

Abstract
It has been long thought that hyper-activation of N-methyl-D-aspartate (NMDA) receptors underlies neurological decline after traumatic brain injury. However, all clinical trials with NMDA receptor antagonists failed. Since NMDA receptors are down-regulated from 4h to 2 weeks after brain injury, activation at 24h, rather than inhibition, of these receptors, was previously shown to be beneficial in mice. Here, we tested the therapeutic window, dose regimen and mechanism of action of the NMDA receptor partial agonist D-cycloserine (DCS) in traumatic brain injury. Male mice were subjected to trauma using a weight-drop model, and administered 10 mg/kg (i.p) DCS or vehicle once (8, 16, 24, or 72h) twice (24 and 48h) or three times (24, 48 and 72h). Functional recovery was assessed for up to 60 days, using a Neurological Severity Score that measures neurobehavioral parameters. In all groups in which treatment was begun at 24 or 72h neurobehavioral function was significantly better than in the vehicle-treated groups. Additional doses, on days 2 and 3 did not further improve recovery. Mice treated at 8h or 16h post injury did not differ from the vehicle-treated controls. Co-administration of the NMDA receptor antagonist MK-801 completely blocked the protective effect of DCS given at 24h. Infarct volume measured by 2,3,5-triphenyltetrazolium chloride staining at 48h or by cresyl violet at 28d was not affected by DCS treatment. Since DCS is used clinically for other indications, the present study offers a novel approach for treating human traumatic brain injury with a therapeutic window of at least 24h.
Keywords: Glutamate, N-methyl-D-aspartate (NMDA) receptor, neuroprotection, head injury
1. INTRODUCTION
In the past few decades, hyperactivation of the NMDA receptors by extracellular excitatory amino acids such as glutamate had been implicated in the cellular events leading to neuronal death and decline in function following traumatic or ischemic brain injury (e.g. Faden et al., 1989; Hayes et al., 1988). Acute increases in extracellular glutamate, detected in both experimental brain trauma models and in human patients, may lead to over-stimulation of glutamate receptors, culminating in neuronal damage (Alessandri et al., 1996; Baker et al., 1993; Brown et al., 1998; Lynch and Dawson 1994, Palmer et al., 1993, Panter and Faden 1992). Agents modulating glutamate transmission were developed and numerous experimental treatments for neurological diseases were based on NMDA receptor antagonists (Tolias et al., 2004, Beauchamp et al., 2008, Kalia et al., 2008), that could theoretically ameliorate the harmful effects of excessive glutamate. However, data obtained from various brain trauma and ischemia models, retinal exposure to glutamate and endotoxin (LPS)-induced neuroinflammation, showed a loss of NMDA receptors, evident a few hours after injury and lasting for 24h or more (Biegon et al., 2004, Biegon et al., 2002, Friedman et al., 2001, Grossman et al., 2003, Miller et al., 1991, Sihver et al., 2001). Loss of NMDA receptors should lead to hypo-activation of these receptors. Indeed, using a mouse model of closed head injury we showed that hyperactivation of the NMDA receptors is short-lived (< 1h) and is followed by profound and long-lasting hypofunction (>7days). Moreover, stimulation of the native receptor using the full agonist NMDA at 24h post injury was actually neuroprotective (Biegon et al., 2004) and so was DCS, the partial agonist of the NMDA receptor-associated glycine site (Temple and Hamm, 1996).
Activity-dependent synaptic processes that modify the strength of hippocampal glutamatergic synapses are known as long-term potentiation (LTP) and are critical to spatial learning and memory. The hippocampus regulates the storage of information and requires activation of the NMDA receptors (Malenka and Nicoll, 1999). We previously tested DCS in the closed head injury model and found that in addition to improved functional recovery, a single dose of DCS 24h after injury significantly restored hippocampal LTP in the CA1 region that was blunted in vehicle-treated mice. In addition, DCS restored the level of brain-derived neurotrophic factor (BDNF), a critical regulator of LTP, which is reduced after closed head injury (Yaka et al., 2007).
The drug DCS, a well characterized chemical agent established in clinical use as an antimicrobial drug (Heifets 1994) in some infectious diseases, is also used as a “cognitive enhancer” in several chronic neurological disorders, including Alzheimer's disease and schizophrenia (Duncan et al. 2004; Heresco-Levy et al., 2002; Laake et al., 2002). Therefore, if found to be beneficial in experimental traumatic brain injury, DCS could readily be translated from the laboratory to the bedside.
The present study was designed to determine: 1) the extent to which early or late administration of DCS could be beneficial following trauma, thus defining its therapeutic window, 2) the possible benefits of multiple doses of DCS following traumatic brain injury, 3) the effect of treatment on early and late lesion volume, and finally 4) the role of the interaction of DCS with the NMDA receptors.
2. MATERIALS AND METHODS
2.1 Animals and drugs
The study was approved by the Institutional Animal Care Committee of The Hebrew University of Jerusalem. Male Sabra mice, 8–12 weeks old, Hebrew University strain used for the study were kept under controlled temperature and light conditions with food and water available continuously. D-cycloserine, DCS, (Sigma, St. Louis, MO, USA) and MK801 (Sigma) were dissolved in saline and doses of 10mg/kg and 1mg/kg, respectively, were given intraperitoneally, (i.p.). The DCS dose was selected based on a dose response study (5,10,30 mg/kg) performed before our previous study (Yaka et al. 2007). Control animals received an identical volume of vehicle (saline).
2.2 Closed head injury model
Experimental closed head injury was induced using a weight-drop device as previously described (Biegon et al., 2004). Briefly, after induction of isoflurane anesthesia, a midline longitudinal scalp incision was made, the skin was retracted and the skull exposed. The left anterior frontal area was identified and a Teflon tipped cone (2mm diameter) was placed 1mm lateral to the midline in the mid-coronal plane. The head was held in place manually and a 75g weight was dropped from a height of 18cm, resulting in a focal injury to the left hemisphere. After trauma, the mice received supporting oxygenation with 95% oxygen for up to 2 min and were returned to their cages.
2.3 Experimental groups
DCS or vehicle was administered to different groups of animals at various time periods post closed head injury. The first set of experiments was designed to determine whether a single administration of DCS at time points earlier or later than 24h would also exert beneficial effect. Thus, four groups of mice were treated with a single dose given at 8 h (n=7) or at 16 h (n=8) for the early time points and at 24 h or 72 h (n=8) for the later post closed head injury time. The second set of experiments was designed to explore the question of multiple dosing. Two groups of mice were treated either twice (n=10), at 24 and 48 h, or three times (n=11), at 24, 48 and 72 h. Control groups received the vehicle (n=14) at the same time points as the drug. The severity of injury was assessed 1h after closed head injury and the neurobehavioral recovery of the mice was followed for 63 or 21 days in the different groups. To investigate the effect of DCS on infarct volume, two groups of mice (n=5/group) treated with vehicle or DCS at 24h post closed head injury were sacrificed 24h later, that is, 48h after injury, and infarct volume was assessed by staining with 2,3,5-triphenyltetrazolium chloride (TTC). To investigate the late effect of DCS treatment on lesion volume, vehicle- and DCS-treated mice were sacrificed at 28 days post injury and brain sections were stained with cresyl violet. To determine whether activation of the NMDA receptor is involved in mediating the neuroprotective effect of DCS, the NMDA antagonist MK-801 was injected (1 mg/kg, i.p) 24 h after injury, alone or along with DCS. Thus, four groups of mice (n=10–12/group) were subjected to a similar severity of injury and 24h later saline (vehicle control), DCS, MK-801 or DCS+MK801 were injected. The functional recovery of these mice was evaluated for up to 28 days.
2.4 Neurobehavioral evaluation
The Neurological Severity Score (NSS; Table 1) assesses the neurobehavioral status of mice based on the presence of some reflexes and the ability to perform various motor tasks, including beam balance, beam walking and spontaneous locomotion (Beni Adani et al., 2001). Scores range from zero in healthy uninjured animals to a maximum of 10, indicating severe neurological dysfunction, with failure in all tasks. The NSS obtained 1h post trauma reflects the initial severity of the injury and is inversely correlated with the neurological outcome (Tsenter et al., 2008). To ensure similar initial severity values in the different groups after closed head injury, mice with similar initial scores were assigned to the experimental (n>8 mice) and the control arms (n>8 mice) of the study. All the groups were evaluated 1h after closed head injury and for up to 63 days. Blinded evaluation of the animals was carried out throughout the experimental periods. The extent of recovery (ΔNSS) was calculated as the difference between the NSS at 1h post closed head injury and at any subsequent time point. Thus, a positive DNSS reflects recovery, 0 reflects no change, and a negative DNSS reflects neurological deterioration.
Table 1.
Neurological Severity Score for head injured mice
| TASK | NSS |
|---|---|
| Presence of mono- or hemiparesis | 1 |
| Inability to walk on a 3cm wide beam | 1 |
| Inability to walk on a 2cm wide beam | 1 |
| Inability to walk on a 1cm wide beam | 1 |
| Inability to balance on a round stick (0.5 cm wide) | 1 |
| Inability to balance on a 1cm wide beam | 1 |
| Failure to exit a 30cm diameter circle (for 2 min) | 1 |
| Inability to walk straight | 1 |
| Loss of seeking behavior | 1 |
| Loss of startle behavior | 1 |
| MAXIMUM TOTAL | 10 |
One point is awarded for failure to perform a task.
NSS at 1 h in the range of 8–10: severe CHI
2.5 Evaluation of infarct volume
At 48h after trauma and 24h after treatment with vehicle or DCS, the mice were sacrificed and their brains were sectioned (2 mm thickness) using a brain mold. The slices were placed in 24-well tissue culture plate containing 2% 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate-buffered-saline (PBS) and incubated for 1h at 37°C, allowing the TTC to stain vital tissue. Then, 4% paraformaldehyde (PFA) in PBS was added to each well and incubated overnight. PFA was removed by three PBS washes. Sections were visualized and photographed using a Stemi SV11 Stereoscope (Zeiss, Jena Germany) equipped with a digital photo-camera (Coolpix 4500, Nikon, Tokyo, Japan). Lesion volume was calculated as the unstained (white) area relative to the uninjured contra-lateral hemisphere area, so as to neutralize the effect of edema formation on the injured hemisphere volume. Calculations were made using commercial ImageJ 1.40 software and relative lesion areas from all brain sections were grouped to provide lesion volume, representing the percentage of dead tissue within the injured hemisphere.
2.6 Evaluation of long-term tissue loss
Brains of animals killed 28 days after trauma were frozen and cryosectioned in the coronal plane (10 micron thickness, cutting temperature −15°C) from the olfactory tubercule to the cerebellum. Sections were collected every 100 microns, thaw-mounted onto coated slides and stained with 0.5% cresyl violet. The area of the hemispheres ipsilateral and contralateral to the trauma was measured using ImageJ software and the percent difference between the two was calculated to estimate long-term tissue loss.
2.7 Statistical Analysis
The data are expressed as the mean ± S.E.M. A Microsoft Excel package was used to determine statistical significance. At each of the post injury days on which NSS was evaluated, significance was determined using the nonparametric Mann-Whitney test for the ΔNSS values on that day. Student's t-test was used for comparing infarct volume. The effect of DCS on mean hemisphere areas on the traumatized and contralateral side was analyzed by 2 way ANOVA (treatment × side) and percent tissue loss was compared using Student's t-test.
3. RESULTS
3.1 Effect of time of administration of DCS on functional recovery
The earlier findings of faster and better recovery of motor and memory functions after a single administration of DCS at 24h (Yaka et al 2007) prompted us to examine the therapeutic window for drug administration. Therefore, four groups of mice were treated with DCS at 8 or 16h post injury – for the earlier time points, and 24 or 72h for the later periods. Control mice were treated with vehicle at the same time points. In contrast to the beneficial effects previously observed after treatment at 24h, administration of DCS at 8 or 16h post trauma did not improve the neurological outcome compared with that of the control, vehicle-treated animals (Fig. 1A). However, when given at 24 or 72h post injury, a single dose of DCS facilitated neurological recovery from day 7 as compared with their respective controls (Fig. 1B). Interestingly, even when given as late as 72h post injury DCS exerted a significant effect (P<0.05 vs saline-treated mice) on days 7–21 post injury, similar to that obtained after treatment at 24h.
Figure 1.

Effect of DCS dose on functional recovery after head injury. The Neurological Severity Score (NSS) tests reflexes, alertness, coordination and motor abilities. The extent of recovery is calculated as the difference between NSS at 1h and that at any other time: ΔNSS(t) = NSS (1h) – NSS (t). A. DCS was given at 8 or 16h. B. DCS was given 24 or 72h after injury. Vehicle was given at the same intervals. *P<0.05 DCS given at 24h or 72h vs saline, given at the same time (Mann-Whitney non-parametric test)
3.2 Effect of multiple doses of DCS on functional recovery
We next investigated whether DCS administration of two doses at 24 and 48h (DCS-2, Fig 2) or three doses (24, 48 and 72h, DCS-3 Fig 2) post closed head injury would confer greater protection than a single dose at 24h. All DCS-treated mice, whether treated 2 or 3 times, displayed similar enhanced functional recovery compared with the vehicle-treated controls. This beneficial effect, already noticed 2 days post trauma, was significant for both groups from day 7–35 (P<0.05) and even more significant thereafter, and sustained for up to 63 days. There was no difference between the double and triple dose regimens in the degree of functional recovery, and it was comparable with that reported earlier (Yaka et al., 2007).
Figure 2.
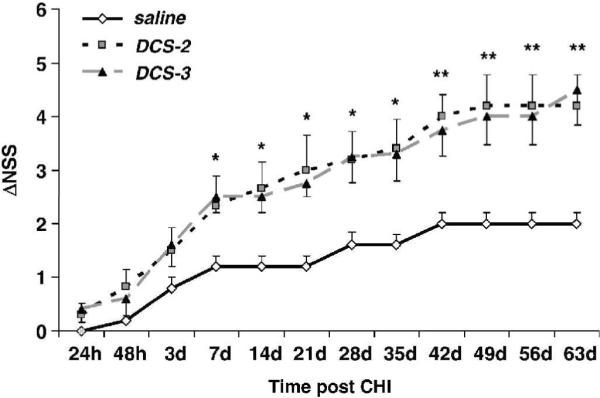
The effect of multiple doses of DCS on functional recovery. DCS was given twice (DCS-2), at 24 and 48h, or three times (DCS-3, 24, 48 and 72h) after closed head injury. *P<0.05 and **P<0.01, both treatment groups vs saline (Mann-Whitney non-parametric test).
3.3 Effect of DCS on infarct volume and long term tissue loss
The hemispheric lesion volume (indicated by arrows), estimated by TTC staining 48h after injury (24h after DCS administration) as 15.5 ± 1.5 and 17.5 ± 2.0% in the saline- and DCS-treated mice, respectively, is shown in Fig. 3. The values were not significantly different, indicating that the drug did not reduce infarct volume. In animals treated with vehicle and killed 28 days later, the contused hemisphere area was 77.7 ± 2.7% that of the contralateral hemisphere at the coronal plane representing the focus of the trauma (i.e. ~22% loss of tissue). DCS treatment had no effect on this parameter (77.2 ± 2.2%, p=0.9, n.s.), as seen in Fig. 4.
Figure 3.

The effect of DCS on infarct volume, assessed 24 h after drug administration (48 h after closed head injury). A. Brain section immersed in 2% TTC in PBS and photographed using a Stemi SV11 Stereoscope (Zeiss, Germany) and digital photocamera Coolpix E990 (Nikon, Japan). Black arrow indicates infracted area. B. Area of hemispheres ipsilateral and contralateral to the trauma was measured and percent hemispheric infarct volume was quantified using the Scion Image-Release Beta 4.0.2 program.
Figure 4.

Histology at impact level 28 days after injury. Cryostat sections were stained with cresyl violet. Note cavitation on left (ipsilateral side) and ventricular enlargement on right (contralateral) side (A). The area of the hemispheres ipsilateral and contralateral to the trauma was measured using Image J software and the percent difference between the two was calculated to estimate long-term tissue loss (B).
3.4 Reversal of the effect of DCS on functional recovery by MK-801
In the next set of experiments we addressed the question whether the effect of DCS is indeed mediated via stimulation of the NMDA receptors. We assumed that if that was the case, the beneficial effect of DCS should be abolished by an antagonist given at the same time. We chose the functional antagonist MK-801. Fig. 5 shows that the beneficial effect of DCS alone from day 7 to 21 post injury was significantly abolished, when MK-801 was given at the same time (P<0.05), with a tendency towards an even worse outcome. When given 24h after injury, MK-801 alone not only inhibited the spontaneous recovery of the mice but even caused further deterioration of their neurological status.
Figure 5.
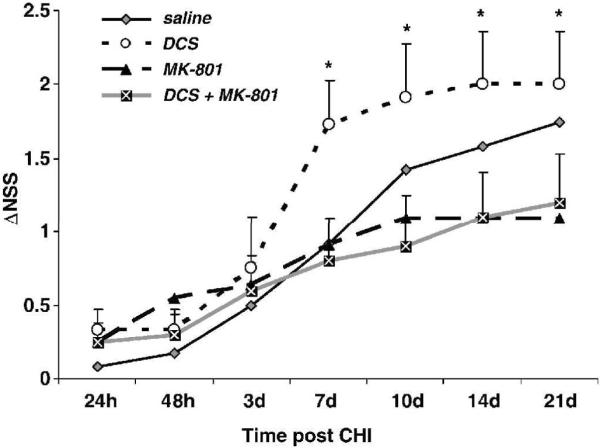
MK-801 reversal of the DCS effect on functional recovery. Mice were treated 24h after closed head injury with vehicle, MK-801, DCS or both drugs together. NSS was followed for 3 weeks after injury. *P<0.05 vs DCS+MK-801 group.
4. DISCUSSION
One of the features of NMDA receptor function is the requirement for more than one agonist to activate the receptor and open its channel (Danysz and Parsons, 1998). Binding of a co-agonist to its specific binding site is an obligatory step for receptor activity, and it has been found that this site exerts a regulatory role. It has been suggested that D-serine acts as an endogenous co-agonist, alongside with glycine (for review, see Wolosker 2007). We selected DCS as a novel candidate for traumatic brain injury treatment because it has been shown to improve cognitive function in various animal models (Temple and Hamm, 1996; Andersen et al., 2002; Schneider et al., 2000). Moreover, DCS has a good safety profile, and it is already used in humans for several different indications (Duncan et al., 2004; Laake et al., 2002, Nitsche et al., 2004). These properties could significantly facilitate the clinical application of DCS in traumatic brain injury patients.
The results of the present study corroborate our recent report (Yaka et al., 2007) on the benefit afforded by the NMDA receptor co-agonist DCS after brain trauma. After previously showing that a single administration of DCS at 24 h post injury restores motor function as well as LTP even two weeks after drug administration, we now examined the therapeutic window and dose regimen for this drug. We show that prolonged, multiple-injection paradigms are not necessary to elicit the benefit of DCS treatment in closed head injury, and it appears that even a short treatment course (a single injection) given one or three days after injury is sufficient to provide a long-term functional benefit. Moreover, when treatment was begun at earlier time points, i.e. 8 or 16h post injury, DCS was not effective in ameliorating the neurobehavioral deficits. Our results agree on the whole with those of Temple and Hamm (1996), who found that DCS significantly attenuated memory deficits in injured rats. However, the studies differ in the species used, dose and treatment protocol. Temple and Hamm treated injured rats once daily on days 1–15 and found that only a dose of 30mg/kg, but not 10mg/kg, was effective, whereas we found that the 10mg/kg dose, given to mice as a single injection, yielded a significant effect (preliminary, unpublished data, Yaka et al., 2007 and present study). This difference may be due to the different animals used in the studies (rats vs mice), and to the different design of the study, as Temple and Hamm did not resort to the single administration regimen.
Traumatic brain injury, the leading cause of mortality and disability among young people in the Western world (see, Thurman et al., 1999, Myburgh et al., 2008, Koskinen et al., 2008) triggers a large, transient increase in excitatory amino acid efflux (especially glutamate) in the brain of experimental animals and in human subjects (Bullock et al., 1998, Palmer et al., 1993). When glutamate, with glycine as cofactor, binds at the extracellular site of the NMDA receptors, the channel is opened, and allows a massive calcium influx. In turn, the increased intracellular calcium concentration initiates several excitotoxic and inflammatory cascades, culminating in neuronal death. Thus, many treatment paradigms in laboratory animals targeted this NMDA receptor hyperactivation and showed that competitive or noncompetitive inhibitors of the receptor improved the outcome in animal models of traumatic brain injury and stroke. The positive results from animal models led to the development and clinical testing of several NMDA receptor antagonists in traumatic brain injury and stroke (Lees 1997). Unfortunately, all of the NMDA receptor antagonists studied to date (Selfotel, Cerestat, D-CPPene, CP-101) have not proved efficient in large controlled clinical trials (Fisher 1998., Maas et al,1999, Narayan et al, 2002). Furthermore, some of these trials had to be discontinued prematurely because of increases in mortality and morbidity in the drug arm of the trials. Importantly, in laboratory animals, NMDA receptor antagonists appeared to be beneficial only when administered prior to the trauma or within a short therapeutic window post injury, such that treatment initiation more than 30–60 minutes after injury was no longer protective (e.g. Chen et al, 1991, Panter and Faden 1992, Di and Bullock 1996, Kroppenstedt et al, 1998, Rod and Auer 1989, Shapira et al, 1990).
In contrast to the early hyperactivation, the prolonged loss of NMDA receptor function provides a wide therapeutic window for the treatment of neurobehavioral deficits after head injury. As we show here, a single injection given as late as 72 h post traumatic brain injury, improves neurobehavioral function during a 1–2 month follow-up. We previously raised the possibility that closed head injury could affect the number of NMDA receptors, their function, or both, in a region-specific manner, and showed alterations in the expression and synaptic distribution of NMDA receptor subunits as early as 15 min following closed head injury (Shcumann et al., 2008). The duration of these alterations is now under investigation. We also suggested that tyrosine phosphorylation of NR2B may contribute to these alterations in NMDA receptor function or signaling pathways in the posttraumatic brain and may be related to pathogenic events leading to neurological deficits (Shcumann et al,. 2008). It appears that at the late time points (24–72h), when the NMDA receptors are down-regulated, their modulation by the co-agonist DCS may prime them and “reboot” the glutamate neurotransmission system to its physiological level. It is not clear whether this effect is associated with restoration of the subunit composition. . The finding that DCS was ineffectual when given at 8 or 16h may suggest that only after a certain degree of hypo-activation or alteration of the subunit population has been reached in the injured brain, is such an effect possible.
There is some evidence that DCS has a bell-shaped dose response curve and may have opposite effects on NMDA receptor channel opening at low or high doses (Andersen et al., 2002, Emmet et al., 1991). To substantiate the mechanism by which DCS exerts its effect, the NMDA receptor open-channel antagonist MK-801 was injected 24 h after injury, alone or with DCS. As previously shown (Biegon et al., 2004), functional recovery after treatment with MK-801 alone was slower than in the vehicle-control mice, indicating that blockade of the physiological glutamate activity at the NMDA receptors at a time point when the receptor is already down -regulated is harmful rather than beneficial. Moreover, MK-801, when given together with DCS, abolished the latter's beneficial effect, even though it binds to distinct sites of the NMDA receptor complex. These results show that the effect of DCS is indeed due to receptor activation, and agree with those of Barth and his colleagues (Barth et al., 1990, Hoane et al., 2000), who administered MK801 to brain- lesioned animals a few days after rather than shortly after injury, and reported exacerbation and prolongation of the neurological deficits. The lack of DCS effect on early infarction and tissue loss further supports the notion that the beneficial effects of the treatment are related to recovery of activity at the glutamatergic/NMDA receptor synapse, possibly related to increased BDNF (Yaka et al., 2007) rather then prevention of early neuronal death. This phenomenon (i.e. functional improvement unrelated to reduction in infarct size) has also been reported by others in animal models and human studies of traumatic brain injury and stroke (Ji et al., 2009, Mark et al., 2008, Belayev et al., 2007, Kokiko et al., 2006).
In conclusion, the current lack of specific therapy for traumatic brain injury, the need for developing new therapeutics for this indication, along with the good safety profile of DCS and its wide use in humans, contribute to the importance and clinical relevance of our present study. The present findings, demonstrating accelerated functional recovery by activation of the hypo-activated NMDA receptor ion channels in a clinically-relevant model of head injury could significantly expedite the clinical application of DCS in human head trauma patients.
Acknowledgements
This study was supported by NIH grant 1R01NS050285 (A. Biegon) and, in part, by a grant from the IDF and Israel Ministry of Defense (E. Shohami). E. Shohami is the incumbent of the Dr. Leon and Dr. Mina Deutsch Chair in Psychopharmacology at The Hebrew University, and a member of the Brettler Center for Research in Molecular Pharmacology and Therapeutics, The Hebrew University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alessandri B, Landolt H, Langemann H, Gregorin J, Hall J, Gratzl O. Application of glutamate in the cortex of rats: a microdialysis study. Acta Neurochir Suppl. 1996;67:6–12. doi: 10.1007/978-3-7091-6894-3_2. [DOI] [PubMed] [Google Scholar]
- Andersen JM, Lindberg V, Myhrer T. Effects of scopolamine and D-cycloserine on non-spatial reference memory in rats. Behav Brain Res. 2002;129:211–216. doi: 10.1016/s0166-4328(01)00318-7. [DOI] [PubMed] [Google Scholar]
- Baker AJ, Moulton RJ, MacMillan VH, Shedden PM. Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J Neurosurg. 1993;79:369–372. doi: 10.3171/jns.1993.79.3.0369. [DOI] [PubMed] [Google Scholar]
- Barth TM, Grant ML, Schallert T. Effect of MK801 on recovery from sensorimotor cortex lesion. Stroke. 1990;11:153–157. [PubMed] [Google Scholar]
- Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury - where is the “golden bullet”? Mol. Med. 2008;14:731–740. doi: 10.2119/2008-00050.Beauchamp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steindart R, Brenneman DE, Eizenberg O, Trembovler V, Shohami E. A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther. 2001;296:57–63. [PubMed] [Google Scholar]
- Biegon A, Alvarado M, Budinger TF, Grossman R, Hensley K, West MS, Kotake Y, Ono M, Floyd RA. Region Selective Effects of Neuroinflammation and Antioxidant Treatment on Peripheral Benzodiazepine Receptors and NMDA Receptors in the Rat Brain. J. Neurochem. 2002;82:1–11. doi: 10.1046/j.1471-4159.2002.01050.x. [DOI] [PubMed] [Google Scholar]
- Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Obenaus A, Zhao W, Saul I, Busto R, Wu C, Vigdorchik A, Lin B, Ginsberg MD. Experimental intracerebral hematoma in the rat: characterization by sequential magnetic resonance imaging, behavior, and histopathology. Effect of albumin therapy. Brain Res. 2007;1157:146–155. doi: 10.1016/j.brainres.2007.04.077. [DOI] [PubMed] [Google Scholar]
- Brown JI, Baker AJ, Konasiewicz SJ, Moulton RJ. Clinical significance of CSF glutamate concentrations following severe traumatic brain injury in humans. J Neurotrauma. 1998;15:253–263. doi: 10.1089/neu.1998.15.253. [DOI] [PubMed] [Google Scholar]
- Bullock R, Zauner A, Woodward JJ, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89:507–518. doi: 10.3171/jns.1998.89.4.0507. [DOI] [PubMed] [Google Scholar]
- Chen M, Bullock R, Graham DI, Frey P, Lowe D, McCulloch J. Evaluation of a competitive NMDA antagonist (D-CPPene) in feline focal cerebral ischemia. Ann Neurol. 1991;30:62–70. doi: 10.1002/ana.410300112. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev. 1998;50:597–664. [PubMed] [Google Scholar]
- Di X, Bullock R. Effect of the novel high-affinity glycine-site N-methyl-D-aspartate antagonist ACEA-1021 on 125I-MK-801 binding after subdural hematoma in the rat: an in vivo autoradiographic study. J Neurosurg. 1996;85:655–661. doi: 10.3171/jns.1996.85.4.0655. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Szilagyi S, Schwartz MP, Bugarski-Kirola D, Kunzova A, Negi S, Stephanides M, Efferen TR, Angrist B, Peselow E, Corwin J, Gonzenbach S, Rotrosen JP. Effects of D-cycloserine on negative symptoms in schizophrenia. Schizophr Res. 2004;71:239–248. doi: 10.1016/j.schres.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Emmett MR, Mick SJ, Cler JA, Rao TS, Iyengar S, Wood PL. Actions of D-cycloserine at the N-methyl-D-aspartate-associated glycine receptor site in vivo. Neuropharmacology. 1991;30:1167–1671. doi: 10.1016/0028-3908(91)90161-4. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Fisher M. The travails of neuroprotective drug development for acute ischemic stroke. Eur Neurol. 1998;40:65–66. doi: 10.1159/000007959. [DOI] [PubMed] [Google Scholar]
- Friedman LK, Ginsberg MD, Belayev L, Busto R, Alonso OF, Lin B, Globus MY. Intraischemic but not postischemic hypothermia prevents non-selective hippocampal downregulation of AMPA and NMDA receptor gene expression after global ischemia. Brain Res Mol Brain Res. 2001;86:34–47. doi: 10.1016/s0169-328x(00)00252-7. [DOI] [PubMed] [Google Scholar]
- Grossman R, Shohami E, Alexandrovich A, Yatsiv I, Kloog A, Biegon A. Increase in Peripheral Benzodiazepine Receptors and Loss of Glutamate NMDA receptors in a mouse model of closed head injury: A Quantitative autoradiographic study. NeuroImage. 2003;20:1971–1981. doi: 10.1016/j.neuroimage.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Hayes RL, Jenkins LW, Lyeth BG, Balster RL, Robinson SE, Clifton GL, Stubbins JF, Young HF. Pretreatment with phencyclidine, an N-methyl-D-aspartate antagonist, attenuates long-term behavioral deficits in the rat produced by traumatic brain injury. J Neurotrauma. 1988;5:259–274. doi: 10.1089/neu.1988.5.259. [DOI] [PubMed] [Google Scholar]
- Heifets LB. Antimycobacterial drugs. Semin Respir Infect. 1994;9:84–103. [PubMed] [Google Scholar]
- Heresco-Levy U, Kremer I, Javitt DC, Goichman R, Reshef A, Blanaru M, Cohen T. Pilot-controlled trial of D-cycloserine for the treatment of post-traumatic stress disorder. Int J Neuropsychopharmacol. 2002;5:301–307. doi: 10.1017/S1461145702003061. [DOI] [PubMed] [Google Scholar]
- Hoane MR, Barbay S, Barth TM. Large cortical lesions produce enduring forelimb placing deficits in untreated rats and treatment with NMDA antagonists or antioxidant drugs induces behavioral recovery. Brain Res Bull. 2000;53:175–186. doi: 10.1016/s0361-9230(00)00327-0. [DOI] [PubMed] [Google Scholar]
- Ji S, Kronenberg G, Balkaya M, Furber K, Gertz K, Kettenmann H, Endres M. Acute neuroprotection by pioglitazone after mild brain ischemia without effect on long-term outcome. Exp Neurol. 2009;216:321–328. doi: 10.1016/j.expneurol.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokiko ON, Murashov AK, Hoane MR. Administration of raloxifene reduces sensorimotor and working memory deficits following traumatic brain injury. Behav Brain Res. 2006;170:233–240. doi: 10.1016/j.bbr.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Koskinen S, Alaranta H. Traumatic brain injury in Finland 1991–2005: A nationwide register study of hospitalized and fatal TBI. Brain Inj. 2008;22:205–214. doi: 10.1080/02699050801938975. [DOI] [PubMed] [Google Scholar]
- Kroppenstedt SN, Schneider GH, Thomale UW, Unterberg AW. Protective effects of aptiganel HCl (Cerestat) following controlled cortical impact injury in the rat. J Neurotrauma. 1998;15:191–197. doi: 10.1089/neu.1998.15.191. [DOI] [PubMed] [Google Scholar]
- Laake K, Oeksengaard AR. D-cycloserine for Alzheimer's disease. Cochrane Database Syst Rev. 2002:CD003153. doi: 10.1002/14651858.CD003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR. Cerestat and other NMDA antagonists in ischemic stroke. Neurology. 1997;49:S66–69. doi: 10.1212/wnl.49.5_suppl_4.s66. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Dawson TM. Secondary mechanisms in neuronal trauma. Curr Opin Neurol. 1994;7:510–516. doi: 10.1097/00019052-199412000-00007. [DOI] [PubMed] [Google Scholar]
- Maas AI, Steyerberg EW, Murray GD, Bullock R, Baethmann A, Marshall LF, Teasdale GM. Why have recent trials of neuroprotective agents in head injury failed to show convincing efficacy? A pragmatic analysis and theoretical considerations. Neurosurgery. 1999;44:1286–1298. [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mark VW, Taub E, Perkins C, Gauthier L, Uswatte G. MRI infarction load and CI therapy outcomes for chronic post-stroke hemiparesis. Restor Neurol Neurosci. 2008;26:13–33. [PubMed] [Google Scholar]
- Miller LP, Lyeth BG, Jenkins LW, Oleniak L, Panchision D, Hamm RJ, Phillips LL, Dixon CE, Clifton GL, Hayes RL. Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 1991;526:103–107. doi: 10.1016/0006-8993(90)90254-9. [DOI] [PubMed] [Google Scholar]
- Myburgh JA, Cooper DJ, Finfer SR, et al. Epidemiology and 12-month outcomes from traumatic brain injury in Australia and New Zealand. J Trauma. 2008;64:854–862. doi: 10.1097/TA.0b013e3180340e77. [DOI] [PubMed] [Google Scholar]
- Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W. Consolidation of human motor cortical neuroplasticity by D-cycloserine. Neuropsychopharmacology. 2004;29:1573–1578. doi: 10.1038/sj.npp.1300517. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Panter SS, Faden AI. Pretreatment with NMDA antagonists limits release of excitatory amino acids following traumatic brain injury. Neurosci. Lett. 1992;136:165–168. doi: 10.1016/0304-3940(92)90040-e. [DOI] [PubMed] [Google Scholar]
- Rod MR, Auer RN. Pre- and post-ischemic administration of dizocilpine (MK-801) reduces cerebral necrosis in the rat. Can J Neurol Sci. 1989;16:340–344. doi: 10.1017/s031716710002919x. [DOI] [PubMed] [Google Scholar]
- Schumann J, Alexandrovich AG, Biegon A, Yaka R. Inhibition of NR2B Phosphorylation Restores Alterations in NMDA Receptor Expression and Improves Functional Recovery following Traumatic Brain Injury in Mice. J. Neurotrauma. 2008;25:945–957. doi: 10.1089/neu.2008.0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JS, Tinker JP, Van Velson M, Giardiniere M. Effects of the partial glycine agonist D-cycloserine on cognitive functioning in chronic low dose MPTP-treated monkeys. Brain Res. 2000;860:190–194. doi: 10.1016/s0006-8993(00)02036-9. [DOI] [PubMed] [Google Scholar]
- Shapira Y, Yadid G, Cotev S, Niska A, Shohami E. Protective effect of MK-801 in experimental brain injury. J. Neurotrauma. 1990;7:131–139. doi: 10.1089/neu.1990.7.131. [DOI] [PubMed] [Google Scholar]
- Sihver S, Marklund N, Hillered L, Langstrom B, Watanabe Y, Bergstrom M. Changes in mACh, NMDA and GABA(A) receptor binding after lateral fluid-percussion injury: in vitro autoradiography of rat brain frozen sections. J Neurochem. 2001;78:417–423. doi: 10.1046/j.1471-4159.2001.00428.x. [DOI] [PubMed] [Google Scholar]
- Temple MD, Hamm RJ. Chronic, post-injury administration of D-cycloserine, an NMDA partial agonist, enhances cognitive performance following experimental brain injury. Brain Res. 1996;741:246–251. doi: 10.1016/s0006-8993(96)00940-7. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- Tolias CM, Bullock MR. Critical Appraisal of Neuroprotection Trials in Head Injury: What Have We Learned? NeuroRx. 2004;1:71–79. doi: 10.1602/neurorx.1.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E. Dynamic Changes in the Recovery after Traumatic Brain Injury in Mice: Effect of Injury Severity on T2- Weighted MRI Abnormalities, Motor and Cognitive Function. J Neurotrauma. 2008;25:324–333. doi: 10.1089/neu.2007.0452. [DOI] [PubMed] [Google Scholar]
- Wolosker H. NMDA receptor regulation by D-serine: new findings and perspectives. Mol Neurobiol. 2007;36:152–164. doi: 10.1007/s12035-007-0038-6. [DOI] [PubMed] [Google Scholar]
- Yaka R, Biegon A, Grigoriadis N, Simeonidou C, Grigoriadis S, Alexandrovich AG, Matzner H, Schumann J, Trembolver V, Tsenter J, Shohami E. D-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J. 2007;21:2033–2041. doi: 10.1096/fj.06-7856com. [DOI] [PubMed] [Google Scholar]