

Abstract
The development of a versatile method for the preparation of 5 to 8-membered hydroxy lactams, involving the iodine(III)-mediated oxamidation of unsaturated O-alkyl hydroxamates is described. This transformation, which is believed to proceed through the intermediacy of singlet nitrenium and bicyclic N-acyl-N-alkoxyaziridinium ions, is in most of the 22 cases examined, both stereospecific and/or highly regioselective.
The reaction of singlet nitrenium ions1 with alkenes has long been known to proceed in stereospecific fashion to generate aziridinium ions.2 However, in comparison to the reaction of carbenes, their isoelectronic congeners, this process has until recently, received scant attention,3–5 despite the growing importance of both aziridines and aziridinium ions in organic synthesis.6 While this reflects the harsh conditions traditionally required for formation of these reactive electrophiles, the iodine(III)-mediated oxidation of O-alkyl hydroxamates 1 offers convenient access to O-stabilized nitrenium ions (7 in Scheme 1).1b Having successfully employed the cyclization of these species with arenes as a route to azaspiranes,7 we became intrigued by the possibility that intramolecular addition of nitrenium ions generated from unsaturated hydroxamates 1 would not only yield bicyclic N-acyl-N-alkoxyaziridinium ions 4, but through concerted ring opening of these products, offer a means to accomplish cyclofunctionalization. Herein, we report the successful development of this reaction as a highly versatile method for the preparation of 5 to 8-membered hydroxy lactams 3.8,9
Scheme 1.

Our preliminary studies focused on substrate 1a which, upon treatment with phenyliodine(III) bis(trifluoroacetate) (PIFA) in CH2Cl2, smoothly underwent cyclization to form anti-addition product 2a in high yield (eq 1). In light of the lability of this ester, a methanol-ammonia quench was employed to remove the trifluoroacetate group and provided α-hydroxyalkyl lactam 3a as a single diastereomer in excellent yield. Importantly, we have found that addition of trifluoroacetic acid to the oxamidation reaction significantly accelerates this process and, in most cases, improves its efficiency (Table 1). That acid catalysis plays a significant role in the formation of 2a is also apparent from the inhibitory effect of acid scavengers and the failure of both PhI(OAc)2 and Pb(OAc)4 to mediate cyclofunctionalization.10,11
Table 1.
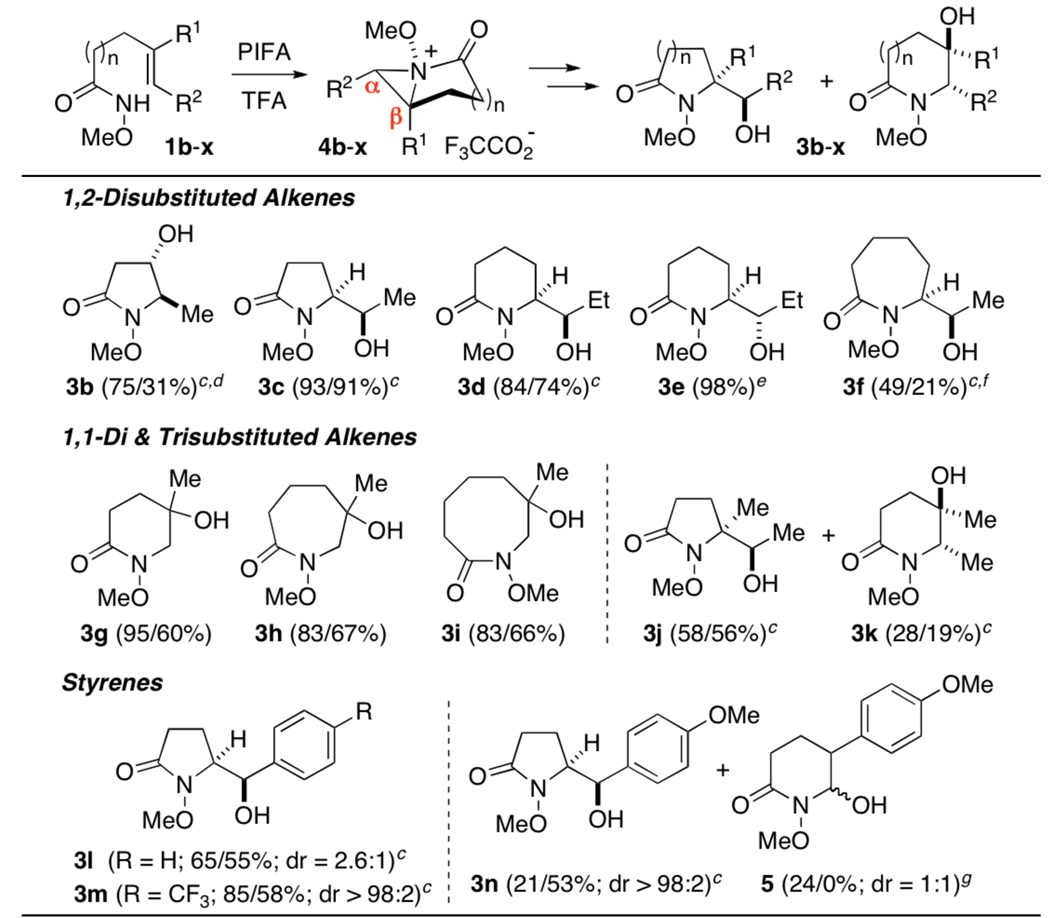 |
Conditions: 1, PIFA (1.2 equiv), TFA (1.0/0.0 equiv), CH2Cl2 (0.15 M), 0 °C; then NH3-MeOH, 20 min.
Isolated yields of oxamidations conducted with and w/o TFA, after purification by flash chromatography.
Generated from E-1.
Workup via hydrazinolysis.
Generated from Z-1.
An azocan-2-one, resulting from β-opening, was also isolated as a single diastereomer in 18/10% yield.
See supporting information for the possible origins of rearrangement product 5.
 |
(1) |
Employing these optimal conditions, a range of unsaturated hydroxamates was screened. As is apparent from Table 1, this reaction is characterized by broad substrate scope and, in most cases, is both stereospecific and highly regioselective. In the case of 1,2-disubstituted alkenes, concerted ion pair collapse of bicyclic aziridinium ions 4c–e occurs solely at the less encumbered α position to yield α-hydroxyalkyl lactams, while bicyclo[2.1.0] ion 4b (n = 0) undergoes ring expansion to form 3b. The isomer outcome of this process is predictable, and is highlighted by the formation of diastereomers 3d and 3e from the corresponding E- and Z-alkenes. Aziridinium ions generated from 1,1-disustituted substrates 1g–i, in contrast, undergo opening at the β position (4, R1 ≠ H), suggesting that substitution occurs with significant charge separation.12 Although the cyclization of trisubstituted alkene 1j proceeded with modest regioselectivity, the stereospecificity associated with formation of 3j and 3k is remarkable given the highly-substituted nature of the bicyclo[3.1.0] ion (R1/R2 = Me, n = 1) from which these products arise. Non-synchronous ring opening was observed with styrene 1l, which afforded a mixture of oxamidation products. Significantly, the presence of a p-CF3 group in 1m serves to disfavor carbocation formation, which is thought to be the origin of this loss of diastereoselectivity.
Extension of our methodology to cyclic substrates was also successful and provides stereocontrolled entry to a number of functionalized tricyclic systems, including 6-azabicyclo[2.2.2] and [3.2.1]octanes (Table 2). Notwithstanding the modest selectivity observed during the formation of 3q and 3r, which reflects the sterically non-biased nature of the bridged aziridinium ion in this case, the regioselectivity of ring opening can be controlled effectively through introduction of substituents, as in the case of 3s, or via steric effects, both proximal and distal, as observed with 3t–u. The exclusive formation of 3w implies that electron-withdrawing groups have an inhibitory effect on proximal ring opening,13a while 3x is believed to arise through interception of the aziridinium ion by the neighboring exo-C3 acetoxy group:13b selective hydrolysis of the resulting 1,3-dioxenium ion accounts for the 1,2-acetyl migration observed in this case.
Table 2.
Oxamidation of Cyclic O-Alkyl Hydroxamatesa
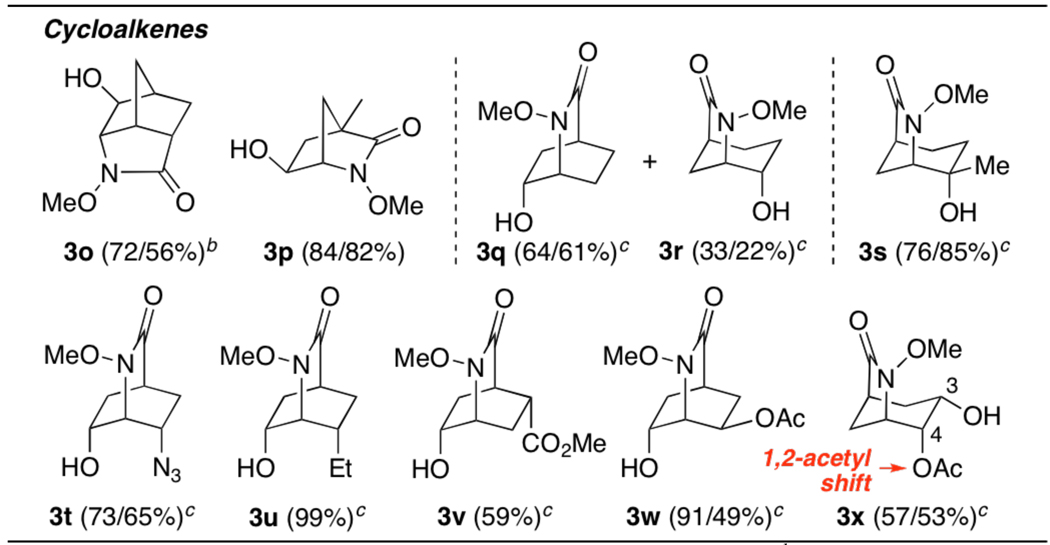 |
Conditions and yields: see Table 1, footnotes a/b.
Ester hydrolysis carried out with aq. NaHCO3.
Oxamidation conducted at 40 °C.
In order to account for our observations, we have developed the mechanistic rationale outlined in Scheme 1. In this case, cyclization begins with ligand substitution of PIFA by 1a to form amido-λ3-iodane 6,14 which undergoes ligand coupling,15 to N,N-bisheteroatom-substituted (anomeric) amide 8,16a or fragmentation to yield singlet nitrenium ion 7. Intramolecular cycloaddition of this electrophile, via transition state 9, would generate 10, and subsequently 2a. Alternatively, 10 may arise from 8, through an SN2-like reaction, which although prohibited in conventional amides, is feasible at the pyramidalized N-center of anomeric amides, as seminal studies by Glover have revealed.16b However, in light of Glover’s observation that these atypical amides also undergo AAl1 acid-catalyzed solvolysis to form nitrenium ions,16c and in consonance with the effect of TFA upon oxamidation, we believe that conversion of 8 to 10 is more likely to proceed via an SN1 process, where rapid and reversible protonation of 8 precedes ionization to 7.17
While the precise location of the N-electrophile on the continuum between 7 and 8 awaits determination, and may prove to be substrate dependant, the absence of syn-oxamidation and lactone products in these reactions implies that cyclization does not occur via addition of PIFA to the double bond and subsequent nucleophilic displacement of an aryl-λ3-iodanyl group.18
Lactam rings are embodied within a wealth of physiologically active natural products and pharmaceutical agents and, as a result, methods that facilitate the preparation of these saturated N-heterocycles are of great importance. In this context, the intramolecular alkene oxamidation method described herein represents a remarkably versatile method to access this important class of targets.19 Mechanistic studies and the application of this chemistry to natural product synthesis are currently underway.
Supplementary Material
Acknowledgment
This work was supported by National Institutes of Health (GM59517).
Footnotes
Supporting Information Available: Characterization, procedures and stereochemical assignments. This material is free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For reviews, see: Falvey DE. In: Reactive Intermediate Chemistry. Moss RA, Platz M, Jones M, editors. Hoboken, N.J.: Wiley-Interscience; 2004. p. 593. Kikugawa Y. Heterocycles. 2009;78:571.
- 2.For a theoretical treatment of this process, see: Hopkinson AC, Lien MH, Csizmadia IG, Yates K. Theor. Chim. Acta. 1980;55:1.
- 3.For the intermolecular reaction of O-stabilized nitrenium ions with alkenes, see: Vedejs E, Sano H. Tetrahedron Lett. 1992;33:3261. Rudchenko VF, Ignatov SM, Kostyanovsky RG. Chem. Commun. 1990:261. Hoffman RV, Christophe NB. J. Org. Chem. 1988;53:4769.
- 4.For the reaction of N-stabilized nitrenium, or diazenium, ions with alkenes, see: Tsukamoto M, Murata K, Sakamoto T, Saito S, Kikugawa Y. Heterocycles. 2008;75:1133. Urry WH, Szecsi P, Ikoku C, Moore DW. J. Am. Chem. Soc. 1964;86:2224.
- 5.For the reaction of aryl nitrenium ions with alkenes, see: Tellitu I, Urrejola A, Serna S, Moreno I, Herrero MT, Domínguez E, SanMartin R, Correa A. Eur. J. Org. Chem. 2007:437. doi: 10.1021/jo062320s. Takeuchi H, Koyama K, Mitani M, Ihara R, Uno T, Okazaki Y, Kai Y, Kasai N. J. Chem. Soc., Perkin Trans. 1. 1985;4:677.
- 6.For a review, see: Hu XE. Tetrahedron. 2004;60:2701.
- 7.(a) Wardrop DJ, Basak A. Org. Lett. 2001;3:1053. doi: 10.1021/ol015626o. [DOI] [PubMed] [Google Scholar]; (b) Wardrop DJ, Zhang WM. Org. Lett. 2001;3:2353. doi: 10.1021/ol0161514. [DOI] [PubMed] [Google Scholar]; (c) Wardrop DJ, Zhang WM, Landrie CL. Tetrahedron Lett. 2004;45:4229. [Google Scholar]; (d) Wardrop DJ, Burge MS. Chem. Commun. 2004:1230. doi: 10.1039/b403081h. [DOI] [PubMed] [Google Scholar]
- 8.Taken in part from the Ph.D. thesis of R.E.F. Chicago, IL: UIC; 2003. [Google Scholar]
- 9.For selected intramolecular alkene oxaminations, see: Donohoe TJ, Callens CKA, Thompson AL. Org. Lett. 2009;11:2305. doi: 10.1021/ol900631y. Fuller PH, Kim J-W, Chemler SR. J. Am. Chem. Soc. 2008;130:17638. doi: 10.1021/ja806585m. Cochran BM, Michael F. Org. Lett. 2008;10:5039. doi: 10.1021/ol8022165. Alexanian EJ, Lee C, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690. doi: 10.1021/ja051406k.
- 10.These reagents mediate hydrazine formation: De Almeida MV, Barton DHR, Bytheway I, Ferreira JA, Hall MB, Liu W, Taylor DK, Thomson L. J. Am. Chem. Soc. 1995;117:4870.
- 11.For the Pb(IV)-mediated formation of bridged aziridines, see: Hu H, Faraldos JA, Coates RM. J. Am. Chem. Soc. 2009;131:11998. doi: 10.1021/ja9044136.
- 12.For the β-opening of bicyclic aziridinium and epoxonium ions, see: Leonard NJ, Dixon DB. J. Org. Chem. 1970;35:3483. Wan S, Gunaydin H, Houk KN, Floreancig PE. J. Am. Chem. Soc. 2007;129:7915. doi: 10.1021/ja0709674.
- 13.(a) Hu XE. Tetrahedron Lett. 2002;43:5315. [Google Scholar]; (b) Williams DR, Osterhout MH, McGill JM. Tetrahedron Lett. 1989;30:1331. [Google Scholar]
- 14.Loudon GM, Radhakrishna AS, Almond MR, Blodgett JK, Boutin RH. J. Org. Chem. 1984;49:4272. [Google Scholar]
- 15.Oae S, Uchida Y. Acc. Chem. Res. 1991;24:202. [Google Scholar]
- 16.(a) Glover SA. Adv. Phys. Org. Chem. 2008;42:35. [Google Scholar]; (b) Glover SA, Rauk A. J. Chem. Soc., Perkin Trans. 2. 2002:1740. [Google Scholar]; (c) Glover SA, Hammond GP. J. Org. Chem. 1998;63:9684. [Google Scholar]
- 17.That the approach of the alkene to the N-center, in this case, may be intimately involved with, and assist in, the departure of the protonated trifluoroacetate group is not discounted.
- 18.See for an example of this process, see: Celik M, Alp C, Coskun B, Gültekin MS, Balci M. Tetrahedron Lett. 2006;47:3659.
- 19.Reductive cleavage of N-methoxy lactams to the corresponding N-H lactams can be accomplished with range of reducing agents, including Mo(CO)6 (ref. 7). For experimental details, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.