

Abstract
Alpha-synuclein (α-syn) aggregation is a neuropathological hallmark of many diseases including Dementia with Lewy Bodies (DLB) and Parkinson’s Disease (PD), collectively termed the α-synucleinopathies. The mechanisms underlying α-syn aggregation remain elusive though emerging science has hypothesized that the interaction between cholesterol and α-syn may play a role. Cholesterol has been linked to α-synucleinopathies by recent work suggesting cholesterol metabolites appear to accelerate α-syn fibrilization. Consistent with these findings, cholesterol-lowering agents have been demonstrated to reduce α-syn accumulation and the associated neuronal pathology in vitro. In this context, this study sought to investigate the in vivo effects of the cholesterol synthesis inhibitor lovastatin on α-syn aggregation in two different transgenic (Tg) mouse models that neuronally over express human α-syn. Lovastatin-treated mice displayed significantly reduced plasma cholesterol levels and levels of oxidized cholesterol metabolites in the brain in comparison to saline-treated controls. Immunohistochemical analysis demonstrated a significant reduction of neuronal α-syn aggregates and α-syn immunoreactive neuropil in the temporal cortex of lovastatin-treated Tg mice in comparison to saline-treated α-syn Tg controls. Consistently, immunoblot analysis of mouse brain homogenates showed a reduction in levels of total and oxidized α-syn in lovastatin-treated α-syn Tg mice in comparison to saline-treated α-syn Tg controls. The reduced α-syn accumulation in lovastatin-treated mice was associated with abrogation of neuronal pathology. The results from this study demonstrate that lovastatin administration can reduce α-syn aggregation and associated neuropathology and support the possibility that treatment with cholesterol-lowering agents may be beneficial for patients with PD and/or DLB.
Keywords: Cholesterol inhibitors, Parkinson’s disease, Dementia with Lewy bodies, neurodegeneration, statins
INTRODUCTION
Parkinson’s Disease (PD) and Dementia with Lewy Bodies (DLB) are common neurodegenerative disorders of the aging population. DLB is defined as dementia occurring concurrently, or before parkinsonism (McKeith, et al., 2005). Both PD and DLB are amongst a group of diseases collectively referred to as the α-synucleinopathies which are characterized by widespread α-synuclein (α-syn) accumulation in cortical and subcortical regions (Spillantini, et al., 1997, Wakabayashi, et al., 1997, Waxman and Giasson, 2009).
Whilst the precise role of α-syn remains to be determined, it is known to be abundantly present at the synapse (Iwai, et al., 1995), where it has been proposed to be involved in synaptic vesicle trafficking and interactions with lipid rafts (Fortin, et al., 2004). In diseases such PD and DLB, α-syn, a natively unfolded protein, is found in oligomeric and fibrillar forms and as neuronal aggregates that have been linked to the neurodegeneration observed in these disorders (Uversky and Eliezer, 2009, Uversky, et al., 2005, Walsh and Selkoe, 2004, Waxman and Giasson, 2009). Interest in the connection between cholesterol and α-syn has been prompted by recent studies demonstrating that they may interact when performing functions related to vesicular trafficking (Lotharius and Brundin, 2002), and that oxidized cholesterol metabolites are detected in the cell membrane and are involved in α-syn oligomerization and fibrilization in α-synucleinopathies, (Bieschke, et al., 2006, Bosco, et al., 2006). In addition, recent epidemiological reports have shown the incidence of PD to be increased in those who intake high levels of cholesterol in their diet (Hu, et al., 2008) and there is some evidence suggesting that the use of cholesterol-lowering drugs such as the statins is associated with decreased risk of PD (Huang, et al., 2007).
A number of studies have investigated the effect of cholesterol-reducing agents on α–syn pathology, recently methyl-ß-cylclodextrin, a molecule that extracts cholesterol from the cell membrane, has been reported to reduce α-syn accumulation both in vitro and in vivo (Bar-On, et al., 2006). Lovastatin, as other statins, reduces cholesterol levels by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the enzyme that converts HMG-CoA to mevalonate, which is the rate-limiting step in the biosynthesis of cholesterol (Alberts, et al., 1980). Lovastatin has previously been shown to dramatically reduce α-syn accumulation in vitro in B103 neuroblastoma cells as well as in primary human neurons and to contribute to overall cell viability as evidenced by increased cell adhesion and neurite outgrowth (Bar-On, et al., 2008). As lovastatin crosses the blood brain barrier once ingested (Saheki, et al., 1994) it is a reasonable candidate for in vivo studies directed at investigating the disease modifying effects of cholesterol lowering drugs in models of α-synucleinopathy.
In this context, this study sought to investigate the in vivo effects of lovastatin on α-syn aggregation in vivo in two Tg mouse lines, each over expressing human α-syn under the control of different neuronal promoters. These mice (collectively termed the α-syn Tg mice) have been previously characterized and been shown to model some aspects of DLB and PD (Fleming, et al., 2004, Masliah, et al., 2000, Rockenstein, et al., 2002). We report a reduction in α-syn aggregation and accumulation of oxidized α-syn and an abrogation of neuronal pathology in lovastatin-treated α-syn Tg mice in comparison to saline-treated controls, highlighting a possible therapeutic relevance for lovastatin in the treatment of α-synucleinopathies.
METHODS
Animals, Treatment and Detection of Cholesterol Levels
For this study, heterozygous Tg mice expressing human α-syn under the regulatory control of the platelet-derived growth factor-β (PDGFβ) or the mThy1.2 neuronal promoter were used. These animals were selected because they display abnormal accumulation of α-syn and develop α-syn-immunoreactive inclusion-like structures in the brain (Masliah, et al., 2005, Masliah, et al., 2000, Masliah, et al., 2001, Rockenstein, et al., 2002). Furthermore, these animals also display neurodegenerative and motor deficits that mimic certain aspects of PD (mThy1.2) and DLB (PDGFβ) (Fleming, et al., 2004, Rockenstein, et al., 2002).
Animals were treated daily via lovastatin gavage 100 mg/kg (20mg/ml in 1% methocellulose) or vehicle for 2 months and sacrificed. A total of 12 animals from each genotype were used and began treatment at 10 months, an age at which extensive behavioral deficits and α-syn accumulation can be observed: 12 non transgenic (Non Tg) animals (n = 6 saline, n = 6 lovastatin), 12 PDGFβ α-syn Tg animals (n = 6 saline, n = 6 lovastatin) and 12 mThy1.2 α-syn Tg animals (n = 6 saline, n = 6 lovastatin) were used. All procedures were completed under the specifications set forth by the Institutional Animal Care and Use Committee. Quantification of plasma cholesterol was done using the Cholesterol/Cholesterol Ester Quantification Kit (BioVision, Mountain View, CA) colorimetric method. The plasma was collected and assayed in triplicate for cholesterol only (not cholesterol esterase).
Quantification of Oxidized Cholesterol Metabolites
Hydrazine derivatization of aldehydes with fluorescent hydrazones, that were separated and quantified by reverse phase HPLC, served to assess the levels of oxidized cholesterol metabolites in mouse brains, as described previously for human tissue (Bosco, et al., 2006). Here, brain tissue from each mouse treated with saline or lovastatin was weighed (0.1-0.03g) and homogenized in 1ml KH2PO4 (pH7.4) using a tissue homogenizer (Tissue Tearor, Biospec Products Inc, Bartlesville, OK). An additional 1ml of KH2PO4 was used to fully recover tissue homogenate from the vessel and homogenizer, followed by addition of 3ml of methanol. The samples were vortexed prior to the addition of 6ml chloroform for extraction. 10μl of a 2.5μM 7-keto-cholesterol dansyl hydrazone solution in isopropanol was added to each sample, which served as an internal standard used in the analysis to correlate for extraction efficiency. Samples were vigorous vortexed and centrifuged at 3,000 rpm for 5 minutes in a Centra CL2 bench centrifuge (IEC, Needham heights, MA). The organic phase was recovered by pipetting, evaporated and then resuspended in 100μl conjugation solution consisting of 100μM dansyl hydrazine and 100μN H2SO4 in isopropanol. Samples were incubated at 37°C for 48h in the dark facilitating hydrazone attachment of the fluorescent dansyl substrate to the reactive aldehyde group on oxidized cholesterol.
Samples were loaded on to a CogentO UDC-cholesterol column (100A 4μm 4.6mm × 250mm MicroSolve Technologies Corp, Long Branch, NJ) connected to a Hitachi L-7485 fluorescent detector (excitation 360nm, emission 534nm) (Hitachi Ltd, Tokyo, Japan) pumped by a Hitachi D-7000 series HPLC and evaluated by Hitachi Chromatography Data station Software version 4.0. Separations were performed at a flow rate of 1ml/min with an isocratic mobile phase of 80% acetonitrile/20% H20 with 0.04% H2PO4.
Brain tissues from each animal were analyzed in triplicate and concentrations of aldol dansyl hydrazone (a reaction product of dansyl hydrazine and oxidized cholesterol) and 10μl of the 2.5μM 7-keto-cholesterol dansyl hydrazone solution (internal standard) were also analyzed on the same day to generate a standard curve. Retention times were 13.5 and 17.5 minutes, respectively, and ratios of the standard/internal standard peak areas were plotted against the known concentrations. The acquired ratio of the peak areas with retention times corresponding to aldol dansyl hydrazone and the internal standard form the brain tissue samples were applied to the standard curve function. Values are presented as levels of oxidized cholesterol per gram of brain tissue.
The putative aldol dansyl hydrazone from mouse brain (13.5 minute retention time) was collected, evaporated and re-dissolved in methanol and analyzed by ESI-TOF mass spectrometry confirming the expected mass of 666.4 m/z, equivalent to the exact mass of aldol dansyl hydrazone.
Immunohistochemical Analysis of α-Syn Accumulation and Neurodegeneration
Brains were removed and divided sagittally. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica, Nussloch, Germany) and placed in cryosolution, whereas the other hemibrain was snap frozen and stored at −70°C for biochemical analysis. Sections were first washed 3 times for 5 minutes each in Phosphate Buffered Solution (PBS). They were then pretreated with 1% triton-X, 3% hydrogen peroxide in PBS for 15 minutes, washed again 3 times for 5 minutes in PBS and blocked in 10% serum matching the animal the secondary antibody was raised in for 1 hour and then washed again 3 times for 5 minutes each in PBS. Sections were then incubated in primary antibodies against mouse anti-NeuN (1:500, Chemicon, Temecula, CA) or rabbit anti-human α-syn (1:500, Chemicon, Temecula, CA) overnight at 4°C. The next day, sections were washed 3 times for 5 min in PBS and then placed in biotinylated secondary antibody (1:100) (Vector Laboratories, Burlingame, CA) for 2 hours. After washing again 3 times in PBS, each of the sections was placed in 20% diaminobenzene (DAB) (Vector Laboratories) for 20 seconds, to ensure uniform DAB-reaction times across sections. Immersing the sections in double distilled H2O halted the reaction with DAB. Sections were then dried, mounted and coverslipped with entillin (Fisher).
Sections for fluorescent immunohistochemistry were incubated with the mouse monoclonal anti-microtubule-associated protein-2 antibody (MAP2, 1:50, Roche Molecular Biochemicals, Indianapolis, IN) overnight at 4°C. Immunofluorescence was then detected with the fluorescein isothiocyanate (FITC)-conjugated anti-mouse secondary antibody (Vector Laboratories). Coverslips were air-dried overnight, mounted on slides with anti-fading media (Vectashield, Vector Laboratories), and blind-coded sections were imaged with the LSCM (Laser scanning confocal microscope, MRC1024, Bio-Rad). The images were analyzed with the Image Quant 1.43 program (NIH), as described previously (Mucke, et al., 1995, Toggas, et al., 1994). Levels of α-syn immunoreactivity in the neuropil were assessed in digital images analyzed with the Image Quant software by selecting and area to exclude cell bodies, setting the threshold levels and expressing the data as corrected optical density.
Stereological analysis
An unbiased stereological estimation of total number of neurons immunolabeled with α-syn or NeuN was performed using an optical fractionator as previously described (Mayhew and Gundersen, 1996). There were generally eight to nine sections in a series. Sampling was performed using the Olympus C.A.S.T.-Grid system (Olympus Denmark A/S, Denmark), using an Olympus BX51 microscope connected to the stage and feeding the computer with distance information in the z-axis. The region of interest was delineated with a 1.25x objective. A counting frame (60%, 35,650μm2) was placed randomly on the first counting area and systemically moved though all counting areas until the entire delineated area had been sampled. Actual counting was performed using a 40x oil objective. Guard volumes (4μm from the top and 4–6μm from the bottom of the section) were excluded from both surfaces to avoid the problem of lost caps, and only the profiles that came into focus within the counting volume (with a depth of 10μm) were counted. The total number of cells was calculated according to the optical fractionator formula (West, et al., 1991).
Fractionation of Mouse Brain Homogenates
Brain homogenates were fractionated by centrifugation into soluble and insoluble fractions as previously described (Ho, et al., 2005). Briefly, brain tissue was homogenized in TNE solution, consisting of 20mM Tris–HCl, 150mM NaCl, 1mM EDTA, 1% NP-40, 5mM 2-mercaptoethanol, 1× protease inhibitor cocktail (Calbiochem, La Jolla, CA) and 1× phosphatase inhibitor cocktail (Calbiochem, La Jolla, CA) containing sodium orthovanadate. To prepare the soluble and insoluble fractions, homogenates were first centrifuged at 5000 × g at 4°C for 10 min to obtain the fraction containing highly NP-40-insoluble material. This insoluble fraction was resuspended in 200μL of modified TNE and resonicated. The supernatant was further centrifuged at 274,000 × g at 4°C to obtain a pellet and the soluble fraction. Samples were stored at −80°C when not in use. Total protein concentration of each fraction was determined using the BCA (bicinchoninic acid) protein assay reagents (Pierce, Rockford, IL) prior to loading the SDS-PAGE gels.
Immunoblot Analysis
Protein levels of α-syn and oxidized α-syn were determined by immunoblot analysis. Twenty micrograms of total protein per mouse were loaded onto 4-12% Bis-Tris (Invitrogen) SDS-PAGE gels, transferred onto Immobilon membranes, incubated with antibodies against total α-syn (1:1000, Chemicon, Temecula, CA) and oxidized α-syn (1:1000, Abcam, Cambridge, MA). After overnight incubation with primary antibodies, membranes were incubated in appropriate secondary antibodies, reacted with ECL (PerkinElmer, Waltham, MA), and developed and analyzed on a VersaDoc gel-imaging machine (Bio-Rad, Hercules, CA). Anti-β-actin (1:1000, Sigma, St. Louis, MO) antibody was used to confirm equal loading.
Statistical Analysis
Differences between groups were tested using one and two factor ANOVA with Fisher PLSD posthoc tests. Additional preliminary analysis between control and treated groups was by unpaired, two-tailed, Student’s t-test. All the results are expressed as mean +/− SEM.
RESULTS
Lovastatin reduced plasma cholesterol and oxidized cholesterol metabolites
Initially, in order to confirm that the lovastatin treatment was effective, blood plasma cholesterol levels were analyzed. This showed a significant reduction in plasma cholesterol levels in all mice treated with lovastatin in comparison to saline-treated controls (Fig. 1A). These results indicate that lovastatin has a similar effect on the levels of cholesterol in the Non Tg and the PDGFβ α-syn Tg mice and the mThy1.2 α-syn Tg mice (hereafter collectively described as the α-syn Tg mice), interestingly the saline-treated PDGFβ α-syn Tg mice displayed higher cholesterol levels compared to the saline-treated Non Tg and mThy1.2 Tg mice.
Figure 1. Characterization of lovastatin activity.
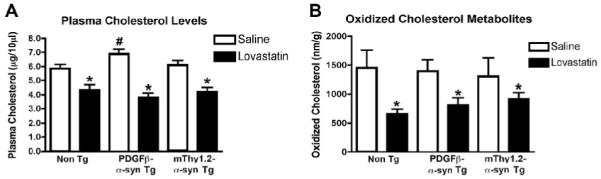
In order to assess the effect of the lovastatin treatment plasma cholesterol levels were analyzed in Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (A).
To assess central effects of lovastatin treatment oxidized cholesterol metabolites were analyzed in the brains of Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (B).
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
Oxidized cholesterol levels were also significantly reduced in the brains of Non Tg and α-syn Tg mice following lovastatin treatment (Fig 1B). There was no increase in oxidized cholesterol levels in saline-treated PDGFβ α-syn and mThy1.2 α-syn Tg mice in comparison to saline-treated Non Tg controls, indicating that α-syn over-expression does not affect the levels of oxidized cholesterol metabolites in the brain.
Lovastatin Treatment Reduced Neuronal α-Syn Accumulation in the Brains of Transgenic Mice
To determine whether α-syn accumulation was reduced in the brains of α-syn Tg mice, immunohistochemical was performed. Consistent with previous studies (Masliah, et al., 2000), we found that both α-syn over expressing Tg mice had widespread α-syn accumulation in neuronal cell bodies of the temporal cortex in comparison to Non Tg controls (Fig. 2A-C, analyzed in G). In contrast, when treated with lovastatin, both lines of α-syn Tg mice, displayed a significant reduction in the number of neurons containing α-syn aggregates (Fig. 2 D-F, analyzed in G). Analysis of the neuropil demonstrated a significant reduction in α-syn immunoreactive neuropil in the lovastatin-treated α-syn Tg mice in comparison to the saline-treated α-syn Tg mice (Fig. 2 B, E an C, F, analyzed in H). Lovastatin had no significant effect on levels of α-syn aggregates or α-syn immunoreactive neuropil in the Non Tg mice (Fig. 2 A, D, analyzed in G and H).
Figure 2. α-syn immunoreactivity in α-syn Tg mice following lovastatin treatment.
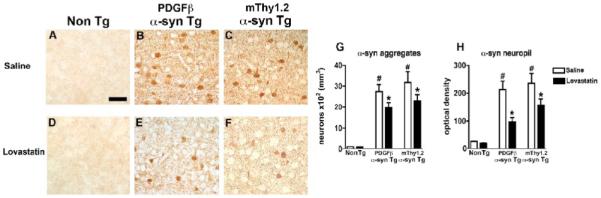
α-syn immunoreactivity was assessed in the temporal cortex of saline-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (A, B and C) and lovastatin-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (D, E, and F).
Quantitative analysis of levels of α-syn aggregates (G) and α-syn immunoreactive neuropil (H) was also conducted.
Scale bar = 50μM
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
Lovastatin Treatment Reduced Accumulation of Total and Oxidized α-Syn in the Brains of Transgenic Mice
In agreement with immunohistochemical studies, immunoblot analysis using an antibody against total α-syn demonstrated a significant reduction in the lovastatin-treated α-syn Tg mice in comparison to saline-treated α-syn Tg mice, in both the soluble (Fig. 3A) and insoluble fractions (Fig. 4A).
Figure 3. Immunoblot analysis of total and oxidized α-syn in α-syn Tg mice following lovastatin treatment – soluble fraction.
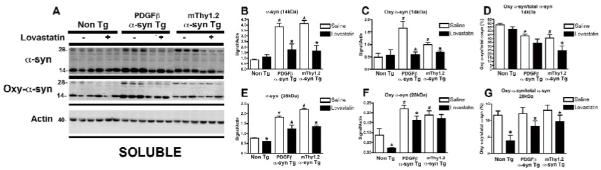
In order to examine the effect of lovastatin on α-syn levels, soluble fractions isolated from mouse brain homogenates were assessed by immunoblot with antibodies against total and oxidized α-syn (A).
Quantitative analysis of bands at 14kDa and 28kDa (corresponding to monomeric and dimeric species of α-syn) were analyzed for total α-syn (B and E) and oxidized α-syn (C and F).
In order to investigate the effect of lovastatin on relative levels of total and oxidized α-syn, ratios were calculated at 14kDa (D) and 28kDa (G).
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
Figure 4. Immunoblot analysis of total and oxidized α-syn in α-syn Tg mice following lovastatin treatment – insoluble fraction.
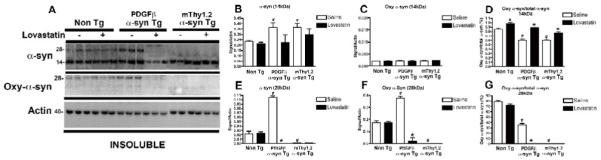
In order to examine the effect of lovastatin on α-syn levels, insoluble fractions isolated from mouse brain homogenates were assessed by immunoblot with antibodies against total and oxidized α-syn (A).
Quantitative analysis of bands at 14kDa and 28kDa (corresponding to monomeric and dimeric species of α-syn) were analyzed for total α-syn (B and E) and oxidized α-syn (C and F).
In order to investigate the effect of lovastatin on relative levels of total and oxidized α-syn, ratios were calculated at 14kDa (D) and 28kDa (G).
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
In the soluble fraction, levels of the α-syn monomer (14kDa) were reduced in the lovastatin-treated α-syn Tg mice in comparison to the saline-treated α-syn Tg mice, as were levels of a higher molecular weight species of α-syn, likely a dimer (28kDa) (Fig. 3A, analyzed in B and E). Lovastatin also appeared to reduce levels of the 28kDa α-syn species in the Non Tg mice. Levels of oxidized α-syn in the soluble fraction were also significantly reduced in the lovastatin-treated α-syn Tg mice in comparison to the saline-treated α-syn Tg mice, represented by a bands at 14 and 28kDa corresponding to monomer and dimer weights (Fig. 3A analyzed in C and F). As expected, saline-treated α-syn Tg mice displayed significantly higher levels of 14kDa and 28kDa α-syn species when compared to saline-treated Non Tg mice. In order to determine the effect of lovastatin on relative levels of oxidized α-syn, the ratio between oxidized α-syn and total α-syn was calculated for each band observed, this demonstrated a significant decease in the ratio of oxidized α-syn to total α-syn at the 28kDa band for the Non Tg and both the α-syn Tg mice and at the 14kDa band for the mThy1.2 α-syn Tg mice in comparison to saline-treated group members (Fig. 3D, G).
In the insoluble fraction, there was a trend towards a decrease in the α-syn 14kDa species in the α-syn Tg mice and a dramatic decrease in the 28kDa α-syn species in the PDGFβ α-syn Tg mice upon lovastatin treatment (Fig. 4A, analyzed in B and E). Levels of the 28kDa species of αsyn were below detection in the mThy1.2 α-syn Tg mice. Lovastatin treatment appeared to have no effect on the levels of total α-syn in the Non Tg mice. Levels of the 28kDa species of oxidized α-syn were dramatically reduced in the PDGFβ α-syn Tg mice (Fig. 4A, analyzed in F) whilst levels of the 28kDa species of mThy1.2 α-syn Tg mice and the 14kDa species of oxidized α-syn were too low to detect by immunoblot, lovastatin had no detectable effect on these levels. Analysis of the relative ratios of oxidized to total α-syn was complicated by the difficulties in detecting some species of total and oxidized α-syn and in the relative sensitivities of the two different antibodies, but, for the 14kDa band at least, it appears that there is a significant increase in the ratios of oxidized to total α-syn for the Non Tg and both the α-syn Tg mice, combined with the results from the soluble fraction this suggests a possible ‘relocation’ of oxidized α-syn from the soluble to the insoluble fraction upon lovastatin treatment.
Lovastatin Reduced Neurodegenerative Pathology in α-Syn Transgenic Mice
In order to determine whether the lovastatin-induced reduction/relocation in α-syn accumulation was associated with an abrogation of neurodegeneration in the α-syn Tg mice, immunohistochemical analysis of the neuronal marker NeuN and the dendritic marker MAP2 was performed. Saline-treated α-syn Tg mice displayed a significant reduction in neuronal density (Fig. 5A-C analyzed in G) and dendritic complexity (Fig. 6A-C, analyzed in G) in the cortex compared to saline-treated Non Tg mice. Lovastatin-treatment attenuated the alterations in the levels of NeuN (Fig. 5 B, E and C, F analyzed in G) and MAP2 immunohistochemistry (Fig. 6 B, E and C,F analyzed in G) in the cortex of the α-syn Tg mice in comparison to saline-treated α-syn Tg mice. No significant differences in NeuN or MAP2 immunoreactivity were observed between saline and lovastatin-treated Non Tg controls (Fig 5A, D, analyzed in G, Fig 6A, D, analyzed in G).
Figure 5. NeuN immunoreactivity to assess neuronal pathology in α-syn Tg mice following lovastatin treatment.
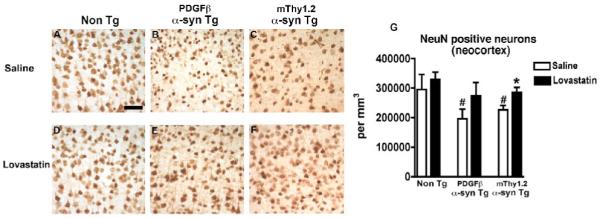
Neuronal density, as evidenced by NeuN immunoreactivity, was examined in the temporal cortex of saline-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (A, B and C) and lovastatin-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (D, E, and F), quantitative analysis (G).
Scale bar = 50μM
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
Figure 6. MAP2 immunoreactivity to assess dendritic pathology in α-syn Tg mice following lovastatin treatment.
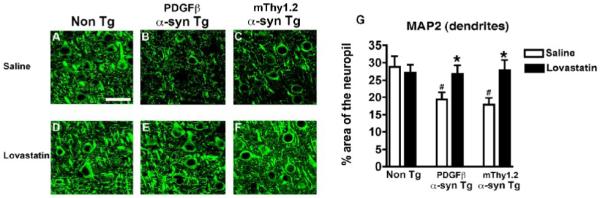
Dendritic complexity, as evidenced by MAP2 immunoreactivity, was examined in the temporal cortex of saline-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (A, B and C) and lovastatin-treated Non Tg, PDGFβ α-syn Tg and mThy1.2 α-syn Tg mice (D, E, and F), quantitative analysis (G).
Scale bar = 50μM
# Indicates a significant difference between saline-treated Non Tg mice and saline-treated α-syn Tg mice (p<0.05, one way ANOVA and post hoc Fisher).
* Indicates a significant difference between saline-treated and lovastatin-treated members of the same genotype (i.e. saline-treated PDGFβ α-syn Tg mice Vs lovastatin-treated PDGFβ α-syn Tg mice) (p<0.05, one way ANOVA and post hoc Fisher).
DISCUSSION
The present study sought to investigate the in vivo use of the cholesterol-lowering agent lovastatin in Tg mice over expressing α-syn. The results from this study demonstrate that lovastatin reduces α-syn accumulation and may lead to a shift in oxidized α-syn from the soluble to the insoluble fraction. This reduction/relocation of α-syn was accompanied by an abrogation of neuronal pathology as evidenced by NeuN and MAP2 immunoreactivity. These results are consistent with previous studies that have demonstrated that the cholesterol modifying agent methyl-ß-cyclodextrin’s ability to reduce the pathology in α-syn Tg mice (Bar-On, et al., 2006) and recent studies demonstrating the effects of statins on levels of oxidized α-syn and α-syn aggregation in vitro (Bar-On, et al., 2008). Additionally, the present study was conducted in two different Tg mouse models, making it unlikely that the results observed were a promoter-specific phenomenon.
The mechanisms through which cholesterol and α-syn interact and how lovastatin might reduce α-syn aggregation and neurodegenerative pathology remain ill defined. Whilst epidemiological evidence has pointed to dietary factors contributing to PD (Hu, et al., 2008), the significance of this is complicated by the fact that circulating peripheral cholesterol cannot cross the blood-brain-barrier (BBB) (Bjorkhem, 2006, Bjorkhem, et al., 2009). A recent study has shed new light on this issue by demonstrating that an oxidized metabolite of cholesterol, 27-hydroxycholesterol (27-OHC), which can cross the BBB, and increase levels of α-syn and induce apoptosis in vitro (Rantham Prabhakara, et al., 2008). Recent work has also reported that oxidized cholesterol metabolites, which can lead to α-syn fibrillization are elevated in α-synucleinopathies (Bieschke, et al., 2006, Bosco, et al., 2006). In light of these results, lovastatin, which itself readily crosses the BBB (Guillot, et al., 1993), may work by reducing the levels of 27-OHC either in the periphery before it crosses the BBB or centrally once it has crossed.
Another possible mechanism linking lovastatin to α-syn modification is oxidative stress. Oxidative stress leads to the generation of nitrative radicals such as NO, O2- and ONOO- which may modify α-syn post-transcriptionally (Good, et al., 1998). Tyrosine residues 39, 125 and 136 on α-syn have all been shown to become nitrated during oxidative stress and this nitration has been linked to α-syn accumulation (Ischiropoulos, 2003, Ischiropoulos and Gow, 2005, Paxinou, et al., 2001, Souza, et al., 2000). Previous studies have shown that statins reduce superoxide production in THP-1 derived monocytes by their action on HMG coenzyme A (Delbosc, et al., 2002). Lovastatin itself is a lipophilic molecule which also exhibits antioxidant behavior and may contribute anti-oxidative effects independent of its ability to inhibit cholesterol synthesis (McTavish and Sorkin, 1991). In MPTP-treated mice, simvastatin was shown to prevent protein tyrosine nitration, dopamine depletion and a reduction in TNF-α levels (Selley, 2005). Collectively, these studies support the hypothesis that statins can inhibit the formation of 3-nitrotyrosine in proteins such as α-syn, which have been linked to progression of DLB/PD. Both of these statin properties, anti-oxidant and cholesterol lowering, might be able to protect against the harmful aspects of DLB/PD.
Taken together, the results from this study and previous studies support the possibility that treatment with cholesterol-lowering agents such as lovastatin could be beneficial to patients with DLB/PD.
ACKNOWLEDGEMENTS
This work was supported by NIH grants AG10435, AG022074, AG18440, AG5131 and NS44233 (to EM) and by a Fellowship from the Short Family to the Stein Institute for Research on Aging (to AOK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J, Springer J. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On P, Crews L, koob AO, Mizuno H, Adame A, Spencer BJ, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05254.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On P, Rockenstein E, Adame A, Ho G, Hashimoto M, Masliah E. Effects of the cholesterol-lowering compound methyl-beta-cyclodextrin in models of alpha-synucleinopathy. J Neurochem. 2006;98:1032–1045. doi: 10.1111/j.1471-4159.2006.04017.x. [DOI] [PubMed] [Google Scholar]
- Bieschke J, Zhang Q, Bosco DA, Lerner RA, Powers ET, Wentworth P, Jr., Kelly JW. Small molecule oxidation products trigger disease-associated protein misfolding. Acc Chem Res. 2006;39:611–619. doi: 10.1021/ar0500766. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260:493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Cedazo-Minguez A, Leoni V, Meaney S. Oxysterols and neurodegenerative diseases. Mol Aspects Med. 2009;30:171–179. doi: 10.1016/j.mam.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, Jr., Lerner RA, Kelly JW. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol. 2006;2:249–253. doi: 10.1038/nchembio782. [DOI] [PubMed] [Google Scholar]
- Delbosc S, Morena M, Djouad F, Ledoucen C, Descomps B, Cristol JP. Statins, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are able to reduce superoxide anion production by NADPH oxidase in THP-1-derived monocytes. J Cardiovasc Pharmacol. 2002;40:611–617. doi: 10.1097/00005344-200210000-00015. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good P, Hsu A, Werner P, Perl D, Olanow C. Protein nitration in Parkinson’s disease. J.Neuropathol.Exp.Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Guillot F, Misslin P, Lemaire M. Comparison of fluvastatin and lovastatin blood-brain barrier transfer using in vitro and in vivo methods. J Cardiovasc Pharmacol. 1993;21:339–346. doi: 10.1097/00005344-199302000-00022. [DOI] [PubMed] [Google Scholar]
- Ho GJ, Hashimoto M, Adame A, Izu M, Alford MF, Thal LJ, Hansen LA, Masliah E. Altered p59Fyn kinase expression accompanies disease progression in Alzheimer’s disease: implications for its functional role. Neurobiol Aging. 2005;26:625–635. doi: 10.1016/j.neurobiolaging.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Hu G, Antikainen R, Jousilahti P, Kivipelto M, Tuomilehto J. Total cholesterol and the risk of Parkinson disease. Neurology. 2008;70:1972–1979. doi: 10.1212/01.wnl.0000312511.62699.a8. [DOI] [PubMed] [Google Scholar]
- Huang X, Chen H, Miller WC, Mailman RB, Woodard JL, Chen PC, Xiang D, Murrow RW, Wang YZ, Poole C. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov Disord. 2007;22:377–381. doi: 10.1002/mds.21290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H. Oxidative modifications of alpha-synuclein. Ann N Y Acad Sci. 2003;991:93–100. doi: 10.1111/j.1749-6632.2003.tb07466.x. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Gow A. Pathophysiological functions of nitric oxide-mediated protein modifications. Toxicology. 2005;208:299–303. doi: 10.1016/j.tox.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-Synuclein Immunization in a Mouse Model of Parkinson’s Disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew TM, Gundersen HJ. If you assume, you can make an ass out of u and me’: a decade of the disector for stereological counting of particles in 3D space. J Anat. 1996;188(Pt 1):1–15. [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- McTavish D, Sorkin EM. Pravastatin. A review of its pharmacological properties and therapeutic potential in hypercholesterolaemia. Drugs. 1991;42:65–89. doi: 10.2165/00003495-199142010-00005. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Campbell I. Transgenic models to assess the neuropathogenic potential of HIV-1 proteins and cytokines. Curr.Topics Microbiol.Immunol. 1995;202:187–205. doi: 10.1007/978-3-642-79657-9_13. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. 2001;21:8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantham Prabhakara JP, Feist G, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J Neurochem. 2008;107:1722–1729. doi: 10.1111/j.1471-4159.2008.05736.x. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Saheki A, Terasaki T, Tamai I, Tsuji A. In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
- Selley ML. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005;1037:1–6. doi: 10.1016/j.brainres.2004.02.083. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Toggas S, Masliah E, Rockenstein E, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in □transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Eliezer D. Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr Protein Pept Sci. 2009;10:483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, Lyubchenko YL, Fink AL. Effects of nitration on the structure and aggregation of alpha-synuclein. Brain Res Mol Brain Res. 2005;134:84–102. doi: 10.1016/j.molbrainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett. 1997;239:45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792:616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]