

Abstract
The prospects for using bacterial DNA as an intrinsic probe for HOCl and secondary oxidants/chlorinating agents associated with it has been evaluated using both in vitro and in vivo studies. Single-strand and double-strand breaks occurred in bare plasmid DNA that had been exposed to high levels of HOCl, although these reactions were very inefficient compared to polynucleotide chain cleavage caused by the OH•-generating reagent, peroxynitrite. Plasmid nicking was not increased when intact Escherichia coli were exposed to HOCl; rather, the amount of recoverable plasmid diminished in a dose-dependent manner. At concentration levels of HOCl exceeding lethal doses, genomic bacterial DNA underwent extensive fragmentation and the amount of precipitable DNA-protein complexes increased several-fold. The 5-chlorocytosine content of plasmid and genomic DNA isolated from HOCl-exposed E. coli was also slightly elevated above controls, as measured by mass spectrometry of the deaminated product, 5-chlorouracil. However, the yields were not dose-dependent over the bactericidal concentration range. Genomic DNA recovered from E. coli that had been subjected to phagocytosis by human neutrophils occasionally showed small increases in 5-chlorocytosine content when compared to analogous cellular reactions where myeloperoxidase activity was inhibited by azide ion. Overall, the amount of isolable 5-chlorouracil from the HOCl-exposed bacterial cells was far less than the damage manifested in polynucleotide bond cleavage and cross-linking.
Keywords: bactericidal reactions, cytosine chlorination, DNA-protein complexes, DNA fragmentation, hypochlorous acid, neutrophils
Introduction
One recurrent issue concerning the biochemistry of host cellular defense mechanisms is the relative extent to which oxidative and nonoxidative processes contribute to microbial killing within phagocytes. Current opinion on this subject varies widely; for example, several groups have presented evidence supporting the notion that myeloperoxidase (MPO)1-generated hypochlorous acid (HOCl) is a primary neutrophil microbicide [1–4], whereas others have disputed this evidence, and suggested that MPO functions physiologically as a catalase [5], thereby protecting bactericidal hydrolytic enzymes introduced during phagocytosis from inactivation by respiration-generated H2O2. Evidence also exists for an additional type of nonoxidative mechanism involving other granule-derived antimicrobial proteins that function by disrupting bacterial homeostasis without inactivating specific enzymes [6,7]. Although one might surmise from the very different nature of the chemistries comprising oxidative versus nonoxidative killing that distinguishing between them should be straightforward, the problem is convoluted by their simultaneous participation in intraphagosomal reactions [8]. Thus, for example, the demonstration that neutrophils kill bacteria under conditions where oxidative processes are inhibited cannot be taken as evidence that oxidative mechanisms do not exist. Furthermore, rather than being redundant, the distinct microbicidal mechanisms are likely to be synergistic [4,9,10].
To evaluate the role of HOCl in intraphagosomal killing by neutrophils, one needs to know both the extent of its formation by MPO-catalyzed oxidation of chloride ion and how its subsequent reactions with bacterial and host-derived targets are distributed. With respect to the first question, a recent kinetic modeling study based upon experimentally determined kinetic parameters and phagosomal dimensions predicts that ~90% of the O2 consumed by stimulated neutrophils will be converted to HOCl [11]. In developing the model it was recognized, but not explicitly taken into account, that Cl− could be the limiting intraphagosomal reagent, a condition which would lower the predicted yields. This possibility arises because the amount of extracellular fluid introduced during phagocytosis is very small, hence the amount of Cl− entrapped during formation of the phagosome is less than the amount of O2 consumed by NADPH oxidase-dependent stimulated respiration. Since the HOCl/O2 stoichiometry cannot exceed 1:1 in the coupled NADPH oxidase/MPO catalyzed reaction, i.e., 2H+ + NADPH + O2 + Cl− → NADP+ + HOCl + H2O, chloride ion will be limiting unless it is replentished in subsequent reactions of HOCl or by transmembrane ion transport processes [12–13]. Chemical trapping studies in which chloride is incorporated into stable reaction products are consistent with this viewpoint. Specifically, in studies using fluorescein-conjugated particles as bacterial mimics, it was found that the amount of chloride trapped within the phagosome as mono-, di-, and trichlorofluoresceins corresponded closely to the calculated amount of extracellular Cl− introduced during its formation [3].2 This amount, however, was only ~10% of the O2 consumed during stimulated respiration. In contrast, when soluble agonists were used to trigger extracellular release of MPO, as much as 40% of the O2 consumed could be trapped as taurine chloramine [16]. Under these conditions, where reaction occurs in the extracellular milieu, Cl− is present in large excess and cannot be significantly depleted over the course of the neutrophil respiratory burst.
The chlorofluorescein trapping and O2 consumption measurements indicate that between ~108 and ~109 HOCl molecules per entrapped particle can be generated within phagosomes [3,11]. The lower limit is clearly a conservative estimate because the phagosomal milieu contains numerous targets that react more rapidly than fluorescein with HOCl [17–20]. Prominent among these are biological sulfhydryl and thioether compounds that, unlike fluorescein, undergo net oxidation with regeneration of Cl− as one of the reaction products, which then is available for MPO-catalyzed formation of additional HOCl.3 Attempts to assess the intraphagosomal distribution of HOCl reactions have made use of 13C-labeling [25,26] to determine the relative extent of chlorination of bacterial and host-derived protein tyrosyl groups. Almost all (~94%) of the HOCl consumed in this reaction was trapped by tyrosyl groups on the host cell protein [25], which was primarily a consequence of its greater abundance within the phagosome. Nonetheless, if ~90% of the O2 consumed is converted to HOCl, as implied by the kinetic model [11], then it follows that each bacterium is exposed to ~108 HOCl molecules, a value which is consistent with the fluorescein trapping results [3]. This number is provocative because the amount of HOCl required to kill various strains of bacteria in in vitro assays is also ~108 HOCl/bacterium [3,27,28]. Thus, one might infer that bactericidal amounts of HOCl are generated within neutrophil phagosomes.
Although these studies appear to be mutually self-consistent, there are several indeterminate factors which might impact significantly upon the conclusions. Tyrosine chlorination is a relatively slow reaction [11] and is unlikely to be involved in bacterial killing, which appears to involve very rapidly reacting sites on the bacterial cell envelope [18,29,30]. Furthermore, MPO strongly associates with the bacterial wall [31–33], suggesting that it could direct HOCl toward vulnerable bacterial targets in the phagosome. Kinetic modeling studies (J. L. Cape, unpublished research) suggest that this proximity effect would be important for targets whose bimolecular rate constants exceed 105 M−1 s−1; these include, for example, cysteine sulfhydryl and methionyl thioether groups and iron-sulfur clusters. Finally, HOCl generation within the phagosome is much slower than the microbicidal reactions following in vitro bolus additions of the oxidant [18,34], so that protective responses to oxidative stress engendered in bacteria undergoing phagocytosis [35,36] could render them less susceptible to oxidative killing than implied by the in vitro bactericidal assays. Having available alternative bacterial components as selective probes for HOCl, particularly ones more reactive than tyrosine, could be very useful in describing the events comprising bactericidal action in neutrophils [24]. In the present study, we have considered using both plasmid and genomic bacterial DNA as potential probes. For reasons discussed herein, our results suggest that monitoring polynucleotide strand cleavage or formation of 5-chlorouracil, a stable product of the reaction between HOCl and cytosine, will not prove useful in this capacity; however, the data obtained suggest that DNA-protein cross-linking could be developed as an intrinsic probe. Indeed, the dramatic increase in DNA-protein complexation following exposure to lethal levels of HOCl was unanticipated, and constitutes the most remarkable finding of these studies.
Materials and methods
Materials
The following chemicals and biological materials were obtained from the indicated sources: alkaline phosphatase (Roche Applied Science); ethylenediaminetetraacetic acid (EDTA), nuclease S1, proteinase K, puromycin, sodium thiosulfate, and thymidine phosphorylase (Sigma); nuclease P1 (U.S. Biological); Hoechst 33342 trihydrochloride trihydrate (Molecular Probes); pETBlue plasmid and NovaBlue cells (Novagen). Hypochlorous acid (HOCl) was purified by vacuum distillation of commercial bleach solutions which had been neutralized to pH 7–7.5 by addition of phosphoric acid [37]; reagent concentrations were determined spectrophotometrically as the OCl− ion (ε292 = 350 M−1 cm−1 [38]). Chloramine (NH2Cl) was prepared by flow-mixing equal volumes of 0.1 M NH3 in water with 8.0 mM HOCl in 50 mM phosphate buffer, pH 7.4 through a tangential 12-jet mixer; the spectrophotometrically determined NH2Cl yield was 95% (3.8 mM), based upon a reported value of ε242 = 429 M−1 cm−1 [39]. Peroxynitrous acid (ONOOH) was prepared as alkaline solutions of the sodium salt by a quenched flow-mixing procedure involving reaction between nitrous acid and hydrogen peroxide [40]; reagent concentrations, determined in 10 mM NaOH as the ONOO− anion (ε302 = 1670 M−1 cm−1 [41]), were typically ~100 mM and contained ≤ 20% residual nitrite ion. Other chemicals were best available grade from commercial sources and used as received; except where indicated, reverse osmosis-deioinized water was used in preparation of reagent solutions.
Two cell lines of Escherichia coli (ATCC 11775) and the engineered K-12 strain, NovaBlue, were used to study bacterial genomic and plasmid DNA damage caused by exposure to oxidants (HOCl, ONOOH) or upon phagocytosis by human neutrophils. Most studies on damage to plasmid DNA were made with NovaBlue cells containing pETBlue plasmid because this plasmid is expressed with a significantly higher copy number in this cell line than in ATCC 11775. However, these cells are killed upon exposure to human serum, so that studies with opsonized bacteria and neutrophils required the use of a serum-resistant strain (in this case, E. coli 11775). Both cell lines were typically grown overnight in Luria broth (LB) at 37 °C to stationary phase, washed with 50 mM sodium phosphate buffer, pH 7.4, containing 0.15 M sodium chloride (PBS) and diluted to ~109 cells/mL, as determined by measuring turbidities of the cell suspensions. For in vitro studies on pETBlue damage, the plasmid was isolated from NovaBlue cells as an aqueous solution using a Promega plasmid purification kit and quantitated by gel electrography. Neutrophils were obtained from venous blood drawn from individual donors with informed consent. The cells were isolated by Hypaque-Ficoll density gradient centrifugation following procedures described in the literature [42]; pyrogen-free water (Baxter) was used throughout the preparative stages. The cells were maintained at 4 °C in Hanks balanced salt solution (HBSS) without Ca2+ or Mg2+ containing 20 mM added HEPES, pH 7.4, and used within several hours following isolation. Cell densities were determined by counting aliquots of suspensions in methylene blue with a hemocytometer.
Reactions with chemical oxidants
Bolus additions of sodium peroxynitrite, HOCl or NH2Cl were made at 37 °C to rapidly stirred PBS solutions containing either 100–120 ng purified pETBlue plasmid or ~108 cells/mL bacteria to give final oxidant concentrations of 0.01–1.0 mM. For studies with peroxynitrite, all solutions were purged of CO2 by bubbling with Ar prior to adding the oxidant to minimize CO2-catalyzed decomposition [43]. Any oxidant remaining after incubation at 37 °C for a timed period (usually 10 min for free plasmid, 20 min for E. coli 11775 cells, 4 h for NovaBlue cells) was scavenged by adding an excess amount of sodium thiosulfate. Genomic and plasmid DNA were extracted from the cells using appropriate Promega kits following instructions supplied by the manufacturer. The purified samples were immediately run on 0.8–1% agarose gels using pH 8.3 buffers containing 90 mM Tris, 90 mM boric acid, 2.5 mM EDTA, and ~1 µg/mL ethidium bromide, and were visualized by UV illumination.
Analysis of DNA-protein cross-linked complexes
Covalently linked DNA-protein complexes formed by exposure of cells to oxidants were isolated by precipitation of intracellular protein with sodium dodecyl sulfate and potassium chloride, as originally described by Zhitkovich and Costa [44] and modified by Kulcharyk and Heinecke [45]. The yields of co-precipitated DNA were determined as previously described by fluorescence spectroscopy following addition of Hoechst reagent to proteinase K-solubilized products.
5-Chlorouracil extraction and measurement
Typically, the supernatants from the precipitation step that contained nonconjugated DNA were purified using Qiagen Qiaquick spin columns and combined with the DNA isolated by digestion of the precipitable protein complexes. These samples, containing the total isolable DNA, were vacuum speed-dried, then taken up in 20 mM sodium acetate buffer, pH 5.1, for digestion, derivatization, and mass spectrometric analysis according to explicit procedures described by Jiang and coworkers [40]. Briefly, sequential digestion with nuclease P1 or S1, alkaline phosphatase, and thymidine phosphorylase released 5-chlorouracil, which was then extracted into ethyl acetate and derivatized with 3,5,-bis(trifluoromethyl)benzyl bromide for gas chromatographic-mass spectrometric analysis using an HP 5989A/5980 instrument operated in the negative chemical ionization mode [46].
Reactions with neutrophils
Bacteria isolated from overnight culture were resuspended in HBSS containing 10% autoserum and incubated at 37 °C for 30 min. The opsonized bacteria were then isolated by centrifugation, resuspended in HBSS supplemented with 1µM MgCl2 and 2 µM CaCl2, and mixed with neutrophils in HBSS to give a ~10:1 cell ratio. When desired, 10 mM NaN3 was included to inhibit MPO activity. The bacteria-neutrophil mixtures were then incubated on a rotating tray at 37 °C for 60 min, after which nonadherent bacteria were removed by centrifugation at 100g for 5 min in a swinging bucket rotor, followed by 1–2 washes of the pelleted neutrophils with HBSS. Effective separation of neutrophils and unattached bacteria was verified by visual inspection with an optical microscope. The isolated neutrophils were then lysed by mixing 100 µL of the HBSS suspension with 300 µL of 1% SDS. DNA released into the thick lysate was sheared by 5–6 passes through a 21-gauge needle, followed by grinding five times in a 2 mL dounce homogenizer, with addition of HBSS as necessary to dilute the suspension. Phagocytosed bacteria were then pelleted by centrifugation at 12K and 10,000 g for 20 min. The pellet was resuspended in 20 mM Tris/Cl, pH 7.5, containing 1 mM PMSF to inhibit proteases and 2% SDS to lyse the cells. The bacterial DNA was sheared by passage through a 21-gauge needle and DNA-protein complexes were precipitated with KCl as described above [44,45]. Soluble DNA was isolated from the supernatants using Qiaquick spin columns and the purified material was added to the DNA-protein complexes for digestion, derivatization, and mass spectral analyses.
Results
Phosphodiester strand cleavage in plasmid DNA
Exposure of ~120 ng isolated pETBlue plasmid to concentration levels of HOCl as high as 250 µM showed no damage to the polynucleotide chain, as determined by electrophoresis on 0.8% agarose gels (Fig. 1). However, at 500 µM HOCl the number of single-strand breaks (or “nicks”) achieved was ~50%, as determined by the appearance of the circular form of the plasmid; a small amount of the linear form, corresponding to double-strand breaks, could also be detected. Exposure to higher levels of HOCl led to complete loss of the native supercoiled form of the plasmid with proportionate increases in the circular and linear forms; at the highest HOCl concentration investigated (1 mM), the amount of recovered plasmid was noticeably diminished, suggesting that, at this level of damage, fragmentation of the plasmid DNA may have been extensive. Identical results were obtained when the exposure time was increased from 10 min to 20 min. No oxidant-induced nicking was observed in plasmid that had been exposed to NH2Cl at concentration levels as high as 3 mM.
Fig. 1.
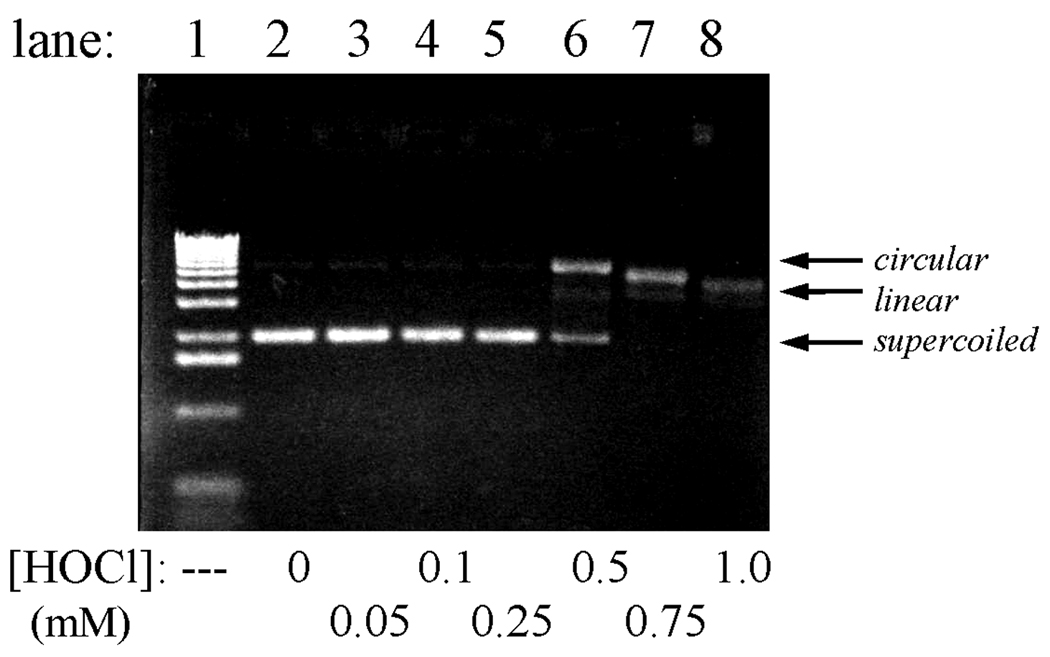
Cleavage of pETBlue plasmid DNA by HOCl. Conditions: 120 ng of purified plasmid was incubated at 37 °C with the indicated amounts of HOCl in 10 µL PBS for 10 min, after which excess Na2S2O3 was added to quench any remaining oxidant and the samples were immediately run on 0.8% agarose as described in Experimental Procedures. Lane 1 shows a standard 1 kB DNA ladder.
A comparative study was made using the peroxide, ONOOH. This oxidant is representative of the reactivities of several reactive oxygen species, including OH• generated by ionizing radiation and Fenton oxidants generated by the reaction of metal ions (Fe(II) and Cu(I)) with H2O2 [47]. Several researchers have reported that ONOOH induces strand breaks, both in isolated plasmid DNA and in genomic DNA within cells (reviewed in [48]). Exposure of the plasmid to ONOOH led to dose-dependent appearance of both single-strand and double-strand breaks. No damage was observed when peroxynitrite solutions were allowed to decay before mixing with the plasmid, or when 25 mM NaHCO3 was added to the reaction medium.4 Addition of 0.1 mM bathocuproine sulfonate (BCS), a Cu(I) chelator, provided partial protection from both single-strand and double-strand cleavage, although addition of 1 mM diethylenetriaminepentaacetic acid or desferrioxamine, both Fe(III) chelators, afforded protection only against double-strand cleavage. Specifically, under the experimental conditions ~50% single-strand cleavage occurred with addition of 5–15 µM peroxynitrite, but required ~100 µM when BCS was present (Fig.2); double-strand cleavage typically became significant after exposure to 100–200 µM peroxynitrite, but was markedly diminished at all concentrations studied (≤ 800 µM) in BCS-containing solutions. Deliberate addition of 10 µM Cu(NO3)2 did not affect the results, although increasing the Cu(II) concentration to 100 µM promoted double-strand cleavage. Pretreating peroxynitrite reagent solutions with MnO2 to remove adventitious unreacted H2O2 [40] had no effect upon the extent of cleavage. Representative gels showing damage observed in the presence of BCS and at elevated Cu2+ levels are shown in Fig. 2; these gels indicate the lower and upper limits of damage, respectively, that were observed. Additional gels illustrating the effects described above are available in Appendix A.
Fig. 2.
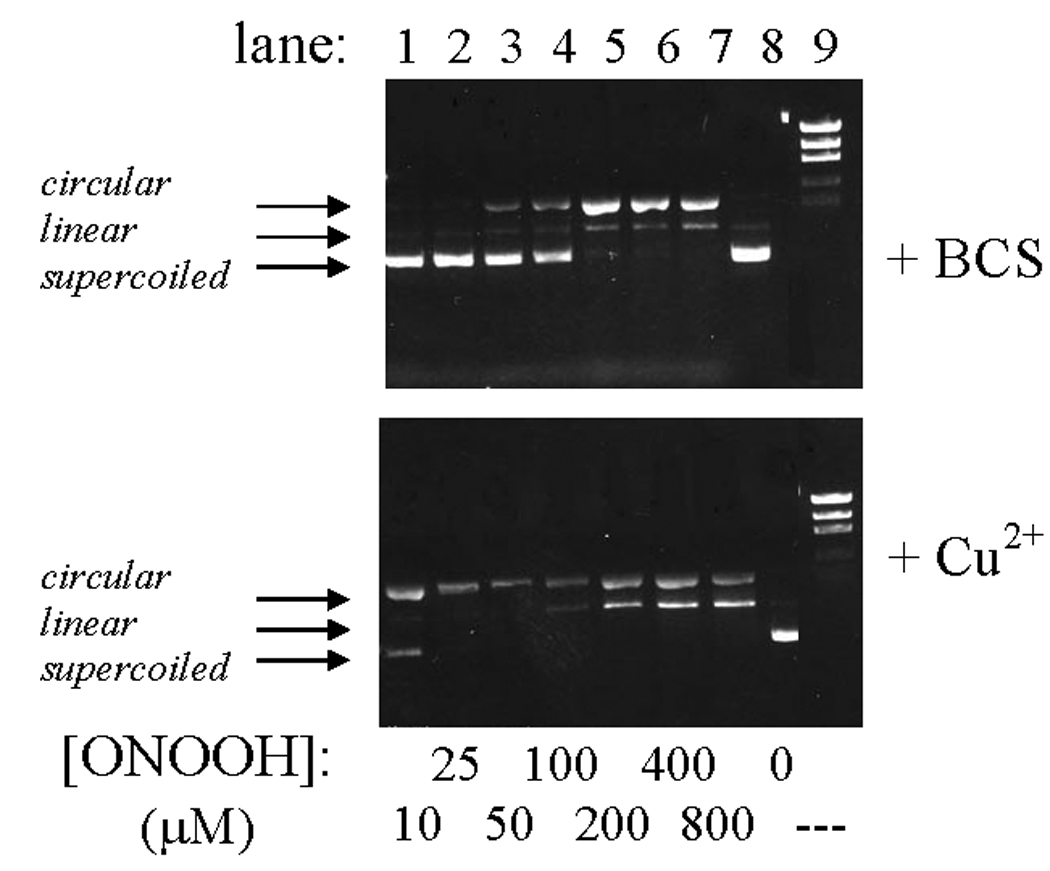
Cleavage of pETBlue plasmid DNA by ONOOH. Conditions: 100–120 ng plasmid in 10 µL phosphate buffer, pH 7.4, mixed at ambient temperature with the indicated amounts of peroxynitrite, then run immediately on 0.8% agarose. Upper gel: 0.1 mM added bathocuproin sulfonate (BCS); lower gel: 0.1 mM added Cu(NO3)2. Lane 9 contains the 1 kB DNA ladder.
Modification of plasmid and genomic DNA in E. coli exposed to oxidants
Unlike the in vitro reactions described above, plasmid nicking did not increase when pETBlue-transformed NovaBlue cells were exposed to HOCl. Rather, the amount of recoverable plasmid decreased in a dose-dependent manner (Fig. 3, Figs 5b) and was completely lost at the highest doses used (0.25–1.0 mM). Under the experimental conditions, LD90 was ~10 µM, as determined by pour-plate analyses of colony-forming units. Similar results were obtained when ONOOH was used as the oxidant, although in this case, single-strand breaks were evident at intermediary oxidant levels (Fig. 4). As discussed below, these differences may reflect the greater capacity for ONOOH to induce cleavage of the polynucleotide chains (cf., Fig. 1, Fig. 2). Based upon the limited in vitro reactivity of HOCl toward purified plasmid (Fig.1), it is unlikely that the loss of recoverable plasmid in the intracellular reactions is due to fragmentation arising from direct reaction with the oxidant.
Fig. 3.
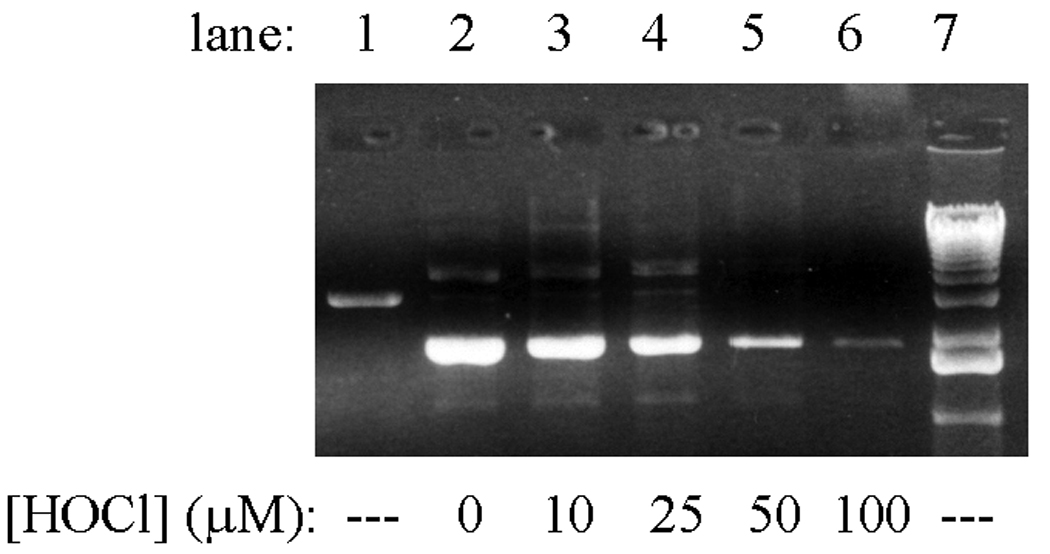
Loss of recoverable pETBlue plasmid DNA from transformed NovaBlue cells following incubation with HOCl. Conditions: ~1×108 E. coli/mL in 10 mL PBS incubated at 37 °C with the indicated amounts of HOCl for 4 h, after which a 10-fold excess of Na2S2O3 was added to remove remaining oxidant, the plasmid was isolated, resuspended in H2O, and run on 0.8% agarose as described in Experimental Procedures. Lane 1 shows purified pETBlue that had been digested with EcoR1, a restriction enzyme which catalyzes a single double-strand cut in the plasmid. Lane 7 contains the 1 kB DNA ladder. No DNA was recovered from cells exposed to HOCl at higher concentrations ([HOCl] = 250–1000 µM).
Fig. 5.

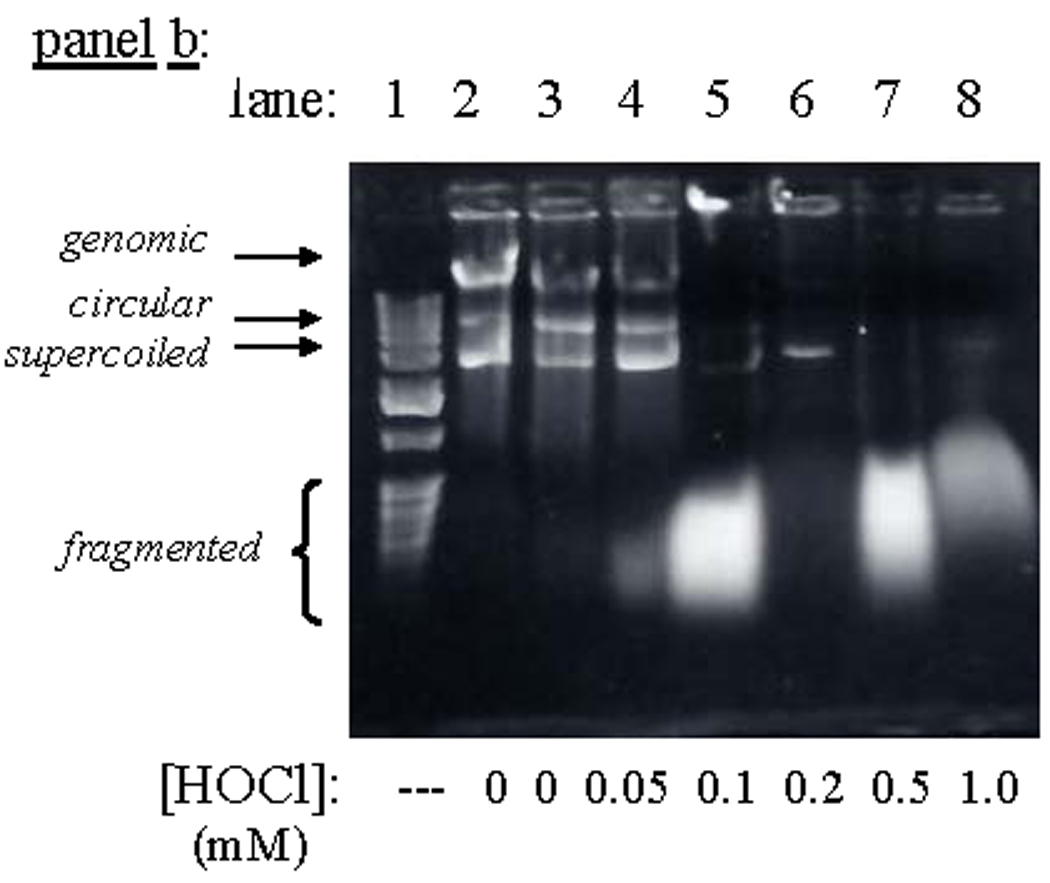
Degradation of DNA in NovaBlue cells following exposure to HOCl. Conditions: E. coli suspended at the indicated cell densities in 50 mM phosphate, pH 7.4, incubated 15 min at ambient temperature, then oxidant quenched with excess Na2S2O3, and the DNA isolated and run on 0.8% agarose. Panel a: 2.4×108 plasmid-free cells/mL, for which LD90 ~38 µM (color photograph) ; panel b: 5×107 pETBlue transformed cells/mL, for which LD90 ~8 µM.
Fig. 4.

Reaction of plasmid DNA in pETBlue-transformed NovaBlue cells exposed to ONOOH. Conditions: ~3×109 cells/mL in 50 mM phosphate, pH 7.4, under an argon atmosphere mixed with the indicated amounts of peroxynitrite at ambient temperature, after which the plasmid was isolated and run on 0.8% agarose.
Genomic DNA isolated from E. coli 11775 and NovaBlue cells underwent dramatic changes in electrophoretic properties upon exposure to HOCl concentrations that exceeded LD90 by 3–4 fold. As illustrated in Fig. 5 for NovaBlue cells, the band attributable to native DNA was diminished and a broad rapidly-moving band appeared, which consisted of ~200–500 bp fragments, as gauged from the 1 kb DNA ladder. These results suggest that treatment with lethal doses of HOCl induced extensive fragmentation of genomic DNA into lower molecular weight polymers. For example, these agarose gels looked very similar to those reported for DNA that had been fragmented by exposure to high doses of UV irradiation and subjected to further cleavage of single-strand breaks by nuclease S1 [54].
The amount of precipitable DNA-protein complexes also underwent dose-dependent increases after exposure of E. coli 11775 to HOCl in amounts comprising 2–5×LD90, but then declined upon exposure to higher doses of HOCl. Data obtained after 6 h incubation is shown in Fig. 6a; the same trend was observed for shorter incubation periods, although the total yield was less. The rate of formation of precipitable DNA was very slow, so that yields obtained upon exposure to a given amount of HOCl increased with time over the entire temporal range (23 h) that was investigated. As shown in Fig. 6b, exposure to 50 µM HOCl led to 5-fold accumulation of complexes over background levels. The amount of complex isolated was dose-dependent; specifically, exposure to 75 µM HOCl led to a net 12-fold increase after 23 h, whereas exposure to 25 µM HOCl gave a 2-fold increase within the same time frame (data not shown).
Fig. 6.
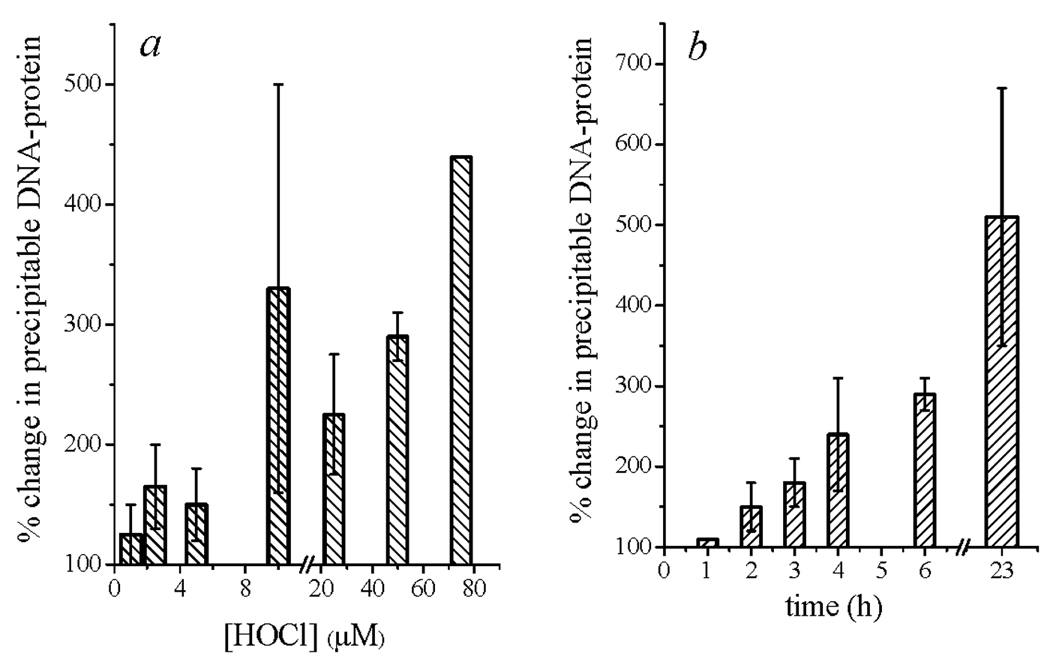
HOCl-induced DNA-protein cross-linking in E. coli 11775. Conditions: ~1×108 cells/mL incubated 6 h in PBS at 37 °C with the indicated concentrations of HOCl (panel a) or incubated with 50 µM HOCl for the indicated times (panel b), after which the protein was precipitated and analyzed for DNA content as described in Experimental Procedures. Under these conditions, LD90 ~15 µM. Error limits are given as the average deviation from the mean value of 2–3 experiments.
Intracellular formation of 5-chlorouracil
5-Chlorouracil has been shown to be a stable marker for intracellular chlorination of cytosine in DNA, as well as the pyrimidines, cytosine and uracil, and the corresponding deoxyribose nucleosides [46,55,56]. A useful protocol for quantitation of 5-chlorocytosine in DNA as 5-chlorouracil has been developed that entails sequential digestions by nucleases to give the free nucleotides, dephosphorylation with alkaline phosphatase to the corresponding nucleosides, and release of the free base pyrimidines with thymidine phosphorylase [46]. During these digestions, the relatively unstable 5-chlorocytosine is deaminated to 5-chlorouracil. The free bases are then isolated by extraction and, following derivatization, analyzed by mass spectrometry. One should note that in vitro assays of reactions of DNA with HOCl have shown that, among the nuclear base moieties, cytosine was by far the preferred target site, forming ~30-fold more monochlorinated product than adenine, the next reactive base [47]. Consequently, the measured 5-chlorouracil content can be taken as a good approximation to the total extent of reaction of HOCl with bacterial DNA.
We have adapted these methods to probe the extent of cytosine chlorination in DNA isolated from HOCl-exposed E. coli. In these studies, nucleotides obtained from the soluble extracted DNA and precipitable DNA-protein complexes were combined for analysis so that the total isolable DNA pool was examined. The relative amounts of 5-chlorouracil measured for stationary-phase E. coli 11775 exposed to 1–75 µM HOCl for 6 h are shown in Fig. 7. The large uncertainties in the plot reflect the low levels of isolated compounds, which varied widely in individual experiments. However, for each run, the amount of 5-chlorouracil detected in HOCl-treated bacteria was always greater than that measured in the corresponding HOCl-negative controls. Two additional trends are evident from the data. First, levels of cytosine chlorination recorded at sublethal doses of HOCl were comparable to those observed when cells were exposed to 2–6 times LD90; second, the chlorouracil yield appeared to pass through a minimum at LD90 (~12 µM). Additional studies using ~108 cells/mL E. coli 11775 carrying the pETBlue plasmid consistently showed a small elevation of plasmid 5-chlorouracil (averaging ~1.8-fold) when exposed to 1–50 µM HOCl (data not shown).
Fig. 7.
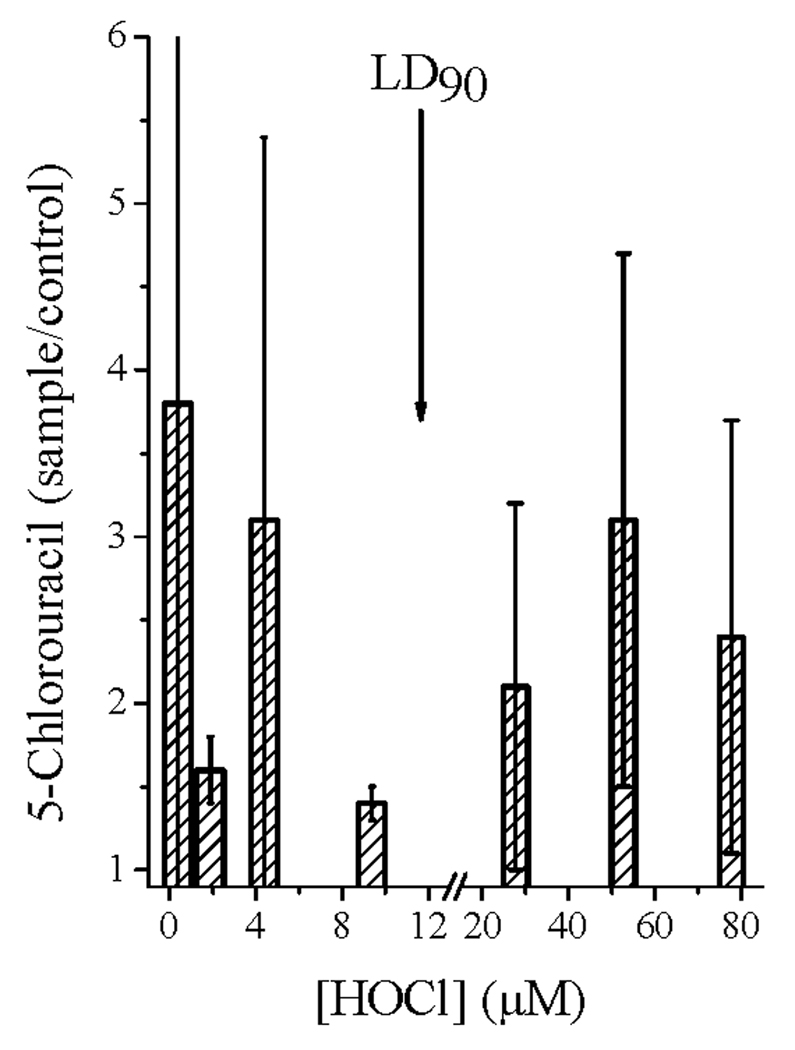
5-Chlorouracil isolated from genomic DNA in HOCl-treated E. coli 11775. Conditions: ~8×107 cells/mL incubated 6 h in PBS at 37 °C with the indicated concentrations of HOCl, after which the DNA was isolated and analyzed mass spectrometrically for 5-chlorouracil content as described in Experimental Procedures. The arrow indicates the HOCl concentration corresponding to LD90 (12 µM). Data are averages of 3–6 experiments; error limits are average deviations from the mean values.
DNA chlorination within neutrophil phagosomes
The persistent accumulation of 5-chlorouracil above background levels in E. coli suggested that these organisms might be used as probes for microbial chlorination within activated neutrophils. Comparisons were therefore made of the 5-chlorouracil content in the DNA of phagocytosed E. coli isolated from fully competent neutrophils with those of E. coli from neutrophils whose MPO had been inhibited by addition of 10 mM sodium azide to the medium. At these concentration levels, azide ion effectively blocks HOCl production within the neutrophil, but does not otherwise impair respiratory activation or phagocytosis [3,20]. No 5-chlorouracil could be detected in the bacterial plasmid DNA isolated from either MPO-competent or MPO-inhibited neutrophils. This result is consistent with extensive earlier studies indicating that phagocytosis of plasmid-bearing bacterial minicells caused no detectable physical or mutagenic damage to the plasmid [57]. However, the 5-chlorouracil content of bacterial genomic DNA isolated from MPO-competent neutrophils was occasionally found to be slightly higher than that from the MPO-deficient controls. Specifically, in a total of four experiments using different individual donors, the ratios of mass spectral peak intensities were 1.7 ± 0.1, 1.5 ± 0.2, 1.0 ± 0.2, and 0.75 ± 0.1. The 5-chlorouracil content of neutrophil DNA following phagocytosis was also determined in one experiment, for which the ratio of normal to azide-inhibited cells was found to be 1.4 ± 0.4. From these data, it seems evident that mass spectrometric determination of nuclear base chlorination cannot be used as a reliable index of intraphagosomal chlorination.
Discussion
DNA strand cleavage by HOCl
In general, HOCl is unreactive toward simple sugars [17], as well as polysaccharides within more complex biopolymers [58]. This lack of reactivity is also reflected in minimal damage to the polyphosphoribose backbone of plasmid DNA observed upon exposure to high concentration levels of HOCl (Fig. 1). This behavior contrasts dramatically with the extensive strand cleavage observed with ONOOH at comparable concentrations (Fig. 2), as has been extensively documented for this oxidant [59], as well as other reactive oxygen species [47,48]. Consistent with this chemical behavior, it has also been noted that HOCl does not induce detectable strand breaks in the genomic DNA of phagocytes [60–62]. A plausible explanation for these differing reactivities is that, whereas HOCl functions primarily as a two-electron oxidant [18], ONOOH rapidly reacts with CO2 in physiological media to form the one-electron oxidants, CO3•− and NO2• [43,49,50]. DNA strand scission occurs primarily by either H-atom abstraction from the deoxyribose unit [63] or reactions initiated by one-electron oxidation of nuclear bases [59]. HOCl is incapable of H-atom abstraction and oxidation of nuclear bases by HOCl involves a one-to-two electron redox noncomplementarity that imposes a high energetic barrier for this reaction. Specifically, one electron reduction of HOCl must generate either OH• or Cl• radicals as products; consequently, this half-reaction is energetically highly unfavorable. In contrast, no such contribution to the activation barrier exists for one-electron oxidants, including the reactive oxygen species that have been shown to effectively cleave the polynucleotide chain [47].
The partial protection toward ONOOH afforded by chelating ligands that inhibit Fenton catalysis was unanticipated because, unlike H2O2, ONOOH undergoes spontaneous homolysis of its O-O bond to generate OH• in a reaction that is metal ion-independent [49,64]. One possibility is that the ligands simply scavenged the OH• generated by ONOOH decay by preferentially reacting with it. However, the high incidence of double-strand breaks suggests the alternative possibility that ONOOH might function as a classical peroxide in Fenton reactions, particularly when catalyzed by copper ions. Double-strand breaks are characteristic of Fenton reactions [65] and thought to arise from “site-specific” reactions directed by coordination of the metal ion catalyst to DNA [66]. Hypochlorous acid has been proposed to react analogously to H2O2 in iron-mediated Fenton reactions [67], that is, to generate OH• or hypervalent one-electron iron oxidants. These observations raise the possibility that the single-strand breaks observed in the pETBlue plasmid DNA at very high HOCl concentrations (Fig. 1) were caused by similar reactions catalyzed by adventitious metal ions bound to the DNA.
The extensive cleavage of genomic DNA observed when E. coli were exposed to relatively high concentration levels of HOCl (~5×LD90) contrasts starkly with the in vitro results obtained with purified plasmid. There is no reason to suspect major differences in the reactivity of plasmid and genomic deoxyribose units toward HOCl; consequently, the reactivity differences most likely reflect altered metabolic capabilities of the bacterial cells induced by reaction with HOCl. One manifestation of these changes is massive, irreversible hydrolysis of ATP phosphoanhydride bonds that accompanies destruction of chemiosmotically coupled energy transduction on the bacterial plasma membrane; these changes coincide with cellular death [18,30]. One expected consequence of this damage would be loss of ATP-dependent DNA repair processes which, in turn, might account for DNA strand cleavage. For example, constitutive DNA N-glycosylases and AP endonucleases of the base excision pathways [68] might still cleave DNA at sites of nuclear base damage but, in the absence of ATP, DNA polymerases and DNA ligases would be unable to rebuild the cleaved polymer chain. Cellular death is also accompanied by inhibition of membrane-localized proteins required for de novo DNA synthesis [8,27,29].
DNA-protein cross-linking
HOCl-initiated formation of covalent DNA-protein [45] and protein-protein adducts [9,69)] has been demonstrated for several model systems, as has similar reactions involving covalent modification of proteins by biologically relevant low molecular weight compounds [70,71]. This type of aggregation is not unique to HOCl, but is also commonly seen in cells and solutions of biopolymers exposed to reactive oxygen species [72–74]. The mechanism of HOCl-elicited cross-linking is unknown, but could involve either radical-radical coupling [9,75] or Schiff base formation [69,71,76,77] involving reactive intermediates [70] formed upon decomposition of HOCl-generated chloramines or chloramides. Because HOCl acts primarily as a two-electron oxidant, it seems unlikely that cross-linking would be initiated by one-electron oxidation of nucleobases, as has recently been demonstrated for DNA-histone cross-linking [78]. From the very slow rate of accumulation of the DNA-protein aggregates (Fig. 6b), it is clear that, whatever their nature, these reactions must proceed via the intermediacy of long-lived secondary species. DNA-protein cross-linking is also very slow in model protein-oligonucleotide complexes that have been exposed to either HOCl or the MPO-H2O2-Cl− enzymatic system, typically requiring several hours for completion [45].
Pyrimidine chlorination
The extensive cleavage of polynucleotide chains and lower yields of recoverable DNA-protein complexes observed at HOCl levels above ~5×LD90 are probably interrelated phenomena which, as discussed above, imply that the nuclear bases have been extensively damaged. Halogenation of free nuclear bases to stable products upon exposure to chemical or enzymatic halogenating systems, as well as activated phagocytes containing myeloperoxidase or eosinophil peroxidase [46,55,56,79–82] is well established, and chlorination of cytosine in duplex DNA by HOCl has been directly demonstrated [83]. However, quantitative comparisons suggest that halogenation of cytosine within polynucleotides is less efficient than halogenation of free nuclear bases [46,56,81], so that the dominant pathways for incorporation of halogenated bases into DNA and RNA may involve cellular uptake and enzymatic modification of the monomeric precursors, rather than direct reaction with HOCl [46,80,82]. Our results appear consistent with this overall view. Specifically, the surprisingly large amount of 5-chlorouracil found in genomic DNA at sub-lethal doses of HOCl could arise by enzymatic incorporation during DNA biosynthesis or repair. This capability is then lost at ~LD90, corresponding to a minimal yield of 5-chlorouracil (Fig. 7); further increases in chlorinated product at higher doses then involve direct reaction with DNA. The abrupt onset of fragmentation (Fig. 5) is also characteristic of HOCl-inflicted damage to other biomolecules in cells and is thought to arise because HOCl is highly selective for its targets [11,18]. In this case, the onset may occur when all biological targets more reactive than the nuclear bases on DNA have been consumed, so that these now are among the most reactive remaining target sites.
Potential for development of bacterial DNA as an intraphagosomal probe for HOCl
As outlined in the Introduction, each phagocytosed bacterium is exposed to 108–109 MPO-generated HOCl molecules, the lower and upper limits being set by the actual amount of chlorine trapped by endogenous probes and the amount of O2 consumed in stimulated respiration, respectively. This level of exposure within the phagosome is equivalent to bolus addition of between 17 and 170 µM HOCl to 1×108 E. coli/mL in suspension. The measured LD90 of 108 E. coli/mL is ~15 µM, so that the dose levels used in the in vitro experiments were equivalent to those generated within the phagosome. The LD90 for other cell densities of bacteria scales proportionately with density.
Of the various reactions investigated, perhaps the most useful for adaptation as a probe of direct intraphagosomal chlorination is DNA-protein cross-linking. This reaction was relatively sensitive to exposure of E. coli to HOCl, particularly when the reaction was allowed to develop over an extended post-exposure time period (Fig. 6). Although this reaction is not selective for HOCl in the sense that numerous reactive oxygen species also induce DNA-protein cross-linking [54,72–74], its kinetics, which appears to involve very slowly reacting HOCl-derived intermediates, may provide a basis for discrimination between HOCl and other oxidants as the source. An additional possibility is presented by the extensive fragmentation of genomic DNA observed at dose levels that were 3–4×LD90, although the origins of these reactions must first be identified.
Detection of elevated levels of 5-chlorouracil in DNA is not diagnostic of direct intraphagosomal reactions of HOCl or chloramines with bacteria. As previously discussed, the relatively high level of incorporation into bacterial genomic DNA observed at sub-lethal doses of HOCl (Fig. 7) renders it likely that the low chlorination enhancement measured for DNA from neutrophils with active MPO also arises by precursor incorporation [46]. In this case, the location of the chlorination reaction, that is, phagosomal fluid versus bacterial cytosol, cannot be determined. Consequently, one cannot use this reaction to probe the extent of direct reaction of the bacterium with MPO-generated HOCl.
The exceptionally high HOCl exposure levels required to nick plasmid DNA (Fig. 1), which is equivalent to ≥40×LD90, almost certainly cannot be achieved within the neutrophil phagosome. Simple chloramines, which react analogously to HOCl [18] are expected to be major secondary products [11,84,85], were found to be completely unreactive. Further, exposure of the plasmid-bearing E. coli to HOCl gave no indication of strand cleavage, even at concentration levels that were far in excess of that required to kill the cells (Fig. 3). Thus, it is highly unlikely that simple nicking analyses of plasmid-bearing bacteria could be used as probes for intraphagosomal HOCl formation. Loss of intracellular plasmid coincided with the onset of genomic DNA fragmentation (Fig. 5b), consistent with more extensive fragmentation and/or formation of DNA-protein complexes. Thus, the two forms of DNA appear to react by common mechanisms.
Supplementary Material
Acknowledgments
The authors are grateful to Michael J. Smerdon and Angela Hintz (Washington State University), Qing Jiang (Purdue University), Jay Heinecke (University of Washington Medical School) and Henry Rosen (University of Washington Medical School) for helpful discussions, and to Wei Wang and William F. Siems (Washington State University) for assistance in agarose gel and mass spectrometric analyses, respectively. This research was supported by a grant (AI-15834) from the National Institute of Allergy and Infectious Diseases.
Appendix A. Supplementary data
Agarose gels of plasmid DNA exposed to ONOOH under various medium conditions can be found in the online version at doi --.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: BCS, bathocuproine sulfonate; GFP, green fluorescent protein; HBSS, Hanks balanced salt solution; LB, Luria broth; MPO, myeloperoxidase; PBS, phosphate-buffered saline; PMSF, phenylmethanesulfonyl fluoride.
Based upon estimates from electron micrographs [14], the average diameter of a neutrophil phagosome is ~1.5 µm. If one assumes spherical geometry, the corresponding volume of a phagosome containing a 1.0 µm diameter particle is ~1.2×10−12 mL; ~108 chlorine atoms trapped by the particle corresponds to ~1.7×10−13 mmols Cl−, requiring an apparent intraphagosomal concentration of ~0.14 M. The measured concentration of Cl− occluded within the phagosomes of human neutrophils scales linearly with the extracellular concentration [15]; at the concentration levels present in serum, the levels extrapolated from these data would be ~0.1 M.
A pertinent example is methionine, which is rapidly oxidized by HOCl to give as products methionine sulfoxide [22] and dehydromethionine [21], a cyclized compound containing a covalent bond between the methionyl sulfur and amido nitrogen atoms. These reactions are thought to proceed via intermediary formation of a chlorosulfonium cation [23], which subsequently undergoes nucleophilic attack by either solvent or the α-amino group with release of chloride, as illustrated below:

Methionine sulfoxide has recently received attention as a potential biomarker of intraphagosomal bactericidal reactions involving neutrophil-generated HOCl [24].
Carbon dioxide in the bicarbonate buffer forms an adduct with ONOO− that rapidly decomposes via O-O bond homolysis to carbonate radical (CO3•−) and nitrogen dioxide (NO2•) [43,49,50]. Unlike OH•, carbonate radical does not abstract H atoms, but selectively oxidizes guanine bases, forming primarily spiroiminodihydantoins and intrastrand cross-linked base pairs [51–53]; NO2• is unreactive toward polynucleotides. Radical recombination leads to net isomerization of ONOO− to NO3− with regeneration of CO2. Since NO3− is unreactive under the in vitro experimental conditions, this CO2− catalyzed reaction effectively protects the DNA from oxidative damage.
References
- 1.Klebanoff SJ. In: Inflammation: Basic Principles and Chemical Correlates. Gallin JI, Goldstein IM, Snyderman R, editors. Vol. 988. New York: Raven Press; pp. 391–444. [Google Scholar]
- 2.Hampton MB, Kettle AJ, Winterbourn CC. Infect. Immun. 1996;64:3512–3517. doi: 10.1128/iai.64.9.3512-3517.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Q, Griffin DA, Barofsky DF, Hurst JK. Chem. Res. Toxicol. 1997;10:1080–1089. doi: 10.1021/tx9700984. [DOI] [PubMed] [Google Scholar]
- 4.Hirche TO, Gaut JP, Heinecke JW, Belaaouaj A. J. Immunol. 2005;174:1557–1565. doi: 10.4049/jimmunol.174.3.1557. [DOI] [PubMed] [Google Scholar]
- 5.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 6.Lehrer RI. Nat. Rev. Microbiol. 2004;2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- 7.Elsbach P, Weiss J. Rev. Infect. Dis. 1983;5:843–853. doi: 10.1093/clinids/5.5.843. [DOI] [PubMed] [Google Scholar]
- 8.Rosen H, Michel BR. Infect. Immun. 1997;65:4173–4178. doi: 10.1128/iai.65.10.4173-4178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers MCM, Winterbourn CC. Arch. Biochem. Biophys. 1991;285:53–59. doi: 10.1016/0003-9861(91)90327-f. [DOI] [PubMed] [Google Scholar]
- 10.Weiss J, Kao L, Victor M, Elsbach P. Infect. Immun. 1987;55:2142–2147. doi: 10.1128/iai.55.9.2142-2147.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winterbourn CC, Hampton MB, Livesy JH, Kettle AJ. J. Biol. Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 12.Painter RG, Valentine VG, Lanson NA, Jr, Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G, Wang G. Biochemistry. 2006;45:10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G. J. Leuk. Biol. 2008;83:1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozenberg-Arska M, Salters MEC, van Strijp JAG, Geuze JJ, Verhoef J. Infect. Immun. 1985;50:852–859. doi: 10.1128/iai.50.3.852-859.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Painter RG, Wang G. Anal. Chem. 2006;78:3133–3137. doi: 10.1021/ac0521706. [DOI] [PubMed] [Google Scholar]
- 16.Weiss SJ, Klein R, Slivka R, Wei M. J. Clin. Invest. 1982;70:598–607. doi: 10.1172/JCI110652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pattison DI, Davies MJ. Chem. Res. Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 18.Hurst JK, Barrette WC., Jr CRC Crit. Rev. Biochem. Mol. Biol. 1989;24:271–328. doi: 10.3109/10409238909082555. [DOI] [PubMed] [Google Scholar]
- 19.Hampton MB, Kettle AJ, Winterbourn CC. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- 20.Hurst JK, Albrich JM, Green TR, Rosen H, Klebanoff S. J. Biol. Chem. 1984;259:4812–4821. [PubMed] [Google Scholar]
- 21.Peskin AV, Winterbourn CC. Free Radical Biol. Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 22.Peskin AV, Turner R, Maghzad GJ, Winterbourn CC, Kettle AJ. Biochemistry. 2009;48 doi: 10.1021/bi901266w. DOI: 10.1021/bi901266w. [DOI] [PubMed] [Google Scholar]
- 23.Armesto XL, Canle ML, Fernandez MI, Garcia MV, Santaballa JA. Tetrahedron. 56:1103–1109. [Google Scholar]
- 24.Rosen H, Klebanoff SJ, Wang Y, Brot N, Heinecke JW, Fu X. Proc. Natl. Acad. Sci. USA. 2009;106 doi: 10.1073/pnas.0909464106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman ALP, Hampton MB, Senthilmohan R, Winterbourn CC, Kettle AJ. J. Biol. Chem. 2002;277:9757–9762. doi: 10.1074/jbc.M106134200. [DOI] [PubMed] [Google Scholar]
- 26.Rosen H, Crowley JR, Heinecke JW. J. Biol. Chem. 2002;277:30463–30468. doi: 10.1074/jbc.M202331200. [DOI] [PubMed] [Google Scholar]
- 27.McKenna SM, Davies KJA. Biochem. J. 1988;254:685–692. doi: 10.1042/bj2540685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King DA, Hannum DM, Qi J-S, Hurst JK. Arch. Biochem. Biophys. 2004;423:170–181. doi: 10.1016/j.abb.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Rosen H, Orman J, Rakita RM, Michel BR, VanDevanter DR. Proc. Natl. Acad. Sci. USA. 1990;87:10048–10052. doi: 10.1073/pnas.87.24.10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurst JK, Lymar SV. Acc. Chem. Res. 1999;32:520–528. [Google Scholar]
- 31.Selvaraj RJ, Zgliczynski JM, Paul BB, Sbarra AJ. J. Infect. Dis. 1978;137:481–485. doi: 10.1093/infdis/137.4.481. [DOI] [PubMed] [Google Scholar]
- 32.Ramsey PG, Martin T, Chi E, Klebanoff SJ. J. Immunol. 1982;128:415–420. [PubMed] [Google Scholar]
- 33.Miyasaki KT, Zambon JJ, Jones CA, Wilson ME. Infect. Immun. 1987;55:1029–1036. doi: 10.1128/iai.55.5.1029-1036.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hampton MB, Vissers MC, Winterbourn CC. J. Leuk. Biol. 1994;55:147–152. doi: 10.1002/jlb.55.2.147. [DOI] [PubMed] [Google Scholar]
- 35.Staudinger BJ, Oberdoerster MA, Lewis PJ, Rosen H. J. Clin. Invest. 2002;110:1151–1163. doi: 10.1172/JCI15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dukan S, Touati D+ J. Bacteriol. 1996;178:6145–6150. doi: 10.1128/jb.178.21.6145-6150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palazzolo AM, Suquet C, Konkel ME, Hurst JK. Biochemistry. 2005;44:6910–6919. doi: 10.1021/bi047342s. [DOI] [PubMed] [Google Scholar]
- 38.Morris JC. J. Phys. Chem. 1966;70:3798–3805. [Google Scholar]
- 39.Thomas EL, Grisham MB, Jefferson MM. Methods Enzymol. 1986;132:569–586. doi: 10.1016/s0076-6879(86)32042-1. [DOI] [PubMed] [Google Scholar]
- 40.Saha A, Goldstein S, Cabelli D, Czapski G. Free Radic. Biol. Med. 1998;24:653–659. doi: 10.1016/s0891-5849(97)00365-1. [DOI] [PubMed] [Google Scholar]
- 41.Hughes MN, Nicklin HG. J. Chem. Soc. A. 1968:450–452. [Google Scholar]
- 42.Rosen H, Michel BR, Chait A. J. Immunol. Methods. 1991;144:117–125. doi: 10.1016/0022-1759(91)90237-a. [DOI] [PubMed] [Google Scholar]
- 43.Lymar SV, Hurst JK. J. Am. Chem. Soc. 1995;117:8867–8868. [Google Scholar]
- 44.Zhitkovich A, Costa M. Carcinogenesis. 1992;13:1485–1489. doi: 10.1093/carcin/13.8.1485. [DOI] [PubMed] [Google Scholar]
- 45.Kulcharyk PA, Heinecke JW. Biochemistry. 2001;40:3648–3656. doi: 10.1021/bi001962l. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Q, Blount BC, Ames BN. J. Biol. Chem. 2003;278:32834–32840. doi: 10.1074/jbc.M304021200. [DOI] [PubMed] [Google Scholar]
- 47.Kennedy LJ, Moore K, Jr, Caulfield JL, Tannenbaum SR, Dedon PC. Chem. Res. Toxicol. 1997;10:386–392. doi: 10.1021/tx960102w. [DOI] [PubMed] [Google Scholar]
- 48.Szabó C, Ohshima H. Nitric Oxide. 1997;1:373–385. doi: 10.1006/niox.1997.0143. [DOI] [PubMed] [Google Scholar]
- 49.Goldstein S, Lind J, Merenyi G. Chem. Rev. 2005;105:2457–2470. doi: 10.1021/cr0307087. [DOI] [PubMed] [Google Scholar]
- 50.Bonini M, Radi R, Ferrer-Sueta G, Da AM, Ferreira C, Augusto O. J. Biol. Chem. 1999;274:10802–10806. doi: 10.1074/jbc.274.16.10802. [DOI] [PubMed] [Google Scholar]
- 51.Joffe A, Geacintov NE, Shafirovich V. Chem. Res. Toxicol. 2003;16:1528–1538. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 52.Crean C, Uvaydov Y, Geacintov NE, Shafirovich V. Nucleic Acids Res. 2008;36:742–755. doi: 10.1093/nar/gkm1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crean C, Geacintov NE, Shafirovich V. Free Radical Biol. Med. 2008;45:1125–1134. doi: 10.1016/j.freeradbiomed.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Angelov D, VYu Stefanovsky, Dimitrov SI, Russanova VR, Keskinova E, Pashev IG. Nucleic Acids Res. 1988;16:4525–4538. doi: 10.1093/nar/16.10.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henderson JP, Byun F, Takeshita J, Heinecke JW. J. Biol. Chem. 2003;278:23522–23528. doi: 10.1074/jbc.M303928200. [DOI] [PubMed] [Google Scholar]
- 56.Masuda M, Suzuki T, Friesen MD, Ravant J-L, Cadet J, Pignatelli B, Nishino H, Ohshima H. J. Biol. Chem. 2001;276:40486–40496. doi: 10.1074/jbc.M102700200. [DOI] [PubMed] [Google Scholar]
- 57.Fox HB, De Togni P, McMahon G, Levy SB, Robinson JS, Karnovsky MJ, Babior BM. Blood. 1987;69:1394–1400. [PubMed] [Google Scholar]
- 58.Hawkins CL, Davies MJ. Free Radic. Biol. Med. 1998;24:1396–1410. doi: 10.1016/s0891-5849(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 59.Burrows CJ, Muller JG. Chem. Rev. 1998;98:1109–1151. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 60.Schraufstätter IU, Browne K, Harris A, Hyslop PA, Jackson JA, Quehenberger O, Cochrane CG. J. Clin. Invest. 1990;85:554–562. doi: 10.1172/JCI114472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schacter E, Beecham EJ, Covey JM, Kohn KW, Potter M. Carcinogenesis. 1988;9:2297–2304. doi: 10.1093/carcin/9.12.2297. [DOI] [PubMed] [Google Scholar]
- 62.Van Rensburg CEJ, Van Staden AM, Anderson R, Van Rensburg EJ. Mutation Research. 1992;265:255–261. doi: 10.1016/0027-5107(92)90054-6. [DOI] [PubMed] [Google Scholar]
- 63.Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 64.Lymar SV, Khairutdinov RF, Hurst JK. Inorg. Chem. 2003;42:5259–5266. doi: 10.1021/ic030104l. [DOI] [PubMed] [Google Scholar]
- 65.Kohen R, Szyf M, Chevion M. Anal. Biochem. 1986;154:485–491. doi: 10.1016/0003-2697(86)90019-9. [DOI] [PubMed] [Google Scholar]
- 66.Chevion M. Free Radic. Biol. Med. 1988;5:27–37. doi: 10.1016/0891-5849(88)90059-7. [DOI] [PubMed] [Google Scholar]
- 67.Folkes LK, Candeias LP, Wardman P. Arch. Biochem. Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 68.Rupp WD. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt FC, editor. Washington, D. C: ASM Press; 1996. pp. 2277–2294. [Google Scholar]
- 69.Hazell LJ, van den Berg JJM, Stocker R. Biochem. J. 1994;302:297–304. doi: 10.1042/bj3020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas EL, Jefferson MM, Grisham MB. Biochemistry. 1982;21:6299–6308. doi: 10.1021/bi00267a040. [DOI] [PubMed] [Google Scholar]
- 71.Hazen SL, Gaut JP, Fong FH, Crowley JR, d’Avignon A, Heinecke JW. J. Biol. Chem. 1997;272:16990–16998. doi: 10.1074/jbc.272.27.16990. [DOI] [PubMed] [Google Scholar]
- 72.Gebicki S, Gebicki JM. Biochem. J. 1999;338:629–636. [PMC free article] [PubMed] [Google Scholar]
- 73.Dizdaroglu M. Mutation Research. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 74.Oleinick NL, Chiu S-M, Ramakrishnan N, Xue L-Y. Br. J. Cancer. 1987;55 Suppl. VIII:135–140. [PMC free article] [PubMed] [Google Scholar]
- 75.Hawkins CL, Davies MJ. Biochem. J. 1998;332:617–625. doi: 10.1042/bj3320617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Murthi KK, Friedman LR, Oleinick NL, Salomon RG. Biochemistry. 1993;32:4090–4097. doi: 10.1021/bi00066a034. [DOI] [PubMed] [Google Scholar]
- 77.Clark RA, Szot S, Williams MA, Kagan HM. Biochem. Biophys. Res. Commun. 1986;135:451–457. doi: 10.1016/0006-291x(86)90015-x. [DOI] [PubMed] [Google Scholar]
- 78.Bjorklund CC, Davis WB. Biochemistry. 2007;46:10745–10755. doi: 10.1021/bi700475b. [DOI] [PubMed] [Google Scholar]
- 79.Takeshita J, Byun J, Nhan TQ, Pritchard DK, Pennathur S, Schwarz SM, Chait A, Heinecke JW. J. Biol. Chem. 2006;281:3096–3104. doi: 10.1074/jbc.M509236200. [DOI] [PubMed] [Google Scholar]
- 80.Henderson JP, Byun J, Mueller DM, Heinecke JW. Biochemistry. 2001;40:2052–2059. doi: 10.1021/bi002015f. [DOI] [PubMed] [Google Scholar]
- 81.Henderson JP, Byun J, Heinecke JW. J. Biol. Chem. 1999;274:33440–33448. doi: 10.1074/jbc.274.47.33440. [DOI] [PubMed] [Google Scholar]
- 82.Henderson JP, Byun J, Williams MV, McCormick ML, Parks WC, Ridnour LA, Heinecke JW. Proc. Natl. Acad. Sci. USA. 2001;98:1631–1636. doi: 10.1073/pnas.041146998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang JI, Sowers LC. Chem. Res. Toxicol. 2008;21:1211–1218. doi: 10.1021/tx800037h. [DOI] [PubMed] [Google Scholar]
- 84.Thomas EL, Grisham MB, Jefferson MM. Methods Enzymol. 1986;132:585–593. doi: 10.1016/s0076-6879(86)32043-3. [DOI] [PubMed] [Google Scholar]
- 85.Grisham MB, Jefferson MM, Melton DF, Thomas EL. J. Biol. Chem. 1984;259:10404–10413. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.