triton X-100 (0.1 % Triton X-100, 0.15 M NaCl, 50 mM Tris pH 7.5, protease inhibitor cocktail one tablet/10 mL (Roche Diagnostics)). By passing through a 1 mL 27-gauge syringe four to six times, the sample were sheared and afterwards centrifuged for one minute at 13,000× g. Samples for native gels were lysed with detergent-free lysis buffer (50 mM Tris/HCl pH 7.4, 175 mM NaCl, 5 mM EDTA pH 8.0, protease inhibitor cocktail one tablet/10 mL. For the native gels, NativeMark and for SDS-PAGE gels SeeBlue Plus Two markers (both Invitrogen) were used.
Using BCA protein assay for determining protein concentrations, 20 μg of each sample was loaded on the gel (Tris-glycine gels, Invitrogen). SDS-PAGE was performed using Tris-Glycine SDS running buffer and SDS-sample buffer (2×, mixed with beta-mercaptoethanol at 1:50). Detergent free Tris-Glycin running buffer and 2× native sample buffer (Invitrogen) were used for Native-PAGE. For immunoblotting, proteins were transferred to PVDF membrane (PerkinElmer, USA). Membranes were blocked in either 5 % milk in Tris-Buffered Saline Tween (TBS-T) or Li-Cor blocking buffer (LI-COR) for one hour at room temperature before being incubated with primary antibodies (mouse anti-alpha-synuclein, 1:1000, BD Transduction; Rabbit anti-gapdh 1:2000; Rabbit anti-luciferase 1:1000) for one to three hours at room temperature or overnight at 4 °C. After three 5 to 10 min washes using TBS-T, membranes were incubated at room temperature for one hour with either HRP-conjugated anti-mouse or anti-rabbit secondary antibodies (1:2000). After three 5 to 10 min TBS-T washes, immunoblots were analyzed using the ECL chemoluminescent detection system (Amersham/GE Healthcare, USA).
Results
Confocal single particle fluorescence analysis of α-synuclein aggregation in the presence of NBB compounds and ferric iron
A panel of three NBB compounds was selected for cell culture studies based on their inhibitory effects on aggregation of prion protein [1], Poly-Q proteins [20] and α-syn [12]. Compounds 293G02 and 301C09 were chosen because they were highly active in our single particle fluorescence aggregation assay (Fig. 1). 293G02 showed a reduction of DMSO induced intermediate I oligomers by 93.3 % (p<0.05) while 301C09 reduced the oligomer fraction by 97.9 % (p<0.05, both n=4). Moreover, 293G02 has turned out to be also active in inhibition of aggregation in cell culture models of prion disease and in a Zebrafish in vivo model of Poly-Q disease, which indicates not only a possible common structure of soluble amyloid oligomers implying a common mechanism of pathogenesis but also that this compound is active also in vivo. Compound 306H03 was chosen because it was also active in the Zebrafish in vivo model, although it had only a weak activity in the in vitro aggregation assay, which may potentially be due to its more lipophilic properties.
Fig 1.

A) SIFT two-dimensional intensity distribution histogram showing the induction of small intermediate I oligomers (1 % DMSO) and larger intermediate II oligomers (1 % DMSO and 10 μM Fe3+) from α-syn as described in Kostka et al. (2008). Treatment with 10 μM 293G02 inhibits the formation of both oligomer species (lower row). B) Inhibition of DMSO-induced oligomer formation by different N′-benzylidene-benzohydrazide (NBB) compounds was quantified by cross-correlation analysis. While 306H03 only showed a minor effect, 301C09 reduced aggregation by 97.9 % and 293G02 by 93.3 % compared to DMSO-treated control samples, respectively. Shown is the mean and standard error (n=4, * p<0.05/student t-test). C) Chemical structures of the three different NBB compounds used in this study.
Analysis of α-synuclein aggregation cell culture
In order to analyze α-synuclein aggregation in living cells, we used the bioluminescent protein-fragment complementation assay (BPCA) as described previously. In order to validate expression of our α-syn-luciferase constructs, we used denatured SDS-PAGE gels as well as native gels. Using SDS-PAGE, we could detect expression of both constructs that were co-transfected as bands of 21 kDa (Syn-hGLuc(1)) and 24 kDa (Syn-hGLuc(2)) (Fig. 2B). In the native gels, oligomerized α-syn could be detected as high-molecular weight (HMW) species on the range of approximately 500 to 700 kDa, indicating formation of higher-order oligomers (Fig. 2A). Interestingly, treatment with 306H03 led to a significant decrease of these HMW-bands (48 % of control cells, normalized for GAPDH expression (Fig. 2C), p<0.005, n=3). A corresponding effect was also observed for the total amount of Syn-hGLuc proteins in denaturing gels indicating that aggregation results in accumulation of these proteins.
Fig 2.

Biochemical analysis of α-syn oligomer formation in compound-treated H4 neuroglioma cells transfected with Syn-hGLuc(1) and Syn-hGLuc(2) constructs by Western blot. A) Formation of higher-order α-syn oligomers can be seen in high molecular weight (HMW) species on the range of 500 – 700 kDa in a native gel. B) Using SDS-PAGE, accumulation of the two α-syn-luciferase fusion proteins can be seen. Notably, accumulation corresponds to the amount of oligomer formation. C) As reference for quantitative analysis, GAPDH expression was analysed using the same lysates.
Luciferase assay
For detailed quantitative analysis of the effect of NBB compounds on aggregate formation we measured luciferase activity in cells co-expressing Syn-hGLuc(1) and Syn-hGLuc(2) (Fig. 3). Compounds 306H03 and 293G02 caused a significant decrease of luciferase activity (51.3 % of control in case of 306H03 treated cells, p<0.005, 62.9 % of control in case of 293G02 treated cells, p<0.01, both n=5) compared to controls treated with DMSO only, which indicates a strong inhibition of oligomer formation. In contrast, compound 301C09 had only a minor effect in this cell culture assay (Fig 3A). To control for potential unspecific effects of NBB compounds on cell viability and/or luciferase activity, we also used cells transfected with full length luciferase. In these experiments, no reduction of luciferase activity was found, which corroborates that the reduced luciferase activity observed in the bioluminescent protein-fragment complementation assay is due to a reduction in the amount of α-syn aggregates (Fig. 3B).
Fig 3.
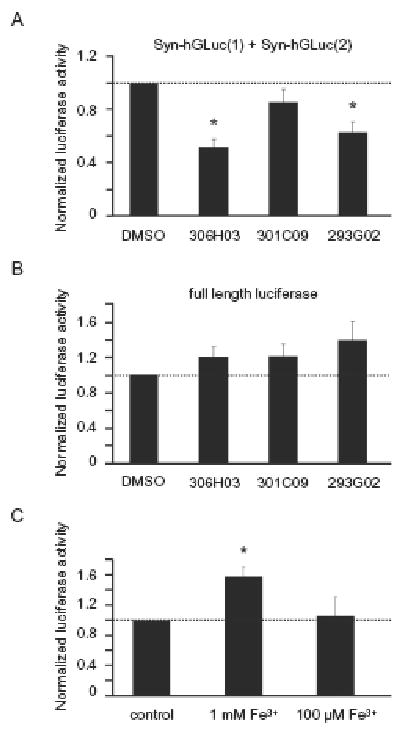
A) Quantitative analysis of the luciferase activity of cells co-transfected Syn-hGLuc(1) and Syn-hGLuc(2) constructs showed a 48.7 % decrease of oligomers in case of 306H03 treated cells (p<0.005) and 37.1 % decrease in case of 293G02 treated cells (p<0.01, both n=5), normalized to control cells. B) In contrast, no reduction of luciferase activity was detected in cells transfected with full length luciferase. C) Cells co-transfected with the Syn-hGLuc proteins and treated with 1 mM FeCl3 showed a 58.6 % (p<0.05) increase of luciferase activity. In all graphs, the mean and standard error is shown.
We furthermore analyzed the effect of iron on the formation of α-syn oligomers using the same approach. A 58.6 % (p<0.05) increase of luciferase activity in the bioluminescent protein-fragment complementation assay was detected at 1 mM FeCl3, indicating an increased formation of α-syn oligomers. At lower concentrations of ferric iron, luciferase activity was comparable to untreated cells (Fig. 3C).
Toxicity assay
As both compound 306H03 and compound 293G02 reduced α-syn aggregation in our cell culture model, we also investigated, whether inhibition of oligomer formation was correlated with an effect of cell viability. For cytotoxicity assay, we measured the release of adenylate kinase (AK) from cells into the media. Two different treatment paradigms were used. In the early treatment paradigm, cells were already treated with the compounds prior to transfection, whereas in the late treatment paradigm, treatment was started two hours after transfection.
Whereas in the late treatment paradigm neither 293G02 nor 306H03 showed an effect on AK release, in the early treatment paradigm a significant reduction of AK-release by 38.1 % (p<0.05, n=4) in cells treated with 293G02 was observed. In this assay, 306H03 showed only a minor effect (Fig. 4).
Fig 4.

Effect of NBB compounds on cytotoxicity was quantified by measuring the release of adenylate kinase. A) A reduction of AK release by 38.1 % (p<0.05, n=4) compared to control cells could be achieved by 293G02 when compounds were added prior to transfection (“early treatment paradigm”, see materials and methods). B) In contrast, when compounds were added 2 h post transfection (“late treatment paradigm”) no effect was observed. In all graphs, the mean and standard error is shown.
Discussion
Protein aggregation and amyloid formation are the key molecular events in a number of human diseases such as Alzheimer's disease, Poly-Q disease, prion disease, and Parkinson's disease (PD) [11]. For PD, a crucial role of α-syn aggregates in disease pathogenesis is suggested by a wealth of evidence including i) the consistent detection of deposits in affected brain areas [22] that also correlates with the stage of the disease [3], ii) mutations in the α-syn gene in certain patients suffering from familial PD [19] and iii) experimental evidence from studies in vitro [10], in cell culture [18] as well as in animal models including drosophila- [4] and mouse- models [14]. Recent research suggests that oligomeric aggregation intermediates rather than mature amyloid fibrils represent the principal toxic aggregate species, which is essential in the pathogenesis of neurodegeneration and a potential therapeutic target in these diseases [10, 12]. Recently, we reported the effect of NBB compounds as aggregation inhibitors in several in vitro [1] and in vivo models [20] for prion disease and Poly-Q disease as well as for an in vitro model of α-syn aggregation [12]. New chemical compounds that target the formation of pathological protein aggregates may provide a useful experimental tool to investigate the functional role of these aggregates in vivo and can represent a starting point for the development of new disease-modifying drugs. Therefore, we wanted to study the effect of NBB compounds on α-syn aggregation and aggregate toxicity in cell culture. As a suitable cell culture assay, we chose the bioluminescent protein-fragment complementation assay (BPCA) due to its ability to directly analyze oligomer formation in cells and to quantify the effect of potential inhibitory compounds [16, 18].
While some of the NBB compounds showed an inhibitory effect on the formation of oligomers, a converse effect was seen in the presence of 1 mM ferric iron. The fact that ferric iron increases α-syn aggregation in our cell culture model (Fig. 3C) is consistent with our previous single molecule results obtained in vitro [12]. This indicates that similar influencing factors can be observed in both models, which further validates the BPCA approach used by us.
The NBBs used in this study satisfy Lipinski's “rule of five” for drug-likeness [13] by having fewer than five H-bond donors, 10 H-bond acceptors, a molecular weight smaller than 500, and a calculated Log P smaller than 5, which in principle predicts good absorption or permeation in vivo.
Interestingly, for the three closely related NBB compounds used, differences in activity were found that indicate that inhibitory activity of these compounds in cell culture depends both on direct molecular potency and on bioavailability. It is well known that permeation through the cell membrane and metabolization by the cells are important factors in living cells that may interfere with the activity of compounds that are active inhibitors of protein aggregation in vitro. This can be observed in the case of 301C09, which is highly efficient in the single particle model, but not in BPCA. On the other hand, more lipophilic compounds that may have comparatively low solubility and activity in aqueous buffers may turn out to have relatively high activity in cell culture. Most likely, the presence of a chloride group in compound 306H03, rather than hydroxyl groups, enhances cell permeability by decreasing the polar surface area (TPSA) from 81.92 Å2 (293G02) to 41.46 Å2 (306H03) and increasing the hydrophobicity (cLogP) from 3.31 (293G02) to 4.94 (306H03). Similar to our previous findings in regard to Poly-Q aggregation [20], also in regard to aggregation of α-syn, 306H03 was significantly less efficient than 293G02 in the single particle in vitro assay, but showed strong activity in living cells.
The most consistent inhibitory effect both in vitro and in cell culture was found for 293G02 which was also the most active compound in vitro and in vivo in regard to prion inhibition [1]. 293G02 significantly and consistently inhibited the formation of oligomers in the single particle SIFT assay and in the bioluminescent protein-fragment complementation assay in cell culture. Moreover, inhibition of α-syn oligomer formation was paralleled by a reduction in cytotoxicity in an early treatment paradigm (Fig. 4A). This suggests that toxic α-syn oligomers are present in our cell culture model and that pharmacological inhibition of oligomer formation can reduce toxicity.
Thus, this approach provides a suitable model system for the development of new drug candidates and allows to characterize compounds in regard to inhibition of toxic oligomer species in the cell. Accordingly, the combination of high-throughput SIFT screening [12] and hit validation by BPCA provides a valid approach to the development of new drug candidates. Moreover, NBB compounds such as 293G02 may provide useful tool compounds to dissect the role of toxic oligomer species in cell culture models and in vivo. In this context it is important to note that recent studies using monoclonal antibodies indicated the presence of similar oligomer-specific structural epitopes in oligomers derived from different proteins [9]. This is in line with our finding that NBB compounds are efficient inhibitors of the formation of pathological aggregates in different neurodegenerative diseases.
Acknowledgments
This study was supported by the Lüneburg Foundation for Parkinson research and NIH NS038372 to BTH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bertsch U, Winklhofer KF, Hirschberger T, Bieschke J, Weber P, Hartl FU, Tavan P, Tatzelt J, Kretzschmar HA, Giese A. Systematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets. J Virol. 2005;79:7785–7791. doi: 10.1128/JVI.79.12.7785-7791.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bieschke J, Giese A, Schulz-Schaeffer W, Zerr I, Poser S, Eigen M, Kretzschmar H. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc Natl Acad Sci USA. 2000;97:5468–5473. doi: 10.1073/pnas.97.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 4.Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 5.Fink AL. The aggregation and fibrillation of alpha-synuclein. Acc Chem Res. 2006;39:628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 6.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 7.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kask P, Palo K, Ullmann D, Gall K. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc Natl Acad Sci USA. 1999;96:13756–13761. doi: 10.1073/pnas.96.24.13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 10.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 11.Koo EH, Lansbury PT, Jr, Kelly JW. Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kostka M, Hogen T, Danzer KM, Levin J, Habeck M, Wirth A, Wagner R, Glabe CG, Finger S, Heinzelmann U, Garidel P, Duan W, Ross CA, Kretzschmar H, Giese A. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J Biol Chem. 2008;283:10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 13.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 14.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 15.Michnick SW. Exploring protein interactions by interaction-induced folding of proteins from complementary peptide fragments. Curr Opin Struct Biol. 2001;11:472–477. doi: 10.1016/s0959-440x(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 16.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 17.Munishkina LA, Phelan C, Uversky VN, Fink AL. Conformational behavior and aggregation of alpha-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 18.Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE. 2008;3:e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 20.Schiffer NW, Broadley SA, Hirschberger T, Tavan P, Kretzschmar HA, Giese A, Haass C, Hartl FU, Schmid B. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J Biol Chem. 2007;282:9195–9203. doi: 10.1074/jbc.M607865200. [DOI] [PubMed] [Google Scholar]
- 21.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 22.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 23.Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 24.Vekrellis K, Rideout HJ, Stefanis L. Neurobiology of alpha-synuclein. Mol Neurobiol. 2004;30:1–21. doi: 10.1385/MN:30:1:001. [DOI] [PubMed] [Google Scholar]