

Abstract
The effects of amyloid-β are extremely complex. Current work in the field of Alzheimer disease is focusing on discerning the impact between the physiological signaling effects of soluble low molecular weight amyloid-β species, and the more global cellular damage that could derive from highly concentrated and/or aggregated amyloid. Being able to dissect the specific signaling events, to understand how soluble amyloid-β induces its own production by upregulating BACE1 expression, could lead to new tools to interrupt the distinctive feedback cycle with potential therapeutic consequences. Here we describe a positive loop that exists between the secretases that are responsible for the generation of the amyloid-β component of Alzheimer disease. According to our hypothesis, in familial Alzheimer disease, the primary overproduction of amyloid-β can induce BACE1 transcription and drive a further increase of amyloid-β precursor protein processing and resultant amyloid-β production. In sporadic Alzheimer disease, many factors, among them oxidative stress and inflammation, with consequent induction of presenilins and BACE1, would activate a loop and proceed with the generation of amyloid-β and its signaling role onto BACE1 transcription. This concept of a signaling effect by and feedback on the amyloid-β precursor protein will likely shed light on how amyloid-β generation, oxidative stress, and secretase functions are intimately related in sporadic Alzheimer disease.
Keywords: Alzheimer disease, amyloid, amyloid-β protein precursor processing, BACE, oxidative stress
Amyloid-β: Functions and Dysfunctions
The amyloid-β peptide (Aβ) is generated following the sequential cleavage of its precursor, the amyloid-β protein precursor (AβPP) by β- and γ-secretase in the amyloidogenic pathway. The non-amyloidogenic pathway, primed by a first cleavage of AβPP by α-secretase, whose identity is still vague, leads to the production of non amyloidogenic C-terminal fragment peptide C83 (Selkoe, 2001, Vassar, 2004). The best candidates to α-secretase function are members of the ADAM family of disintegrin and metalloproteases (Kojro and Fahrenholz, 2005): recent data show how the different expression and integrity of these proteases can modulate the phenotype of Alzheimer disease (AD) mice models (Schmitt et al., 2006, Schroeder et al., 2009). The β-secretase is known to be the β-site AβPP cleaving enzyme I, BACE1 (Hussain et al., 1999, Sinha et al., 1999), a 501 amino acid aspartyl protease widely expressed in brain, that fulfills most of the requirements expected for a candidate β-secretase (Lin et al., 2000, Vassar et al., 1999). The γ-secretase is a multimeric, high molecular weight complex with proteolytic activity, formed by a minimum of four molecules: Presenilin1/2 (PS1/2), Nicastrin, Pen-2 and Aph-1 (De Strooper et al., 1999, Haass and De Strooper, 1999, Selkoe, 2001).
Amyloid-β Functions
As much as a cellular and molecular function for AβPP and its derivatives has been searched for, no clear physiological roles have been fully characterized, and they often mingle with the toxic effects of Aβ. The similarity of AβPP to NOTCH and to its processing strengthens the idea that AβPP and its derivatives may have a signaling role.
Aβ is the subject and object of pathways leading to cell death or survival, where it could play a role not just as a toxic compound, but as a functional signaling intermediate. TrkA is a member of the tyrosine kinase family receptors. Upon binding to its ligand, i.e., NGF, the intracellular C-terminal portion of TrkA phosphorylates and activates the Src homology 2 domain containing protein, which leads to MAPK activation and stimulation of cells growth. It also activates the PLCγ pathway which also leads to MAPK activation as well as PI3K which leads to AKT activation and inhibition of apoptosis (Gomez and Cohen, 1991, Qian et al., 1998, Sawada et al., 2000, Ulrich et al., 1998). Its expression seems reduced in brains of AD patients (Marinelli et al., 1999), more than the physiological age related switch between TrkA and p75NTR would predict. In this picture, BACE1 stabilization and increased Aβ production seem to be a consequence of aging (Costantini et al., 2006). On the other end, Aβ strikes the production of NGF, activation of the TrkA/p75NTR pathways and MAPT (tau) hyperphosphorylation (Bulbarelli et al., 2009). p75NTR, a low affinity receptor for NGF, acts via binding with Trk receptors and mostly leading to cell death and apoptosis (Harrington et al., 2004), and its blockade has been shown to halt Aβ induced-NGF dependent cell death (Yaar et al., 2008, Yaar et al., 2007). The apparent nonsense could be explained by and interplay between neurons and activated NGF-secreting astrocytes, attempting to survive in an amyloid milieu: in “old” hippocampal neurons expressing p75NTR, this would lead to cell death (Saez et al., 2006) and Aβ could be a regulator of the process.
One of the downstream consequences of p75NTR signaling is the activation of NFKB, via p38/MAPK and JNK: although mostly considered to be a surviving pathway (Bui et al., 2002), some authors have proposed that NFKB activation could strike apoptosis in neuroblastoma cells via p53 (Costantini et al., 2005). Furthermore, inflammation and upregulation of Il-6, IL-1, and TNF-α through microglia activation and Aβ also contributes to NFKB activation (Dudal et al., 2004, Jin et al., 2008, Pan et al., 2009).
The MAPK pathway is often involved in Aβ-driven signaling, as downstream of Aβ-RAGE interaction (Yan et al., 2009), TrkA/p75NTR, insulin receptor (Townsend et al., 2007), and many more: in general, the MAPK signaling seems activated in AD (Lagalwar et al., 2006, Lee and Das, 2008, Zhu et al., 2002) and correlates strongly with the oxidative stress in AD models (Tamagno et al., 2003).
Intracellular Aβ accumulation, which may commence way before extracellular, seems to be involved in various types of cellular damage, such as mitochondrial toxicity, proteasome impairment and synaptic damage; p53 expression can also be activated by intracellular Aβ (Ohyagi et al., 2005), leading to apoptosis, and has been reported to be upregulated in AD pathological regions (Hooper et al., 2007) and in Down's syndrome (de la Monte, 1999), although, when mutant PS1 is overexpressed, p53 upregulation seems to depend more on failure of proteasomal degradation than on a transcriptional mechanism [37].
Other signaling pathways that are somehow related to Aβ generation involve NOTCH, which is processed intramembranously by the same PS1-dependent γ-secretase activity, competes with AβPP for this processing and interacts with AβPP itself on the plasma membrane (Berezovska et al., 2001, Oh et al., 2005). Wnt is involved in correct cell development and axon guidance together with β-catenin, APC and GSK3β, and its pathway has been found to be deregulated in AD, by PS mutants and by Aβ itself (Boonen et al., 2009, Magdesian et al., 2008).
As a final example of the complexity of Aβ physiological role, it has been shown that neuronal activity modulates the production and secretion of Aβ (Nitsch et al., 1993); in turn, Aβ can depress this neuronal activity, via glutamatergic receptors, creating a negative feedback loop (Kamenetz et al., 2003, Shemer et al., 2006) that would act as a sort of synaptic homeostasis mechanism, preventing exitotoxicity. As reported in the next paragraphs, this modulation can become detrimental to neurons as quality and quantity of Aβ vary.
Amyloid-β Dysfunctions
According to the amyloid cascade hypothesis of AD, Aβ is considered to be the primary motor of neuronal degeneration, although the pathway leading to neuronal death is much complicated and involves numerous steps (Hardy and Allsop, 1991). In particular, although debated until now, neurofibrillary tangles composed of hyperphosphorylated protein tau are considered a secondary event in the disease progression, a consequence of Aβ toxicity and Aβ plaque formation (Verdile et al., 2004). Amyloid plaques, one of the defining neuropathological characteristics of AD, are neither specific of this condition (Armstrong et al., 1996, Dickson et al., 1992, Yamaguchi et al., 1998) nor are properly “pathogenic”, as they have now come to be considered an end stage of amyloid deposition, representing an inactive reservoirs of species that are in equilibrium with the smaller, putatively neurotoxic assemblies (Hardy and Selkoe, 2002). Although neuronal degeneration occurs in proximity of the amyloid plaques, some studies have suggested that intermediate Aβ aggregates such as protofibrils or simple oligomers are also involved in AD pathogenesis and even appear to be the more dangerous species. Furthermore, in patients dying with AD, there is a relatively weak correlation between the severity of dementia and the density of fibrillar amyloid plaques (Dickson et al., 1995, Katzman, 1986, Terry et al., 1991). More attention has thus been focused on the early stages of amyloid production and on its “maturation” from small soluble molecules, to oligomers and into more and more complex high molecular weight aggregates.
Aβ is a physiological cell product, mainly generated as a 40 amino acids peptide (Aβ1-40), while only about 10% as a longer Aβ1-42 peptide. The latter peptide is proportionally increased in patients affected by AD, both in familial and sporadic cases, and has a greater propensity to aggregate and form oligomers and fibrils (Burdick et al., 1992, Haass et al., 1992, Jarrett et al., 1993, Kumar-Singh et al., 2006). The conformational change from an α-helix into a well organized β-sheet structure is a well known characteristic of Aβ aggregation (Xu et al., 2005). A major role in the aggregation is played by the C-terminus of Aβ and by the hydrophobic core in the group of residues 17–21 (Tycko, 2003). This is in line with the pathogenic role of Aβ42 and other shorter x-42 peptides which maintain a full-length C-terminus. The Aβ pool is also composed by many different N- and C-terminal truncated peptides, identified both in plaques and away from plaques, in soluble forms and in biological fluids (Hardy and Selkoe, 2002, Russo et al., 1997, Russo et al., 2000). It has been pointed out how N-terminal truncated peptides, in particular the pyroglutamate-3-42 peptide, are enriched in AD brains (Harigaya et al., 2000, Russo et al., 2000) early in the pathological process (Teller et al., 1996) and are more prone to aggregate and to exert a toxic effects on the cell [64,65]. Interestingly, BACE1 is known to play a role in generating N-terminal truncated Aβ species, especially if overexpressed in vitro or due to oxidative stress (Borghi et al., 2006, Liu et al., 2002). Some authors argue that N-terminal peptides are actually derived from proteases' activity on the full length 1-x peptides (Sun et al., 2008). However the shorter peptides may be generated, it is clear that different qualities of the Aβ peptides, truncated at both N- and C-terminal, including soluble species and species generated inside the cell, are important in defining their characteristics in terms of aggregation and toxicity (Gong et al., 2003, Hoshi et al., 2003, Kienlen-Campard et al., 2002). Aβ assemblies with different degrees of aggregation, and thus different sizes, have been shown to induce diverse degeneration pathways. The toxic action of Aβ seems not only due to the ability to form aggregates in the extracellular milieu, but also to the presence of small soluble Aβ oligomers inside the cell (Lambert et al., 1998). The end result of Aβ accumulation, be it extra- or intracellular, is the damage to membranes or organelles, which eventually leads to the derangement of the cellular biochemical processes. This, in turn, leads to oxidative stress, inflammation and ultimately to cell death (Chauhan and Chauhan, 2006, Weiner and Frenkel, 2006).
One of the most known and studied effects of Aβ is, in fact, its ability to induce, and be induced by, oxidative stress. Several byproducts of protein, lipid and glucose oxidation are elevated in the brains of patients with AD, and to a lesser extent in the brains of healthy aged controls, as the burden of free radicals builds up proportionally to the duration of the disease (Borghi, 2007, Butterfield et al., 2001, Markesbery and Lovell, 1998, Sayre et al., 1997). Both amyloid deposits and soluble Aβ seem to drive the accumulation of reactive oxygen species (Behl, 2005, Pratico, 2008). Oxidative stress is itself able to induce the increased generation of Aβ species, and AβPP processing (Paola et al., 2000, Tamagno et al., 2005, Tong et al., 2005). An interesting downstream effect of Aβ-induced oxidative stress on lipids is the interference with membrane stability and the generation of calcium flow into the cells, leading to enhanced toxicity for the cell (Kirkitadze and Kowalska, 2005). Furthermore, Aβ can strike the production of pro-inflammatory molecules, such as TNF-α and IL-1β, leading to microglial activation, production of an immune response, and to an enhanced production of AβPP and its processing to generate more Aβ (Akiyama, 2006, Atwood et al., 2003). On the other end, of particular interest is the report that AβPP promoter responds positively to inflammatory mediators, i.e., IL-1, via an IRE/IRP mechanism: inflammation leads, this way, to enhanced AβPP and Aβ production. This effect has been shown to be reversed by iron chelation (Rogers et al., 2002). Metals, in particular Zn and Cu, have indeed been proposed to contribute to Aβ toxicity by increasing its aggregation.
Aβ oligomers have been found to alter memory function in mice models of AD, and the role of soluble oligomers, as opposed to amyloid plaques, has been recognized (Dodart et al., 2002, Lesne et al., 2006). The importance of low molecular weight oligomers is especially evident as they appear necessary and sufficient to alter LTP in vivo and in vitro (Cleary et al., 2005, Dineley et al., 2002, Walsh et al., 2005) and as they appear to actually reduce the density of synapses (Shankar et al., 2007). At the molecular level, different Aβ aggregates act by increasing inward excitatory post-synaptic currents with membrane depolarization (Hartley et al., 1999), through the AMPA and NMDA channels, in different ways (Ye et al., 2003); different types of Aβ assemblies are also able to alter neuronal architecture, cause perturbations in axonal transport and even down-regulate cell surface levels of NMDA receptors (Kelly and Ferreira, 2006, Maloney et al., 2005, Snyder et al., 1994, Tamagno et al., 2006, White et al., 1999, Zhao et al., 2006).
Aβ is thus able, in many of its forms, to induce neurodegeneration. Apoptosis is induced by oligomeric, pre-fibrillar and fibrillar Aβ (Di Carlo, 2009, Dickson, 2004, Glabe, 2001, Oddo et al., 2003). Aβ interacts with molecules outside the cell, like integrins, RAGE or AβPP itself, hypothetically sparking a signal that leads to cell death (Verdier et al., 2004) or acting via intracellular initiated damage, e.g., on mitochondria (Kerr, 2002). It can damage the membrane in diverse ways as well, allowing leakage of ions, with toxic consequences to the cell (Marchesi, 2005, Yu et al., 2006); these include inducing death through a calcium mediated mechanism (Diaz et al., 2009), or disturbing physiological ion exchange (Bores et al., 1998, Wu et al., 1997).
Regardless of the specific pathways, it appears clear that Aβ is intimately involved in a number of cellular signaling events. It is likely that the derangement of these pathways dates much earlier than the time of the clinical presentation of AD, and much earlier than the accumulation of Aβ itself into plaques. Efforts should be made, therefore, to identify the mechanisms that lead, in sporadic AD, to increased Aβ production, and to the effects that this produces. This may allow for much more effective therapeutic/preventive interventions.
BACE1 and its control
BACE1 is the aspartyl protease that is the limiting step in the amyloidogenic process, with an N-terminal signal sequence (residues 1-21) and a pro-peptide domain (residues 22-45) that are removed post-translationally (Bennett et al., 2000, Hussain et al., 1999). There are several indications that that the β-secretase cleavage on AβPP is highly sequence-specific (Cole and Vassar, 2007), and radiosequencing has demonstrated that Aβ isolated from amyloid plaques, as well as that produced in cell lines, predominantly begins at the Asp+1 residue of Aβ, although alternative Aβ species begin at Val-3, Ile-6, and Glu+11 (Cole and Vassar, 2007, Haass et al., 1992, Roher et al., 1993). Over-expression of BACE1 apparently changes this specificity, leading to the preferential production of Aβ species starting at position +11 or +3, and often showing cyclized glutamates residue (pE3-x or pE11-x) (Borghi et al., 2006, Liu et al., 2002, Piccini et al., 2005). The abundance of Aβ3/11-40/42 produced by BACE1 over-expression suggests a possible pivotal role of the N-terminally truncated Aβ species in AD pathogenesis, as they are in fact more prone to aggregation and more resistant to proteolysis (Schilling et al., 2008).
Several reports show that levels of BACE1 protein levels and activity are increased in brains of sporadic and familial AD patients, compared to normal aged controls (Fukumoto et al., 2002, Holsinger et al., 2002, Yang et al., 2003, Zhao et al., 2007). Moreover, BACE1 levels rise following physiological stress or injury, such as oxidative stress (Tamagno et al., 2002), traumatic brain injury (Blasko et al., 2004), ischemia (Wen et al., 2004), hypoxia (Zhang et al., 2007) and energy impairment (Velliquette et al., 2005). These results imply that age-related stress may increase BACE1 levels and drive AD pathogenesis. The exact mechanisms of this up-regulation are not entirely understood, and hypotheses vary from transcriptional, post-transcriptional, translational, post-translational and degradation control.
BACE1 promoter and control pathways
The BACE1 gene spans ∼30kb on human chromosome 11q23.2 and includes 9 exons. BACE1 gene promoter has a complex structure, divided into two distinct promoter regions, carrying several transcription factor binding sites, such as for SP1, AP1, AP2, CREB, glucocorticoid receptor, Zeste, NFkB and GC boxes and CLS sites (Sambamurti et al., 2004), many of which are organized in repeats, typical of an inducible protein. Different signaling pathways, such as JNK/AP1 (Tamagno et al., 2008), NFκB (Buggia-Prevot et al., 2008) and p25/cdk5/STAT3 (Wen et al., 2008) have been suggested to control BACE1 transcription. A strong inflammatory reaction is present in AD brain, and long-term nonsteroidal anti-inflammatory drug (NSAID) use reduces the risk of AD, suggesting that inflammation may play an important role in AD pathophysiology (Akiyama et al., 2000). The BACE1 gene promoter also has a binding site for the transcriptional regulator proliferator-activated receptor γ (PPARγ; (Sastre et al., 2006)). Activation of PPARγ by NSAIDs or PPARγ agonists cause repression of BACE1 gene promoter activity, while pro-inflammatory cytokines that reduce PPARγ levels lead to increased BACE1 mRNA. Thus, the effects of inflammation and NSAIDs on AD may involve, at least in part, the action of PPARγ on BACE1 gene expression (Sastre et al., 2006).
Several research groups have shown that oxidative stress up-regulates the expression of BACE1, even through the activity of the γ-secretase (Buggia-Prevot et al., 2008, Tamagno et al., 2008). Moreover, it has been shown that the presence of oxidative stress is necessary to obtain the increase of BACE1 expression under stress conditions (Jo et al., 2008, Tamagno et al., 2008, Tamagno et al., 2008) and hypoxia inducible factor 1alpha (HIF1α) activation seems to play a role in BACE1 transcription (Guglielmotto et al., 2009).
Post-transcriptional and post-translational control of BACE1 activity
As mentioned above, BACE1 has been found to be transcriptionally regulated by several different mechanisms, involving various transcription factors and pathways (Buggia-Prevot et al., 2008, Cho et al., 2008, Christensen et al., 2004, Rossner et al., 2006, Wen et al., 2008) and, given the complexity and richness in transcription factor recognition sites of its gene promoter, it is likely to be a highly regulated protein (Sambamurti et al., 2004). Additional non-transcriptional mechanisms have been hypothesized to account for increased BACE1 protein levels and activity in sporadic AD.
Tesco and colleagues (Tesco et al., 2007) have demonstrated that the elevated BACE protein levels found in AD patients and animal models of acute brain injury, including ischemia and acute head trauma, may be due to impaired degradation and stabilization of BACE1. This would then lead to increased production of the Aβ peptide, thereby contributing to AD pathogenesis. Since Aβ has also been reported to induce apoptosis, this could result in a vicious cycle that fosters Aβ generation and cell death. In the same study, in vivo and in vitro data implicate GGA3 as the key player in regulating degradation of BACE due to its capacity as a trafficking molecule that delivers BACE to the endosomal-lysosomal system.
Also, maturation of BACE1 may be controlled by PS1 itself (Kuzuya et al., 2007). Likewise, expression of BACE1 antisense transcripts (Faghihi et al., 2008), which respond to cellular stresses and Aβ itself, seem to stabilize BACE1 mRNA, could thus increase its protein expression. Alternative splicing of BACE1 pre-mRNA has been found to be a control system as well (Mowrer and Wolfe, 2008).
Amyloid-β is the center of a loop between BACE1 and γ-secretase
We have extensively analyzed BACE1 transcriptional activation, first as a response to oxidative stress, both in vivo and in vitro. As mentioned, oxidative stress is a recognized activator of BACE1 transcription, possibly through HIF1α (Guglielmotto et al., 2009). We have shown how oxidative stress is an activator of PS1 and PEN2 transcription as well, and how, in an oxidative stress model, the upregulation of BACE1 is dependent on the proper expression of PS1 and AβPP (Tamagno et al., 2008), i.e., on the activity of the γ-secretase on AβPP. This led us to think that either Aβ or the AβPP Intra-Cellular Domain (AICD) could be the molecule responsible for BACE1 induction. Over-expression of mutant PS1 determines an increase of Aβ42 species (Duff et al., 1996) and familial AD cases with PS1 mutations have mostly an increased ratio of Aβ42/40. When we transposed our model onto PS1 mutations, we found that over-expression of PS1 mutants could alone determine an increase of BACE1 expression. Furthermore, this was true in the brain of patients affected with familial AD with PS1 mutations and in PS1 M146V knock in mice. Finally, cells that lacked AβPP did not respond to PS1 mutants as did wild type cells, and cells lacking PS1/2 had lower basal levels of BACE1. These data indicated again that AβPP and PS1 are necessary and sufficient to induce BACE1 transcription. AICD was investigated by means of transient transfections and AICD transgenic mice, but turned out not to be responsible. Instead, treating neuronal and neuroblastoma cells with 1μM soluble Aβ1-42 increased BACE1 transcription, which was blocked if anti Aβ42 antibodies were added to the culture medium (Giliberto et al., 2009).
Discussion
We are essentially describing and proposing a positive loop that exists between the secretases that are responsible for the generation of the amyloid component of AD. According to our hypothesis, in familial AD the primary overproduction of Aβ42 can induce BACE1 transcription, and determine a further increase of AβPP processing and of amyloid production (Figure 1). In sporadic AD, one of many causal factors, such as oxidative stress and inflammation, can determine a primary induction of PS1/Pen2 and of BACE1, and the loop proceeds with the generation of Aβ42 and its signaling to BACE1 transcription. This alone sheds light on how Aβ generation, oxidative stress and secretase functions are intimately related in sporadic AD.
Figure 1. Schematic of the tentative model of Aβ induced activation of BACE1 transcription.
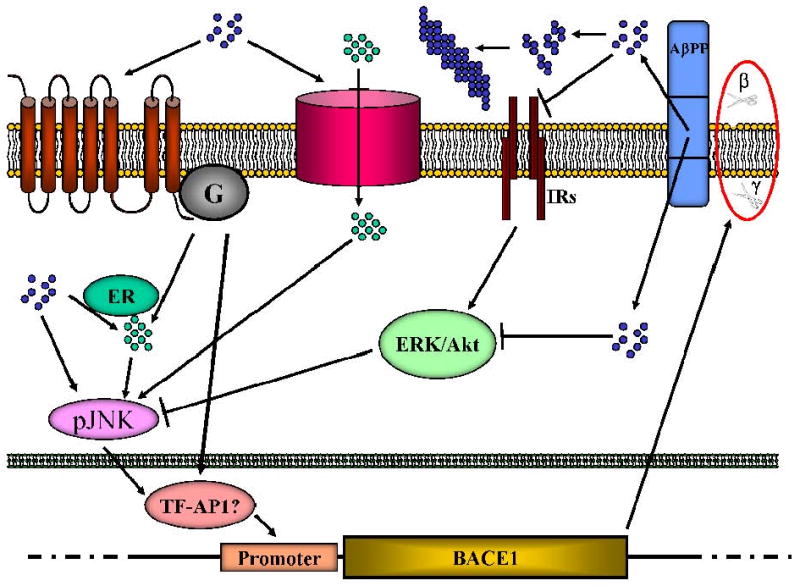
Soluble Aβ (blue dots) could activate G-coupled receptors or ionic (Ca++) channels leading to increase Ca++ entry. The same effect could be achieved intracellularly (green dots) via interaction with the ER, liberating Ca++. This could lead to JNK activation and finally AP1-mediated transcription of BACE1. Inhibition of IRs and Akt pathway by Aβ could liberate the action of JNK on AP1, achieving the same effect. Increased production of BACE1 completes the cycle, enhancing the processing of AβPP (red circle) to form more Aβ. Oligomers and fibrils (aggregated blue dots) are formed as a consequence of enhanced amyloidogenesis, but it is not clear if they have a role in this signalling.
Blue dots: Aβ; green dots: Ca++; arrows: stimulation; blunt arrows: inhibition; IRs: insulin receptors; G: G-proteins and G-coupled receptors; β and γ: β-and γ-secretase cleavages; ER: endoplasmic reticulum.
It is not clear, though, how Aβ could reach BACE1 transcriptional apparatus. As reported, there are numerous signaling pathways that seem to be regulated or at least influenced by Aβ. We have searched for a possible pathway that could induce BACE1 transcription, and believe that JNK signaling fits in the picture, as it is induced both by oxidative stress and by amyloid accumulation (Wang et al., 2004, Yao et al., 2005) and has been proposed as an inducer of BACE1 and PS1 [82,126,146]. Indeed, we have found a significant activation of JNK/AP-1, as expected, in oxidative stress tests in vitro and in vivo, and the transactivation of BACE1 in these models was not seen when JNK function was genetically or pharmacologically eliminated. ERK has been shown to be a negative regulator of γ-secretase activity (Kim et al., 2006), while PS1 seems to induce ERK (Kim et al., 2005). We have found that ERK negatively regulates the basal expression of BACE1 and opposes JNK in the oxidative stress induced-BACE1 upregulation paradigm. EGF, a physiological stimulator of the ERK pathway, is also able to decrease BACE1 levels. Contrary to what was previously reported (Kim et al., 2005), in our system, the inhibition of γ-secretase leads to an activation of ERK1/2 and a deactivation of JNK, and vice versa. Since the Akt pathway was also induced by oxidative stress upon inhibition of γ-secretase, it can be speculated that the pro-apoptotic JNK is opposed by the anti-apoptotic ERK1/2 and Akt and that the γ-secretase plays a role in controlling which pathway the cell follows.
In this picture, Aβ is at the center of a loop that not only determines the upregulation of BACE1 and stimulates amyloidogenic AβPP processing further, but also fosters γ-secretase activation and its role in determining the faith of the cell toward apoptosis.
It remains to be determined how Aβ reaches its target(s) from the plasma membrane or endosomal compartment. Activation of BACE1 seems to depend, at least in part, on JNK/AP-1. How does Aβ activate JNK? Aβ is known to disturb intracellular calcium homeostasis, and JNK could be activated by CaMKII (Wu et al., 2009) or downstream of a PI3K inducing signal which is calcium-mediated (Assefa et al., 1999). NFAT1 has been suggested to regulate BACE1, as it is normally dephosphorylated in Ca++ dependent manner by calcineurin, while it is phosphorylated and inactivated by JNK (Cho et al., 2008, Chow et al., 1997, Villar et al., 2006): in our model, the NFAT family of transcription factors, with the only exception of NFAT2c (Ortega-Perez et al., 2005), would be inactivated by JNK activation, thus would not be able to enter the nucleus and induce BACE1 transcription.
Cell surface receptors may be involved as well. As described above, numerous proteins have been described to interact with Aβ directly or indirectly, suche as AβPP itself, TrkA, p75NTR, some G-proteins, NMDA and AMPA receptors, prion protein (Lauren et al., 2009) and many more. Also, besides being a means of scavenging Aβ from tissues and having a role in directing AβPP processing, LRP family of receptors and apolipoprotein E, of which the E4 allele has a strong linkage with AD, may as well be a way for Aβ to penetrate into the cell (Bu et al., 2006, Jaeger and Pietrzik, 2008) and interact with other still unidentified molecules to strike the signaling pathway that leads to BACE1.
Finally, inhibition of the insulin receptor signaling, and Akt1 activation, by intracellular Aβ is in line with our hypothesis whereas ERK and Akt are opposed to Aβ/JNK signaling in the race to activate BACE1 transcription (Liao and Xu, 2009).
As mentioned above, the effects of Aβ on the cell can be varied and numerous, and are extremely complex. The field must discern if the physiological, signaling effect induced by soluble low molecular weight Aβ species or a more non-specific “damage” that derives from highly concentrated and/or aggregated amyloid results in the clinical and pathological conditions that define AD. Being able to dissect the specific signaling and to understand how soluble Aβ42 induces its own production by up-regulating BACE1 expression would lead to new tools to interrupt the amyloid vicious cycle, with potential therapeutic consequences.
Acknowledgments
This work was supported by the Italian Minister of Health (to M.T.), CARISA Foundation (to M.T.), the National Institutes of Health R01 AG026151 (to M.A.S.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akiyama H. Abeta, tau and alpha-synuclein and glial cells. Nihon Shinkei Seishin Yakurigaku Zasshi. 2006;26:23–31. [PubMed] [Google Scholar]
- 2.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong RA, Cairns NJ, Myers D, Smith CU, Lantos PL, Rossor MN. A comparison of beta-amyloid deposition in the medial temporal lobe in sporadic Alzheimer's disease, Down's syndrome and normal elderly brains. Neurodegeneration. 1996;5:35–41. doi: 10.1006/neur.1996.0005. [DOI] [PubMed] [Google Scholar]
- 4.Assefa Z, Valius M, Vantus T, Agostinis P, Merlevede W, Vandenheede JR. JNK/SAPK activation by platelet-derived growth factor in A431 cells requires both the phospholipase C-gamma and the phosphatidylinositol 3-kinase signaling pathways of the receptor. Biochem Biophys Res Commun. 1999;261:641–645. doi: 10.1006/bbrc.1999.1090. [DOI] [PubMed] [Google Scholar]
- 5.Atwood CS, Perry G, Smith MA. Cerebral hemorrhage and amyloid-beta. Science. 2003;299:1014. doi: 10.1126/science.299.5609.1014b. author reply 1014. [DOI] [PubMed] [Google Scholar]
- 6.Behl C. Oxidative stress in Alzheimer's disease: implications for prevention and therapy. Subcell Biochem. 2005;38:65–78. doi: 10.1007/0-387-23226-5_3. [DOI] [PubMed] [Google Scholar]
- 7.Bennett BD, Denis P, Haniu M, Teplow DB, Kahn S, Louis JC, Citron M, Vassar R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's beta -secretase. J Biol Chem. 2000;275:37712–37717. doi: 10.1074/jbc.M005339200. [DOI] [PubMed] [Google Scholar]
- 8.Berezovska O, Jack C, Deng A, Gastineau N, Rebeck GW, Hyman BT. Notch1 and amyloid precursor protein are competitive substrates for presenilin1-dependent gamma-secretase cleavage. J Biol Chem. 2001;276:30018–30023. doi: 10.1074/jbc.M008268200. [DOI] [PubMed] [Google Scholar]
- 9.Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1) J Neural Transm. 2004;111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- 10.Boonen RA, van Tijn P, Zivkovic D. Wnt signaling in Alzheimer's disease: up or down, that is the question. Ageing Res Rev. 2009;8:71–82. doi: 10.1016/j.arr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Bores GM, Smith CP, Wirtz-Brugger F, Giovanni A. Amyloid beta-peptides inhibit Na+/K+-ATPase: tissue slices versus primary cultures. Brain Res Bull. 1998;46:423–427. doi: 10.1016/s0361-9230(97)00382-1. [DOI] [PubMed] [Google Scholar]
- 12.Borghi R. Evaluation of diffraction catastrophes by using Weniger transformation. Opt Lett. 2007;32:226–228. doi: 10.1364/ol.32.000226. [DOI] [PubMed] [Google Scholar]
- 13.Borghi R, Patriarca S, Traverso N, Piccini A, Storace D, Garuti A, Cirmena G, Odetti P, Tabaton M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer's disease. Neurobiol Aging. 2007;28:1009–1014. doi: 10.1016/j.neurobiolaging.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Bu G, Cam J, Zerbinatti C. LRP in amyloid-beta production and metabolism. Ann N Y Acad Sci. 2006;1086:35–53. doi: 10.1196/annals.1377.005. [DOI] [PubMed] [Google Scholar]
- 15.Buggia-Prevot V, Sevalle J, Rossner S, Checler F. NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J Biol Chem. 2008;283:10037–10047. doi: 10.1074/jbc.M706579200. [DOI] [PubMed] [Google Scholar]
- 16.Bui NT, Konig HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JH. p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem. 2002;81:594–605. doi: 10.1046/j.1471-4159.2002.00841.x. [DOI] [PubMed] [Google Scholar]
- 17.Bulbarelli A, Lonati E, Cazzaniga E, Re F, Sesana S, Barisani D, Sancini G, Mutoh T, Masserini M. TrkA pathway activation induced by amyloid-beta (Abeta) Mol Cell Neurosci. 2009;40:365–373. doi: 10.1016/j.mcn.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 19.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 20.Chauhan V, Chauhan A. Oxidative stress in Alzheimer's disease. Pathophysiology. 2006;13:195–208. doi: 10.1016/j.pathophys.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Cho HJ, Jin SM, Youn HD, Huh K, Mook-Jung I. Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell. 2008;7:137–147. doi: 10.1111/j.1474-9726.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- 22.Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- 23.Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol Cell Biol. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 25.Cole SL, Vassar R. The Alzheimer's disease beta-secretase enzyme, BACE1. Mol Neurodegener. 2007;2:22. doi: 10.1186/1750-1326-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costantini C, Rossi F, Formaggio E, Bernardoni R, Cecconi D, Della-Bianca V. Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. J Mol Neurosci. 2005;25:141–156. doi: 10.1385/JMN:25:2:141. [DOI] [PubMed] [Google Scholar]
- 27.Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de la Monte SM. Molecular abnormalities of the brain in Down syndrome: relevance to Alzheimer's neurodegeneration. J Neural Transm Suppl. 1999;57:1–19. doi: 10.1007/978-3-7091-6380-1_1. [DOI] [PubMed] [Google Scholar]
- 29.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 30.Di Carlo M. Beta amyloid peptide: from different aggregation forms to the activation of different biochemical pathways. Eur Biophys J. 2009 doi: 10.1007/s00249-009-0439-8. in press. [DOI] [PubMed] [Google Scholar]
- 31.Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB. Small molecule blockers of the Alzheimer Abeta calcium channel potently protect neurons from Abeta cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:3348–3353. doi: 10.1073/pnas.0813355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect? J Clin Invest. 2004;114:23–27. doi: 10.1172/JCI22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–298. doi: 10.1016/0197-4580(95)00013-5. discussion 298-304. [DOI] [PubMed] [Google Scholar]
- 34.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 35.Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- 36.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 37.Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol Aging. 2004;25:861–871. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 39.Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, 3rd, Kenny PJ, Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 41.Giliberto L, Borghi R, Piccini A, Mangerini R, Sorbi S, Cirmena G, Garuti A, Ghetti B, Tagliavini F, Mughal MR, Mattson MP, Zhu X, Wang X, Guglielmotto M, Tamagno E, Tabaton M. Mutant presenilin 1 increases the expression and activity of BACE1. J Biol Chem. 2009;284:9027–9038. doi: 10.1074/jbc.M805685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci. 2001;17:137–145. doi: 10.1385/JMN:17:2:137. [DOI] [PubMed] [Google Scholar]
- 43.Gomez N, Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353:170–173. doi: 10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- 44.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S, Danni O, Parola M, Smith MA, Perry G, Tamagno E, Tabaton M. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- 46.Haass C, De Strooper B. The presenilins in Alzheimer's disease--proteolysis holds the key. Science. 1999;286:916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 47.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 48.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 49.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 50.Harigaya Y, Saido TC, Eckman CB, Prada CM, Shoji M, Younkin SG. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer's disease brain. Biochem Biophys Res Commun. 2000;276:422–427. doi: 10.1006/bbrc.2000.3490. [DOI] [PubMed] [Google Scholar]
- 51.Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Morl K, Meyer M, Hempstead BL, Yoon SO, Giehl KM. Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc Natl Acad Sci U S A. 2004;101:6226–6230. doi: 10.1073/pnas.0305755101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 54.Hooper C, Meimaridou E, Tavassoli M, Melino G, Lovestone S, Killick R. p53 is upregulated in Alzheimer's disease and induces tau phosphorylation in HEK293a cells. Neurosci Lett. 2007;418:34–37. doi: 10.1016/j.neulet.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 57.Jaeger S, Pietrzik CU. Functional role of lipoprotein receptors in Alzheimer's disease. Curr Alzheimer Res. 2008;5:15–25. doi: 10.2174/156720508783884675. [DOI] [PubMed] [Google Scholar]
- 58.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 59.Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M, Hyun DH, Park JH, Choi YH, Gwon AR, Camandola S, Cheng A, Cai H, Song W, Markesbery WR, Mattson MP. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer's disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.07.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 62.Katzman R. Alzheimer's disease. N Engl J Med. 1986;314:964–973. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- 63.Kelly BL, Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 64.Kerr JF. History of the events leading to the formulation of the apoptosis concept. Toxicology. 2002;181-182:471–474. doi: 10.1016/s0300-483x(02)00457-2. [DOI] [PubMed] [Google Scholar]
- 65.Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN. Intracellular amyloid-beta 1-42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis. J Biol Chem. 2002;277:15666–15670. doi: 10.1074/jbc.M200887200. [DOI] [PubMed] [Google Scholar]
- 66.Kim MY, Park JH, Choi EJ, Park HS. Presenilin acts as a positive regulator of basal level activity of ERK through the Raf-MEK1 signaling pathway. Biochem Biophys Res Commun. 2005;332:609–613. doi: 10.1016/j.bbrc.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 67.Kim SK, Park HJ, Hong HS, Baik EJ, Jung MW, Mook-Jung I. ERK1/2 is an endogenous negative regulator of the gamma-secretase activity. FASEB J. 2006;20:157–159. doi: 10.1096/fj.05-4055fje. [DOI] [PubMed] [Google Scholar]
- 68.Kirkitadze MD, Kowalska A. Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer's disease. Acta Biochim Pol. 2005;52:417–423. [PubMed] [Google Scholar]
- 69.Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- 70.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 71.Kuzuya A, Uemura K, Kitagawa N, Aoyagi N, Kihara T, Ninomiya H, Ishiura S, Takahashi R, Shimohama S. Presenilin 1 is involved in the maturation of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) J Neurosci Res. 2007;85:153–165. doi: 10.1002/jnr.21104. [DOI] [PubMed] [Google Scholar]
- 72.Lagalwar S, Guillozet-Bongaarts AL, Berry RW, Binder LI. Formation of phospho-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:455–464. doi: 10.1097/01.jnen.0000229236.98124.d8. [DOI] [PubMed] [Google Scholar]
- 73.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee S, Das HK. Inhibition of basal activity of c-jun-NH2-terminal kinase (JNK) represses the expression of presenilin-1 by a p53-dependent mechanism. Brain Res. 2008;1207:19–31. doi: 10.1016/j.brainres.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 76.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 77.Liao FF, Xu H. Insulin signaling in sporadic Alzheimer's disease. Sci Signal. 2009;2:pe36. doi: 10.1126/scisignal.274pe36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu K, Doms RW, Lee VM. Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression. Biochemistry. 2002;41:3128–3136. doi: 10.1021/bi015800g. [DOI] [PubMed] [Google Scholar]
- 80.Magdesian MH, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST. Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem. 2008;283:9359–9368. doi: 10.1074/jbc.M707108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR. Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer's disease. J Neurosci. 2005;25:11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marchesi VT. An alternative interpretation of the amyloid Abeta hypothesis with regard to the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:9093–9098. doi: 10.1073/pnas.0503181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marinelli L, Cammarata S, Nobbio L, Schenone A, Zaccheo D, Angelini G, Tabaton M. Tyrosine kinase A-nerve growth factor receptor is antigenically present in dystrophic neurites from a variety of conditions but not in Alzheimer's disease. Neurosci Lett. 1999;273:67–71. doi: 10.1016/s0304-3940(99)00625-4. [DOI] [PubMed] [Google Scholar]
- 84.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 85.Mowrer KR, Wolfe MS. Promotion of BACE1 mRNA alternative splicing reduces amyloid beta-peptide production. J Biol Chem. 2008;283:18694–18701. doi: 10.1074/jbc.M801322200. [DOI] [PubMed] [Google Scholar]
- 86.Nitsch RM, Farber SA, Growdon JH, Wurtman RJ. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc Natl Acad Sci U S A. 1993;90:5191–5193. doi: 10.1073/pnas.90.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 88.Oh SY, Ellenstein A, Chen CD, Hinman JD, Berg EA, Costello CE, Yamin R, Neve RL, Abraham CR. Amyloid precursor protein interacts with notch receptors. J Neurosci Res. 2005;82:32–42. doi: 10.1002/jnr.20625. [DOI] [PubMed] [Google Scholar]
- 89.Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T. Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer's disease. FASEB J. 2005;19:255–257. doi: 10.1096/fj.04-2637fje. [DOI] [PubMed] [Google Scholar]
- 90.Ortega-Perez I, Cano E, Were F, Villar M, Vazquez J, Redondo JM. c-Jun N-terminal kinase (JNK) positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain. J Biol Chem. 2005;280:20867–20878. doi: 10.1074/jbc.M501898200. [DOI] [PubMed] [Google Scholar]
- 91.Pan XD, Chen XC, Zhu YG, Chen LM, Zhang J, Huang TW, Ye QY, Huang HP. Tripchlorolide protects neuronal cells from microglia-mediated beta-amyloid neurotoxicity through inhibiting NF-kappaB and JNK signaling. Glia. 2009;57:1227–1238. doi: 10.1002/glia.20844. [DOI] [PubMed] [Google Scholar]
- 92.Paola D, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso D, Zaccheo D, Odetti P, Strocchi P, Marinari UM, Tabaton M, Pronzato MA. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- 93.Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, Giliberto L, Armirotti A, D'Arrigo C, Bachi A, Cattaneo A, Canale C, Torrassa S, Saido TC, Markesbery W, Gambetti P, Tabaton M. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- 94.Pratico D. Oxidative stress hypothesis in Alzheimer's disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 95.Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–1029. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 96.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 97.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ, et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J Biol Chem. 1993;268:3072–3083. [PubMed] [Google Scholar]
- 98.Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer's disease. Prog Neurobiol. 2006;79:95–111. doi: 10.1016/j.pneurobio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 99.Russo C, Saido TC, DeBusk LM, Tabaton M, Gambetti P, Teller JK. Heterogeneity of water-soluble amyloid beta-peptide in Alzheimer's disease and Down's syndrome brains. FEBS Lett. 1997;409:411–416. doi: 10.1016/s0014-5793(97)00564-4. [DOI] [PubMed] [Google Scholar]
- 100.Russo C, Schettini G, Saido TC, Hulette C, Lippa C, Lannfelt L, Ghetti B, Gambetti P, Tabaton M, Teller JK. Presenilin-1 mutations in Alzheimer's disease. Nature. 2000;405:531–532. doi: 10.1038/35014735. [DOI] [PubMed] [Google Scholar]
- 101.Saez ET, Pehar M, Vargas MR, Barbeito L, Maccioni RB. Production of nerve growth factor by beta-amyloid-stimulated astrocytes induces p75NTR-dependent tau hyperphosphorylation in cultured hippocampal neurons. J Neurosci Res. 2006;84:1098–1106. doi: 10.1002/jnr.20996. [DOI] [PubMed] [Google Scholar]
- 102.Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- 103.Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sawada J, Itakura A, Tanaka A, Furusaka T, Matsuda H. Nerve growth factor functions as a chemoattractant for mast cells through both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling pathways. Blood. 2000;95:2052–2058. [PubMed] [Google Scholar]
- 105.Sayre LM, Zagorski MG, Surewicz WK, Krafft GA, Perry G. Mechanisms of neurotoxicity associated with amyloid beta deposition and the role of free radicals in the pathogenesis of Alzheimer's disease: a critical appraisal. Chem Res Toxicol. 1997;10:518–526. doi: 10.1021/tx970009n. [DOI] [PubMed] [Google Scholar]
- 106.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, Jagla W, Schlenzig D, Lindner C, Rudolph T, Reuter G, Cynis H, Montag D, Demuth HU, Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 107.Schmitt U, Hiemke C, Fahrenholz F, Schroeder A. Over-expression of two different forms of the alpha-secretase ADAM10 affects learning and memory in mice. Behav Brain Res. 2006;175:278–284. doi: 10.1016/j.bbr.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 108.Schroeder A, Fahrenholz F, Schmitt U. Effect of a dominant-negative form of ADAM10 in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;16:309–314. doi: 10.3233/JAD-2009-0952. [DOI] [PubMed] [Google Scholar]
- 109.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 110.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shemer I, Holmgren C, Min R, Fulop L, Zilberter M, Sousa KM, Farkas T, Hartig W, Penke B, Burnashev N, Tanila H, Zilberter Y, Harkany T. Non-fibrillar beta-amyloid abates spike-timing-dependent synaptic potentiation at excitatory synapses in layer 2/3 of the neocortex by targeting postsynaptic AMPA receptors. Eur J Neurosci. 2006;23:2035–2047. doi: 10.1111/j.1460-9568.2006.04733.x. [DOI] [PubMed] [Google Scholar]
- 112.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 113.Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED, Huffaker HJ, Krafft GA, Holzman TF. Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J. 1994;67:1216–1228. doi: 10.1016/S0006-3495(94)80591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun X, Becker M, Pankow K, Krause E, Ringling M, Beyermann M, Maul B, Walther T, Siems WE. Catabolic attacks of membrane-bound angiotensin-converting enzyme on the N-terminal part of species-specific amyloid-beta peptides. Eur J Pharmacol. 2008;588:18–25. doi: 10.1016/j.ejphar.2008.03.058. [DOI] [PubMed] [Google Scholar]
- 115.Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M. The various aggregation states of beta-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med. 2006;41:202–212. doi: 10.1016/j.freeradbiomed.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 116.Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 117.Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma-and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tamagno E, Guglielmotto M, Giliberto L, Vitali A, Borghi R, Autelli R, Danni O, Tabaton M. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.015. in press. [DOI] [PubMed] [Google Scholar]
- 119.Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 120.Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, Danni O. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp Neurol. 2003;180:144–155. doi: 10.1016/s0014-4886(02)00059-6. [DOI] [PubMed] [Google Scholar]
- 121.Teller JK, Russo C, DeBusk LM, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann DM, Tabaton M, Gambetti P. Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down's syndrome. Nat Med. 1996;2:93–95. doi: 10.1038/nm0196-93. [DOI] [PubMed] [Google Scholar]
- 122.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 123.Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun X, Song W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J Neural Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- 125.Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- 126.Tycko R. Insights into the amyloid folding problem from solid-state NMR. Biochemistry. 2003;42:3151–3159. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- 127.Ulrich E, Duwel A, Kauffmann-Zeh A, Gilbert C, Lyon D, Rudkin B, Evan G, Martin-Zanca D. Specific TrkA survival signals interfere with different apoptotic pathways. Oncogene. 1998;16:825–832. doi: 10.1038/sj.onc.1201842. [DOI] [PubMed] [Google Scholar]
- 128.Vassar R. BACE1: the beta-secretase enzyme in Alzheimer's disease. J Mol Neurosci. 2004;23:105–114. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 129.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 130.Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Verdier Y, Zarandi M, Penke B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J Pept Sci. 2004;10:229–248. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- 132.Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimer's disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50:397–409. doi: 10.1016/j.phrs.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 133.Villar M, Ortega-Perez I, Were F, Cano E, Redondo JM, Vazquez J. Systematic characterization of phosphorylation sites in NFATc2 by linear ion trap mass spectrometry. Proteomics. 2006;6 1:S16–27. doi: 10.1002/pmic.200500407. [DOI] [PubMed] [Google Scholar]
- 134.Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol. 2006;6:404–416. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 137.Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 138.Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I, Maurin T, Wang L, Figueroa H, Herman M, Krishnamurthy P, Liu L, Planel E, Lau LF, Lahiri DK, Duff K. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999;842:439–444. doi: 10.1016/s0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- 140.Wu A, Derrico CA, Hatem L, Colvin RA. Alzheimer's amyloid-beta peptide inhibits sodium/calcium exchange measured in rat and human brain plasma membrane vesicles. Neuroscience. 1997;80:675–684. doi: 10.1016/s0306-4522(97)00053-5. [DOI] [PubMed] [Google Scholar]
- 141.Wu CY, Hsieh HL, Sun CC, Yang CM. IL-1beta induces MMP-9 expression via a Ca(2+)-dependent CaMKII/JNK/c-JUN cascade in rat brain astrocytes. Glia. 2009 doi: 10.1002/glia.20890. in press. [DOI] [PubMed] [Google Scholar]
- 142.Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational transition of amyloid beta-peptide. Proc Natl Acad Sci U S A. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yaar M, Arble BL, Stewart KB, Qureshi NH, Kowall NW, Gilchrest BA. p75NTR antagonistic cyclic peptide decreases the size of beta amyloid-induced brain inflammation. Cell Mol Neurobiol. 2008;28:1027–1031. doi: 10.1007/s10571-008-9298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yaar M, Zhai S, Panova I, Fine RE, Eisenhauer PB, Blusztajn JK, Lopez-Coviella I, Gilchrest BA. A cyclic peptide that binds p75(NTR) protects neurones from beta amyloid (1-40)-induced cell death. Neuropathol Appl Neurobiol. 2007;33:533–543. doi: 10.1111/j.1365-2990.2007.00844.x. [DOI] [PubMed] [Google Scholar]
- 145.Yamaguchi H, Sugihara S, Ogawa A, Saido TC, Ihara Y. Diffuse plaques associated with astroglial amyloid beta protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol. 1998;95:217–222. doi: 10.1007/s004010050790. [DOI] [PubMed] [Google Scholar]
- 146.Yan SD, Bierhaus A, Nawroth PP, Stern DM. RAGE and Alzheimer's Disease: A Progression Factor for Amyloid-beta-Induced Cellular Perturbation? J Alzheimers Dis. 2009;16:833–843. doi: 10.3233/JAD-2009-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 148.Yao M, Nguyen TV, Pike CJ. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J Neurosci. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ye CP, Selkoe DJ, Hartley DM. Protofibrils of amyloid beta-protein inhibit specific K+ currents in neocortical cultures. Neurobiol Dis. 2003;13:177–190. doi: 10.1016/s0969-9961(03)00068-8. [DOI] [PubMed] [Google Scholar]
- 150.Yu HB, Li ZB, Zhang HX, Wang XL. Role of potassium channels in Abeta(1-40)-activated apoptotic pathway in cultured cortical neurons. J Neurosci Res. 2006;84:1475–1484. doi: 10.1002/jnr.21054. [DOI] [PubMed] [Google Scholar]
- 151.Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- 152.Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- 154.Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]