

Abstract
We previously determined that Notch2, and not Notch1 was required for forming proximal nephron segments. The dominance of Notch2 may be conserved in humans, since Notch2 mutations occur in Alagille syndrome (ALGS) 2 patients, which includes renal complications. To test whether mutations in Notch1 could increase the severity of renal complications in ALGS, we inactivated conditional Notch1 and Notch2 alleles in mice using a Six2-GFP∷Cre. This BAC transgene is expressed mosaically in renal epithelial progenitors but uniformly in cells exiting the progenitor pool to undergo mesenchymal to epithelial transition. Although delaying Notch2 inactivation had a marginal effect on nephron numbers, it created a sensitized background in which the inactivation of Notch1 severely compromised nephron formation, function and survival. These and additional observations indicate that Notch1 in concert with Notch2 contributes to the morphogenesis of renal vesicles into S-shaped bodies in a RBP-J dependent manner. A significant implication is that elevating Notch1 activity could improve renal functions in ALGS2 patients. As proof of principle, we determined that conditional inactivation of Mint, an inhibitor of Notch-RBP-J interaction, resulted in a moderate rescue of Notch2 null kidneys, implying that temporal blockage of Notch signaling inhibitors downstream of receptor activation may have therapeutic benefits for ALGS patients.
Keywords: Nephron segmentation, Notch, Mint, RBP-J, Alagille syndrome, Six2
Introduction
The mammalian kidney forms as a result of reciprocal inductive interactions between the metanephric mesenchyme (MM) and the ureteric bud. MM cells undergo mesenchymal to epithelial transition (MET) and through a series of stereotypical morphogenetic events form nephrons composed of distinct tubular segments along a proximal-distal axis. The cellular processes that convert a spherical aggregate of mesenchymal cells into a tubular structure composed of distinct simple epithelial cell types and the molecules that orchestrate these processes are only beginning to be identified (Dressler, 2006). In mice, Notch2, a type-I transmembrane protein, plays an essential role early in nephron segmentation to ensure the emergence of proximal cell fates (Cheng et al., 2007; Kopan et al., 2007). In humans, reduced Notch activity results in Alagille syndrome, observed in patients with one mutant allele of the Notch ligand Jagged1 (ALGS1, (Piccoli and Spinner, 2001)) or the receptor Notch2 (ALGS2, (McDaniell et al., 2006)). The four mammalian Notch genes code for receptors that mediate short-range communication via a conserved mechanism. Notch1, Notch2 and genes coding for their ligands Dll1 and Jag1 are all expressed in the renal vesicle (RV), the first structure to display epithelial characteristics during MET (Chen and Al-Awqati, 2005; Cheng et al., 2007; Piscione et al., 2004). Following ligand binding, a juxtamembrane domain unfolds, permitting metalloproteases to cleave and remove the Notch extracellular domain. Ectodomain shedding is followed by intramembrane cleavage mediated by γ-secretase; this step releases the Notch intracellular domain (N-ICD) to translocate into the nucleus where it associates with the transcription factor RBP-J and recruits the transcription activation machinery (Kopan and Ilagan, 2009). In the nucleus N-ICD may need to compete for binding to RBP-J with ubiquitous co-repressor proteins including Mint (Oswald et al., 2002; Tsuji et al., 2007; Yabe et al., 2007). Although Notch1 and 2 and the ligands Jag1 and Dll1 could act redundantly in kidney development, inactivation of Notch2, γ-secretase or RBP-J in the MM prior to RV formation results in complete absence of glomeruli and proximal tubules (PT), with only distal tubules forming; these structures are formed, however, in the absence of Notch1 (Cheng et al., 2003; Cheng et al., 2007; Wang et al., 2003). This is despite the fact that Notch1 is activated in the RV and N1-ICD remains easily detectable in the presumptive precursors of PT and podocytes within the S-shaped bodies.
Since both N1-ICD and N2-ICD have the same affinity for RBP-J (Del Bianco et al., 2008; Friedmann et al., 2008; Lubman et al., 2007) and over-expression of N1-ICD can drive renal epithelial progenitors to the PT cell fate (Cheng et al., 2007), it remained possible that Notch1 made a contribution to the overall level of canonical Notch signaling within the RV and S-shaped bodies, one that was too small to support nephron segmentation in the absence of Notch2. Alternatively, endogenous Notch1 is activated but incapable of activating the same target genes that Notch2 activates during nephrogenesis at physiologic levels. Finally, Notch1 activation may lag behind Notch2, missing the developmental window (Chen and Al-Awqati, 2005). To test which of these alternatives explains our results, and to delay Notch2 inactivation until after onset of kidney development, we removed conditional Notch2 alleles using the Six2-GFP∷Cre transgene. This allowed nephrons to form with limiting amounts of Notch2, and provided a sensitized, functional background on which we could assess the contribution of Notch1 to kidney development. We determined that Notch1 contributed to nephrogenesis in vivo, albeit to a lesser degree than Notch2. Since Notch1 was capable of activating the same target genes as Notch2, we asked if in principle, methods aimed at augmenting Notch1 signaling could help ALGS patients. We inactivated Mint, an inhibitor of N-ICD mediated transactivation of Notch target genes (Oswald et al., 2002; Tsuji et al., 2007; Yabe et al., 2007) and asked if PT or glomeruli reemerged in the complete absence of Notch2. Modest improvement was indeed seen in Notch2 null kidneys, suggesting that efforts at elevating Notch1 activity in ALGS2 patients with one functional Notch2 allele, or in ALGS1 patients with global reduction in Notch signaling, may have therapeutic benefits.
Materials and Methods
Mice
All experiments involving mice were approved by the Washington University IACUC. The Six2-GFP∷Cretg transgenic mice (Park et al., 2007) were bred with mice with floxed alleles of RBP-J (RBP-J f/f) (Tanigaki et al., 2002), Notch1 (N1 f/f) (Yang et al., 2004), Notch2 (N2 f/f) or both N1f/f;N2f/f. Pax3-Cretg; N2+/f (Cheng et al., 2007) were bred with mice with floxed alleles of Mint (Mintf/f) (Yabe et al., 2007). Compound heterozygotes were bred with mice homozygous for the floxed alleles of the desired genes. All mice used in this study were maintained on mixed backgrounds. Mice and embryos were genotyped following the Universal PCR genotyping protocol (Stratman et al., 2003), primer sequences are available upon request, and their age estimated based on the assumption that noon of the day on which a vaginal plug was observed was embryonic day 0.5 (E0.5). Analysis of serum blood urea nitrogen levels was done at the O'Brein Center renal chemistry core and presented as mg/dL.
Histology and Immunohistochemistry
For analysis of GFP expression pattern the kidneys were dissected and frozen in OCT embedding media on dry ice. Frozen blocks were sectioned at 6 to 12 um thickness and air-dried. The sections were then rinsed in PBS and fixed for five minutes in 4% PFA before visualization of GFP or were stained for Pax2 (1:200, Covance) followed by Cy3 conjugated secondary antibody and then counterstained with DAPI before visualization of fluorescent signals. For visualization of Six2 and GFP, kidneys were fixed in 4% PFA for 30 minutes, and then rinsed in 30% sucrose overnight before embedding in OCT. For all immunohistochemistry kidneys were fixed overnight in Bouin's fixative at 4°C, except 4% paraformaldehyde was used for detection of RBP-J, washed and stored in 70% ethanol prior to embedding in paraffin and sectioning at 5–7 um thickness.
Prior to immunostaining the sections were de-paraffinized, boiled for 20 minutes in Trilogy (Cell Marque) or Antigen Unmasking Solution (Vector Labs H-3300 for detecting RBP-J) and cooled at RT for 20 minutes. Detection of RBP-J and Notch2 required quenching of endogenous hydrogen peroxidase activity with 3% H2O2 treatment for 10 minutes at RT. Sections were then blocked in PBS containing 1% bovine serum albumin (BSA), 0.2% powdered skim milk and 0.3% Triton X-100 for 30 minutes at RT prior to incubation primary antibody overnight in a humidified chamber at 4°C. For anti-RBP-J staining (Cosmo Bio Co., SIM-2ZRBP2, 1:100), the sections were additionally blocked with Avidin and then Biotin (Vector Labs SP-2001) prior to incubation with primary antibody. Tissue sections were then incubated with secondary antibody conjugated with biotin (1:500), then developed using ABC Vectastain kit (Vector Labs, PK-6100) followed by tyramide amplification (PerkinElmer, NEL744001KT).
For detecting Notch2 tissue expression pattern we used an antibody raised against the 258 Carboxyl terminal amino acids of Notch2 (Varadkar et al., 2008) (1:100), tissue sections were permeabilized with 1% Triton X-100 in PBS for 15 minutes at RT prior to blocking with 50mM Tris pH8.0, 150mM NaCl containing 5% donkey serum and 0.05% Tween 20. After overnight incubation with primary antibody overnight the sections were developed by incubating with HRP conjugated secondary antibody and then by tyramide amplification.
Additional primary antibodies and lectins utilized in the study include: Six2 (1:500; HM6123 was a gift from Andrew McMahon, (Kobayashi et al., 2008)), GFP (1:500, AVES Lab. Inc.), anti-beta-gal. (1:5000; Cappel), p21 (1:200; Santa Cruz Sc-6246), NCAM (1:200; Sigma), Jagged-1 (1:100; Santa Cruz Sc-6011), WT-1 (1:100; Santa Cruz Sc-192), and FITC-Conjugated Lotus Lectin (1:200; Vector Labs. Secondary antibodies conjugated with Cy3 (Jackson ImmunoResearch) or Alexa488 (Molecular Probes) were utilized to visualize the binding pattern of primary antibodies.
Estimation of glomerular number and density
The optical fractionator probe of the Stereo Investigator system (MicroBrightField, Inc.) was utilized to estimate glomerular number and kidney volume. Every 20th kidney section was analyzed, and counting was performed under 10× magnification of hematoxylin and eosin stained sections. Glomerular density was calculated by dividing the estimated number of total glomeruli by the estimated total volume for each kidney.
Verification of Notch2 floxed allele deletion
E14.5 Six2-GFP∷ Cretg; N2f/f kidneys were digested by incubating with 2.25% pancreatin and 0.7% trypsin in Ca-Mg free Tyrode's solution (pH 7.4) for 1hour on ice, while periodically disrupting the kidneys by repeated pipetting. DMEM with 10% fetal bovine serum was added and samples were then filtered through a 36 um nylon mesh (Small Parts;#CMN-0035D) and were centrifuged at 800 rpm for 3 minutes. The pellet was resuspended with FACS sorting buffer (containing 3% FCS in PBS with 0.01% azide). GFP+ cells were sorted using a MoFlo high speed flow cytometer (Dako Cytomation, Fort Collins, CO). Genomic DNA was extracted from GFP+ cells using the QIAamp DNA Micro kit (Qiagen). 30ng of genomic DNA was used with the following primers to amplify and detect any undeleted Notch2 floxed alleles: ttcaaccccagataggaagcagctcagctcacag and gtgcactggagttgggggacccataacttcg. Serial dilution of N2+/f genomic DNA with wild type DNA was used to verify that we are able to detect as little as one intact Notch2 floxed allele per 100 genomes.
Statistical analysis
All results are presented as mean ± s.d. In the graphs the height of the bar represents the mean and the error bars represent standard deviation. Two-tailed unpaired t-tests were performed to compare mice with Notch-deficient kidneys to their wild-type littermates. The resulting p values are mentioned in the text and figure legends.
Results
The Six2-GFP∷Cre transgene is expressed mosaically in the renal epithelial progenitor population, with uniform high level of expression in the induced mesenchyme and pre-tubular aggregates prior to RV formation
We have previously observed that canonical Notch2 signaling was required for proximal nephron development during a narrow developmental window at or before the conversion of the renal vesicle (RV) to an S-shaped body (SB), and that Notch1 was dispensable (Cheng et al., 2007). We hypothesized if we could inactivate Notch2 just prior to RV formation, we would create a Notch2-deficient environment in which to test if Notch1 contributed to nephron development. The Six2-GFP∷Cre BAC transgene (Cheng et al., 2007; Humphreys et al., 2008) had unique properties making it ideal for this experiment. This transgene was designed for expression in the nephron progenitor population, within the metanephric mesenchyme (MM) (Kobayashi et al., 2008). The self-renewing progenitors for all renal epithelial lineages, except for the collecting ducts, reside in the mesenchymal cap condensate (MCC) surrounding each ureteric bud tip and can be recognized by the expression of Six2, Cited1 and Pax2 (Boyle et al., 2008; Kobayashi et al., 2008; Mugford et al., 2009; Self et al., 2006). These cells reside in two broad sub-domains: cortical to the branching ureteric bud the `capping metanephric mesenchyme' (CMM; Six2+, Cited1+, Wnt4−) contains a pure stem population, and lateral to it the `induced metanephric mesenchyme (IMM; Six2low, Cited1−, Wnt4+) is a transient nephron precursor population beginning to respond to Wnt9b signals (Mugford et al., 2009). Staining for GFP∷Cre and for endogenous Six2 protein revealed that in the CMM, many Six2-expressing cells did not express GFP∷Cre at E13.5 (arrowheads in Fig. 1A & A'). A higher fraction of IMM cells, lateral to the ureteric bud, stained for both Six2 and GFP (Fig. 1A, A' &A”). Importantly, cells exiting the progenitor pool during MET, which down-regulate Six2 expression (Mugford et al., 2009) retained high levels of GFP∷Cre expression (arrows in Fig.1A &A”). Furthermore, GFP remained detectable in pre-tubular aggregates ventral (medullary) to the branching ureteric bud (asterisks in Fig.1A and A”). Fate mapping of the Wnt4+ population, which includes the IMM and pre-tubular aggregates, revealed that these cells quickly convert to nephron fates and are not the self-renewing nephron progenitors (Kobayashi et al., 2008). Expression of the Six2-GFP∷Cre transgene faded in the RV (asterisks in Fig.1A'). As late as post-natal day one (P1), MCC contained Pax2+, GFP− progenitors (Fig.1B, B'), suggesting that this Cre line would allow for the retention of intact Notch2 floxed alleles (N2f/f) within the renal epithelial progenitor population of Six2-GFP∷Cretg;N2f/f mice. Thus, although some progenitor cells become depleted of floxed alleles within the MCC early on, and would be expected to generate Notch2-depleted descendents throughout nephrogenesis, this mosaically expressed Six2-GFP∷Cre transgene allows many MCC to retain floxed alleles until the end of nephrogenesis. Importantly, due to strong expression of Cre in pretubular aggregates, all cells achieved recombination of floxed alleles by the SB stage as demonstrated by β-galactosidase expression in every SB cell of E13.5 Six2-GFP∷Cretg;Rosa26R kidneys (Fig. 1C, (Kobayashi et al., 2008)).
Figure 1. The Six2-GFP∷Cre transgene is expressed mosaically in renal epithelial progenitor population, with uniform high level of expression in the induced mesenchyme and pre-tubular aggregates prior to RV formation.
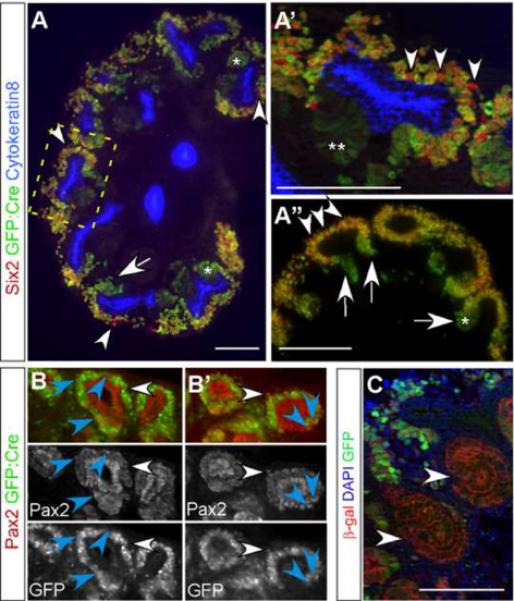
(A-A") Six2-GFP∷Cre transgene (green) is expressed at variable levels within renal epithelial progenitors. Not all Six2+ cells (red) express GFP∷Cre at E13.5 (arrowheads). GFP∷Cre is expressed at higher levels in cells exiting or no longer in the definitive renal epithelial progenitor pool (arrows) and in pretubular aggregates (*) and continues to be expressed in RV (**). A' is a higher magnification of the boxed area in A. (B and B') Co-staining for Pax2 (red) and the Six2-GFP∷Cre transgene (green) reveals that not all mesenchymal cap condensates that are Pax2+ express Cre recombinase (blue arrowheads) at P1. The white arrowheads are placed as positional references and point at cells expressing both Pax2 and GFP. (C) Importantly, due to strong expression of Cre in pre-tubular aggregates, all cells achieved recombination of floxed alleles by the S-shaped stage as demonstrated by β-galactosidase expression in every S-shaped body cell of E13.5 Six2-GFP∷Cretg;Rosa26R kidneys. All cells of the S-shaped bodies (arrowheads) have experienced cre mediated recombination and have turned on β-galactosidase expression in the E13.5. All scale bars are 100um.
Delayed inactivation of Notch2 in renal epithelial progenitors results in sufficient nephron segmentation and proximal tubule formation for survival
Based on our characterization of the Six2-GFP∷Cre BAC transgene, we hypothesized that using this line we could attain genetic inactivation of Notch2 as late as in the pre-tubular aggregates of each round of nephrogenesis, a stage in which all cells are GFP∷Cre positive, while retaining sufficient numbers of stem cells with an intact Notch2 locus for subsequent rounds of nephrogenesis. Six2-GFP∷Cretg;N2+/f male mice were mated with N2f/f females to generate Six2-GFP∷Cretg;N2f/f mice. In contrast to Pax3-Cretg;N2f/f animals that die at birth due to absence of PT and glomeruli (Cheng et al., 2007), Six2-GFP∷Cretg;N2f/f mice had a normal lifespan and were fertile. Histological examination on P1 confirmed that all newborn Six2-GFP∷Cretg;N2f/f kidneys had many mature nephrons containing glomeruli with Wilms' Tumor 1 (WT1) positive podocytes connected to lotus tetragonolobus lectin (LTL)-positive PT (Fig. 2D). In addition, the presence of numerous SB in the cortex at P1 suggested that many Notch2-containing stem cells survived to birth (Fig.2E, F). Nonetheless, examination of several P1 Six2-GFP∷Cretg;N2f/f kidneys revealed many mice (~30% of n=23) that had visibly smaller kidneys when compared with control littermates (n=13; Fig. 2A and B). Smaller kidneys had on average 62% fewer glomeruli than the wild-type littermate kidneys (Fig. 2C; 4950±488 in N2f/f, n=3 versus 1902±1038 in Six2-GFP∷Cretg; N2f/f, n=3, p=0.001), and glomerular density was reduced by 49% (2.4± 0.2 ×10−6 glomeruli/um3 in N2f/f, n=3 versus 1.2± 0.8 ×10−6 glomeruli/um3 in Six2-GFP∷Cretg;N2f/f, n=3, p=0.002). Reduced glomerular number and density would be expected if some stem cells lost Notch2 protein, generating Notch2-deficient RV that failed to form nephrons containing proximal segments; variability in nephron numbers would be expected if the number of stem cells expressing the transgene was variable in this outbred cohort. Dependence of kidney phenotypes on genetic background has been previously reported (McCright et al., 2002). Alternatively, since inherent fluctuations in biochemical reactions will increase the probability of failure when involving low copy number molecules (Levine et al., 2007), limiting amounts of Notch2 could fail to support proximal development in a stochastic manner.
Figure 2. Delayed inactivation of Notch2 in the renal epithelial progenitors results in sufficient nephron segmentation and proximal tubule formation for survival.
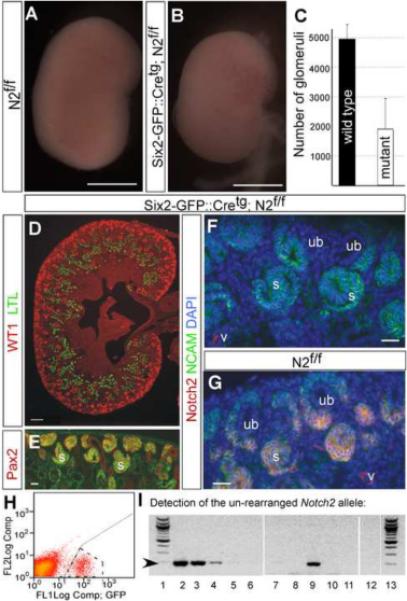
Compared with N2f/f littermate kidneys (A), approximately 30% of post-natal day one Six2-GFP∷ Cretg; N2f/f kidneys (B) are visibly smaller. (C) Graph illustrating the average (height of the bar) number of glomeruli in N2f/f, (n=3) is 4950±488 versus 1902±1038 in the visibly smaller Six2-GFP∷Cretg; N2f/f, (n=3, p=0.001) P1 kidneys. (D) All the kidneys of newborn Six2-GFP∷Cretg; N2f/f mice have many mature nephrons containing podocytes within glomeruli (red) and LTL+ proximal tubules (green). (E) P1 Six2-GFP∷Cretg; N2f/f kidneys contain NCAM+ (green) and Pax2+ (red) S-shaped bodies confirming that nephron segmentation occurred. (F) The kidneys of newborn Six2-GFP∷Cretg; N2f/f mice have no detectable Notch2 protein in the nascent renal epithelial structures, whereas in (G) N2f/f kidneys, Notch2 protein (red) co-localizes with NCAM (green) in the cell membrane of a subset of cap condensate cells and all nascent renal epithelial structures. (H) By FACS we isolated the GFP+ population (boxed by dashed line) from E14.5 Six2-GFP∷ Cretg; N2f/f kidneys. (I) PCR analysis of genomic DNA extracted from GFP+ FACS sorted cells reveals that the Notch2 floxed alleles are inactivated in the GFP+ cells. We determined the lower limit of detection of the Notch2 floxed allele to be one copy per 100 cells. Lanes 1 and 13 contain 100bp DNA ladder (Invitrogen), lanes 2 to 12 contain PCR product, in which the starting template for lanes 2 to 6 contain 10-fold serial dilutions of 30ng of genomic DNA extracted from tail of N2+/f mouse diluted in wild type genomic DNA. The starting template for lane 7 is wild type genomic DNA, for lane 8 is water, for lane 9 is 30ng of genomic N2f/f DNA and for lanes 10, 11 and 12 the starting template was 30ng of genomic DNA extracted from GFP+ kidney cells of three different Six2-GFP∷ Cretg; N2f/f mice. The arrow at the left hand side of gel points at the expected size of the Notch2 floxed allele. We are able to detect a strong PCR product even when only one Notch2 floxed allele is present per 100 genomes (lane 4) but not in the GFP+ cells (lanes 10,11 and 12). Scale bars in A and B are 1mm, scale bar in D is 100um, and scale bars in E,F and G are 20um. UB: ureteric bud, S: S-shaped body, V: blood vessel.
Staining for Notch2 protein in N2f/f littermate kidneys detected protein in the pre-tubular aggregates, the RV and the SB (Fig. 2G). In contrast, staining of Six2-GFP∷Cretg;N2f/f P1 kidneys could not detect Notch2 protein in any nephron precursors (Fig. 2F). To confirm the timing of Notch2 inactivation, we isolated GFP positive cells (enriched for pretubular aggregates; Fig. 1) by FACS sorting from E14.5 Six2-GFP∷Cretg; N2f/f MCC (Fig. 2H). Whereas we could easily detect an intact, floxed Notch2 allele when present at 1 copy per 100 cells (Fig. 2I, lane 4), we could not detect any intact Notch2 alleles in the GFP positive population (Fig. 2I, lanes 10 to 12). This indicates that by MET, we achieved complete inactivation of Notch2.
In conclusion, this particular Six2-GFP∷Cre transgene allowed many progenitors to postpone Notch2 inactivation until MET. Notch2 mRNA/protein synthesized prior to gene inactivation in the Six2-GFP∷Cretg;N2f/f line may allow sufficient - though undetectable - Notch2 to act alone (or in conjunction with other Notch receptors) within the epithelial lineages to support normal segmentation in most of the developing nephrons.
Notch1 is required in Six2-GFP∷Cretg;N2f/f mice for nephrogenesis
The survival of Six2-GFP∷Cretg;N2f/f mice allowed us to ask if Notch1 contributed to PT and podocyte determination in this reduced Notch2 signaling background. If Notch1 failed to compensate for loss of Notch2 because of temporal differences in expression (Chen and Al-Awqati, 2005) or because of qualitative differences between Notch1 and Notch2, then Notch1 would not be expected to activate required Notch2 transcriptional targets, and inactivation of Notch1 in Six2-GFP∷Cretg;N2f/f kidneys should not compromise the kidney any further. Conversely, the dominant role of Notch2 could be due to a quantitative difference between the two receptors. Attaining the level of Notch activity in the RV necessary for nephron segmentation in this sensitized background could require a contribution from Notch1 (Fig. 5B). In this scenario, combined inactivation of Notch1 and Notch2 should further compromise nephrogenesis. Whereas Six2-GFP∷Cretg;N1f/f mice were all born with wild type like kidneys with multiple PT and glomeruli (Fig.3B, A), removal of one Notch1 allele in a Notch2-deficient background severely compromised nephrogenesis (Fig.3C). At P1, 67% of Six2-GFP∷Cretg;N1+/f;N2f/f mice have visibly smaller kidneys than wild type, twice the frequency of what we observed in Six2-GFP∷Cretg;N2f/f mice (31%, Fig. 5A). Only one third of the Six2-GFP∷Cretg;N1+/f;N2f/f mice survived to weaning, and most of these mice died by 6 months of age (Fig. 3D). Survivors displayed a variable reduction in kidney size, the longest surviving mice containing at least one near-normal kidney (Fig. 3E). In extreme cases, only a few glomeruli and PT formed, some of which were dilated (Fig. 3C). Combined complete inactivation of Notch 1 & 2 (Six2-GFP∷Cretg; N1f/f; N2f/f) resulted in pups that died at P1 and all kidneys had few PT with little (or no) evident glomeruli (Fig.5A, Supplementary Fig.S1). Compromised nephron segmentation was evident by the presence of abnormal NCAM+, Pax2+ structures in the nephrogenic zone of Six2-GFP∷Cretg;N1+/f;N2f/f kidneys (Fig. 3H) where comma and S-shaped structures are found in wild type kidneys (Fig. 3G). We analyzed blood urea nitrogen (BUN) levels in surviving Six2-GFP∷Cretg;N1+/f;N2f/f, Six2-GFP∷Cretg; N2f/f, and Six2-GFP∷Cretg;N1f/f and compared it to wild type littermates within the first week after birth. Only Six2-GFP∷Cretg;N1+/f;N2f/f mice had compromised renal function at this age (49.8±35.2 mg/dL, n=14, compared with 25± 3.1 mg/dL in controls, n=19, p=0.0045 ; Fig. 3F).
Figure 5. The specification of proximal nephron cell fates is acutely sensitive to the dosage of canonical Notch1 and Notch2 signaling activity, which allows the removal of repressors of RBP-J-dependent transcription to partially rescue nephrogenesis.
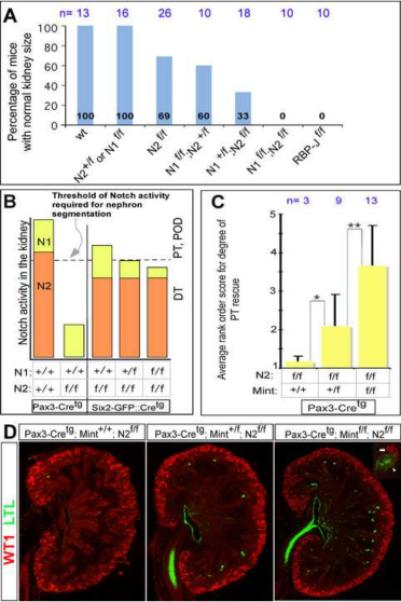
(A) The frequency of animals with normal kidney size, y-axis, inversely correlates with the increasing loss of canonical Notch signaling activity, x-axis. The height of each blue bar represents the frequency of animals of a particular genotype having normal kidney size at P1. The number of mice analyzed per genotype at P1 is indicated next to the genotype. All floxed alleles (f) presented in the graph were inactivated using Six2-GFP∷ Cretg. Wt = wild type littermates. (B) A model of the individual contributions of Notch1 and Notch2 to nephron segmentation based on current and previous observations. Notch2 signaling (orange bar) is critical for reaching the threshold of Notch signaling activity (dashed line) required for sufficient PT and podocyte (POD) development to ensure survival. Inactivation of Notch2 in the Six2-GFP∷Cretg; N2f/f mice creates a sensitized Notch signaling background in which a further reduction in Notch signaling by inactivating Notch1 to create Six2-GFP∷Cretg;N1+/f; N2f/f mice, reveals that both Notch1 and Notch2 are required for PT and POD, in the absence of which only distal tubule (DT) form. (C, D) Progressive loss of Mint on the Pax3-Cretg; N2f/f background results in the reemergence of proximal cell fates. (C) Kidneys from Pax3-Cretg; N2f/f, Pax3-Cretg; Mint+/f; N2f/f, and Pax3-Cretg;Mintf/f; N2f/f were scored on a rank-order scale (see figure S3) for degree of rescue based on the presence of LTL+ PT structures. Decreasing doses of Mint consistently corresponded to a greater degree of PT reemergence. * - P= 0.001. ** - P= 0.0008. (D) Representative (average rank-score) images of sagittal, center cut P0 kidney sections from each of the above genotypes stained with WT1 (red; MM and podocytes) and LTL (green; PT). In some cases we observed WT1+ and LTL+ cells polarized in the same structure in Pax3-Cretg; Mintf/f; N2f/f mice (inset), which we have never observed in Pax3-Cretg; N2f/f animals.
Figure 3. Inactivation of Notch1 in the Notch2-deficient kidneys reveals that Notch1 is required for nephrogenesis, normal renal function and survival.
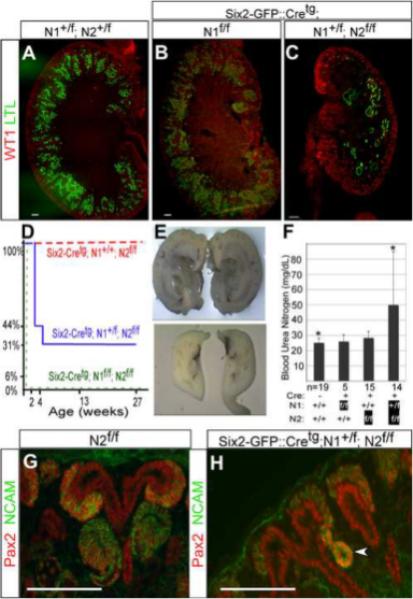
WT1 and LTL staining of post-natal day one (A) wild type (N1+/f; N2+/f), (B) Six2-GFP∷Cretg;N1f/f and (C) Six2-GFP∷Cretg;N1+/f;N2f/f kidneys reveals reduced number of proximal tubules with the inactivation of one Notch1 allele in the Notch2-deficient background. (D) The graph depicts the percentage of mice of a genotype surviving as they age up to six months. Whereas the Six2-GFP∷Cretg; N2f/f mice (n=10) have a normal lifespan (red dashes), the Six2-GFP∷Cretg;N1+/f; N2f/f mice (n=16) have a variably reduced lifespan (blue line) and Six2-GFP∷Cretg;N1f/f;N2f/f mice (n=4) die soon after birth (green dashes). (E) Bisected kidneys from one Six2-GFP∷Cretg;N1+/f; N2f/f mouse that died at six months revealed one hydronephrotic obstructed kidney (top panel) and a second hypoplastic kidney (bottom panel). (F) The blood urea nitrogen (BUN) levels are increased in Six2-Cretg; N1+/f;N2f/f mice within the first week of birth when compared with wild-type littermates (49.8±35.2 mg/dL, n=14, compared with 25± 3.1 mg/dL in controls, n=19, *p=0.0045). The average BUN levels are represented by the height of each bar and one standard deviation by the error bars. (G) In post-natal day one kidneys of N2f/f mice NCAM and Pax2 staining revealed comma and S-shaped structures in the nephrogenic zone. (H) Abnormal nephron segmentation was evident by the presence of abnormal NCAM+, Pax2+ structures (arrowhead) in the nephrogenic zone of Six2-GFP∷Cretg;N1+/f;N2f/f kidneys. All scale bars are 100um.
The data presented above suggest a quantitative difference exists between Notch1 and Notch2 activity during the early stages of nephrogenesis, but demonstrate that both provide the same function. Further support to this hypothesis comes from comparing wild type, Pax3-Cretg;N2+/f (data not shown), or Six2-GFP∷Cretg;N1f/f mice (all wild type in appearance) with Six2-GFP∷Cretg;N1f/f;N2+/f mice. 40% of Six2-GFP∷Cretg;N1f/f;N2+/f mice have visibly smaller kidneys at P1 (n=10) when compared with wild type mice (Fig.5A and Supplementary Fig.S1). This result also demonstrates that like in humans, a single Notch2 allele is not sufficient on its own to provide all RV with the required activity. The dominant function for Notch2 in nephrogenesis is also reflected in the observation that Six2-GFP∷Cretg;N1f/f;N2+/f mice have a milder reduction in nephrogenesis when compared with Six2-GFP∷Cretg;N1+/f;N2f/f mice (Fig.5A and Supplementary Fig. S1).
Inactivation of RBP-J resembles the combined inactivation of Notch1 and Notch2 with a severe reduction in S-shaped body formation and development of proximal tubules and glomeruli
The experiments described above established that Notch1 and Notch2 contribute to nephrogenesis, but did not resolve why Notch1 plays a minor role relative to Notch2. One possibility is that though both receptors contribute to nephron segmentation, they do so in different ways. For instance, only Notch2 functions via RBP-J while Notch1 may function in a RBP-J independent manner. Alternatively, there may be unique inhibitors of N1-ICD that dampen the ability of N1-ICD to associate with RBP-J. To ask if canonical or non-canonical signals were involved, we inactivated RBP-J, the common mediator of the canonical Notch signaling pathway, using the Six2-GFP∷Cretg transgene. If only Notch2 signals via RBP-J, Six2-GFP∷Cretg;RBP-Jf/f mice would be expected to produce a similar phenotype to Six2-GFP∷Cretg;N2f/f mice. If however Notch1 made its contribution to nephron segmentation via RBP-J, then Six2-GFP∷Cretg;RBP-Jf/f mice should have very few nephrons and would be comparable to the combined inactivation of Notch1 and Notch2.
All Six2-GFP∷Cretg;RBP-Jf/f mice die within two days of birth with abnormally small kidneys and compromised vasculature (Fig. 4A versus 4D). A few nephrons containing glomeruli and PT did form (Fig. 4G&H). These few nephrons were capable of filtration and connected to the collecting duct system as the bladders contained a clear filtrate (arrow in Fig. 4D); however, filtration was insufficient, resulting in death. RBP-J is normally expressed in the nucleus of all cells in the nephrogenic zone (Fig. 4B) and the cytoplasm of mature PT at P1 (Fig.4C). Except for a few cells (arrowhead in Fig.4E), most MCC cells Six2-GFP∷Cretg;RBP-Jf/f kidneys were deficient for RBP-J (Fig. 4B versus 4E) at P1, and the few PT that formed did not express RBP-J (Fig. 4F). The presence of a few RBP-J+ MCC is likely due to the mosaic expression of Six2-GFP∷Cretg within the MCC. The cortexes of P1 Six2-GFP∷Cretg;RBP-Jf/f kidneys were devoid of SB and mature nephrons, abundant in wild type littermates (arrows in Fig. 4B). Furthermore, expression of Jag1, a marker for the presumptive PT precursors, was absent from E17.5 Six2-GFP∷Cretg;RBP-Jf/f kidneys except for the spotted expression in RV-like structures (Supplementary Fig. S2). The similarity between Six2-GFP∷Cretg;RBP-Jf/f (Fig. 4) and Six2-GFP∷Cretg;N1f/f;N2f/f kidneys (Supplementary Fig. S1) indicates that both Notch1 and Notch2 signal via RBP-J during nephrogenesis. In aggregate, Notch1 and Notch2 activate the canonical Notch signaling pathway to contribute to PT and glomeruli formation, with partial redundancy and paralog dominance: Notch2 plays a dominant role but Notch1 provides activity via RBP-J, which becomes essential when Notch2 amounts are limiting (Fig. 5A and 5B).
Figure 4. Inactivation of RBP-J resembles the combined inactivation of Notch1 and Notch2 with a severe reduction in S-shaped body formation and development of proximal tubules and glomeruli.
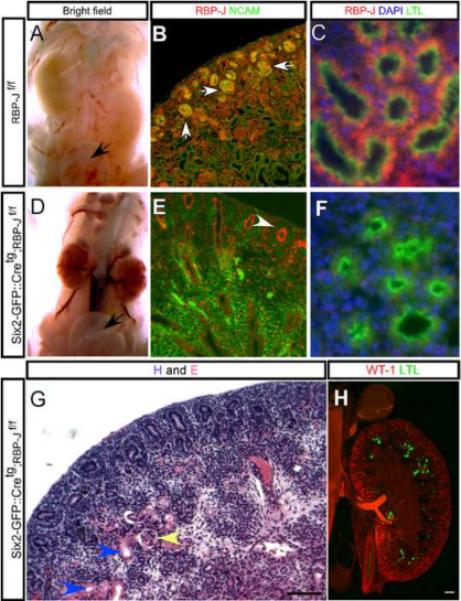
Compared with (A) wild type, RBP-Jf/f mice (D) the Six2-GFP∷ Cretg ; RBP-Jf/f mice die by post-natal day two and though their kidneys are small, managed to produce a clear filtrate present in the bladder (black arrow). (B) In RBP-Jf/f mice, the RBP-J protein (red) is present in the nucleus of all kidney cells at P1 including S-shaped bodies (arrows) which results in (C) mature LTL+ proximal tubules containing RBP-J. (E) In Six2-GFP∷ Cretg; RBP-Jf/f P1 kidneys only a few mesenchymal cap condensate cells still retain RBP-J expression (white arrowhead) and (F) the absence of RBP-J protein in the proximal tubules. (G and H) The Six2-GFP∷ Cretg; RBP-Jf/f kidneys develop very few glomeruli (yellow arrowhead) and proximal tubules (blue arrowheads or green signal). SGC is an abbreviation for Six2-GFP∷ Cretg. Scale bars are 100um.
Removal of Mint, a repressor of RBP-J-dependent transcription, mildly rescues PT formation in Pax3-Cretg;N2f/f kidneys
Since Notch1 is activated at high levels during nephron segmentation relative to other areas in the developing embryo, we wished to examine if, in principle, augmenting Notch1 activity could benefit humans with one functional allele of Notch2 or Jag1. Nuclear N-ICD/RBP-J complexes can be inhibited by several endogenous repressor proteins, including SMRT, CIR, KyoT2 and Mint/SHARP/Spen (Kopan and Ilagan, 2009). Of these, Mint is present in the kidney (Newberry et al., 1999), it antagonizes canonical Notch activity, and its removal was shown to elevate Notch activity in vivo (Kuroda et al., 2003; Tanigaki and Honjo, 2007; Tsuji et al., 2007; Yabe et al., 2007). To ask if reducing inhibitor activity might improve N1-ICD activity, we inactivated Mint in Pax3-Cretg;N2f/f mice. Because Pax3-Cretg;N2f/f mice have no remaining nephrons, and we have shown that Notch1 does contribute to the formation of normal nephrons, any enhancement of N1-ICD activity achieved by removing Mint in these mice should allow at least a few glomeruli and PT to form. Furthermore, PT should be readily detectable given that this background completely lacks them. On the other hand if N1-ICD is functioning at full capacity during nephron segmentation and the limiting factor is the amount of N1-ICD, we would expect no rescue of PT formation in Pax3-Cretg;N2f/f mice. Consistent with the hypothesis that Notch1 activity can be augmented by removal of inhibitors, deletion of conditional Mint alleles on the Notch2 null background (Pax3-Cretg;Mintf/f;N2f/f mice) resulted in a reemergence of some nephrons containing LTL-positive PT structures and WT1-positive cells resembling glomerular podocyte (Fig 5C,D). This “rescue” was consistent, as improvement over Pax3-Cretg;N2f/f mice was observed in 12 of 13 double knockout animals, but variable since not all animals displayed similar number of nephrons. Even the best rescue was not sufficient for a functional kidney, resulting in perinatal death of all Pax3-Cre;Mintf/fN2f/f mice. Interestingly, even inactivation of one copy of Mint resulted in the emergence of some PT in Notch2 mutant mice (Pax3-Cretg;Mint+/f;N2f/f) suggesting a dose response. In order to quantify the degree of PT formation (as marked by LTL+ structures) in Pax3-Cretg;N2f/f mice with progressive loss of Mint, we devised a rank-order score, with no PT (Pax3-Cretg ;N2f/f) defined as 1, and the greatest degree of rescue observed defined as 5 (Supplementary Fig.S3). Four observers blinded to genotype information scored Pax3-Cretg;N2f/f kidneys containing 2,1, or 0 copies of Mint. The scores for each genotype were averaged and the T-test analysis established if the scores were statistically significant. This analysis confirmed a dose-dependent increase in LTL+ PT as more Mint alleles were inactivated in the absence of Notch2 (Fig 5C). Pax3-Cretg;N2f/f (average rank score 1.125) improved with one Mint allele removed (Pax3-Cretg;Mint+/f;N2f/f, average rank score 2.0) and more so with two alleles inactivated (Pax3-Cretg;Mintf/f;N2f/f, average rank score 3.6). This proof of principle experiment demonstrates that N1-ICD activity can be increased to further its contribution to nephrogenesis and implies that the functional activity of Notch1 can be enhanced with reagents that free RBP-J of transcriptional repressors.
Discussion
Paralog dominance: Notch1 and Notch2 contribute jointly but unequally to nephron formation
We established that the BAC Six2-GFP∷Cretg is not expressed in all renal epithelial progenitors; it is expressed at higher levels in induced metanephric mesenchyme and in renal progenitors exiting the stem cell pool, a period during which it completely inactivates floxed alleles. We used this unique tool to generate kidneys with reduced Notch2 signaling by delayed inactivation of Notch2. Although Six2-GFP∷Cretg;N2f/f cells completely lost immunoreactivity to Notch2, the exact timing of Notch2 protein depletion within this population is impossible to determine and is likely variable. This resulted in some nephron loss and in visibly smaller kidneys in 30% of newborn mice without decreasing viability. We present evidence that in this compromised background, Notch1 activity was necessary to maintain proper nephron segmentation and subsequent maturation. Inactivation of one Notch1 allele in Six2-GFP∷Cretg;N1+/f; N2f/f kidneys lowered Notch activity to or below this threshold, significantly reducing the number of nephrons and thus compromising viability in 60% of newborns. Inactivation of Notch1 and Notch2 resembled loss of RBP-J in Six2-GFP∷Cretg; RBP-Jf/f mice and resulted in a fully penetrant, lethal small kidney phenotype. Our analysis of an allelic series progressively reducing canonical Notch signaling capacity during nephrogenesis leads us to propose a model in which each nephron segments properly if overall Notch activity crosses a threshold level in the RV. The Notch2 receptor is dominant (its loss in the MM prior to the onset of kidney development results in complete loss of nephrons) but Notch1 contributes to nephrogenesis (its contribution becomes apparent when Notch2 levels are limiting). In mice, Notch2 heterozygotes are close to the threshold, as evident from the effects of reducing Jag1 (McCright et al., 2002) or Notch1 (this study) in a Notch2 heterozygous background. In humans, where haploinsufficiency of Notch2 or Jagged1 has pathological consequences, the activity levels presumably fall below this threshold (McDaniell et al., 2006) even with a full complement of Notch1. Given that each RV gives rise to exactly one nephron, the number of nephrons that form thus equals the sum of RVs in which overall Notch signals reached this threshold, and accurately reflects the dose of remaining wild-type Notch1 and Notch2 alleles. Why Notch2 is dominant to Notch1 in this process and the molecular basis of this threshold are under investigation.
The phenomenon of Notch paralog dominance occurs in humans, where Notch1 is unable to compensate for mutations in Notch2 that result in juvenile cystic kidney disease, one complication of Alagille syndrome (Lu et al., 2003; McDaniell et al., 2006). In addition to the kidney, Alagille syndrome impacts the heart, liver and craniofacial bone development, all sites where Notch1 and Notch2 overlap but where Notch2 may be the dominant paralog (Geisler et al., 2008). Inversely, loss of Notch1 (Rangarajan et al., 2001), but not Notch2 or Notch3 compromises the skin barrier, resulting in sensitivity to carcinogens; Notch1 activity seems equal to the combined output of Notch2 and Notch3 (Demehri et al., 2009). In addition, Notch1 is the dominant paralog in arterial endothelial cells (Krebs et al., 2000; Vooijs et al., 2007), the aortic valve (Garg et al., 2005) oligodendrocyte (Givogri et al., 2002), in osteoclast progenitors (Bai et al., 2007), and in T-cells, where Notch2 can only drive development if Dll4 is over expressed (Besseyrias et al., 2007).
Elevating Notch transcriptional activity during nephrogenesis in ALGS patients may ameliorate kidney disease
Even in the absence of any obvious developmental malformations, one functional Notch2 allele in Six2-GFP∷Cretg;N1f/f; N2+/f mice is not sufficient to support a normal level of nephrogenesis as normal nephrogenesis required a contribution from Notch1. In humans, the relative contribution of Notch1 must be less than in mice given that mutations in one Notch2 allele compromises kidney development. Alternatively, the proteins produced by mutant Notch2 alleles in ALGS2 patients function as dominant-negative receptors. We determined that Notch1 contributes to normal renal development in mice and may do so in humans where only Notch2 and Jagged1 mutations have been associated with defects in renal development. Notch1 mutations could act as modifiers of ALGS, increasing the severity of Alagille symptoms.
Activation of a single Notch molecule generates a unit of signal in the form of a released intracellular domain; a phenotypic threshold is reached when enough units reach the nucleus to replace the appropriate and sufficient numbers of RBP-J containing transcriptional repressor complexes with transactivating complexes. The factors that could contribute (alone or in combination) to differences between Notch1 and Notch2, and therefore to relative signal `strength', are under investigation in our laboratory. Although developing therapies aimed at elevating Notch1 activity in ALGS is a rationale goal that requires deeper mechanistic understanding to limit untoward effects, a proof of principal experiment was attempted to demonstrate feasibility of this approach. Towards this end, we report that genetically ablating Mint, a known Notch inhibitor (Kuroda et al., 2003; Tanigaki and Honjo, 2007; Tsuji et al., 2007; Yabe et al., 2007), resulted in regaining some nephrons on a Notch2 null background. This result indicates that generation of N1-ICD is not the only factor limiting the contribution of Notch1 to nephrogenesis, and efforts aimed at elevating Notch1 activity transiently downstream of receptor activation (via agonists or inhibition of antagonist) in ALGS2 patients with one functional Notch2 allele, or in ALGS1 patients with global reduction in Notch signaling, may have therapeutic benefits. The next task will be to demonstrate the benefit and safety of such strategies in animal models of ALGS (McCright et al., 2002).
Supplementary Material
Acknowledgements
We thank the histology core of Dept. of Developmental Biology for preparation of tissue sections. We thank the Mouse Genetics Core and the Digestive Diseases Research Core Center (supported by NIH P30DK052574) for the generation of genetically altered mice. We thank Mary Blandford and Tao Shen for assistance with animal husbandry and mouse genotyping. We thank Andrew McMahon for the Six2 antibody. We thank William Eades and Jacqueline Hughes in the Siteman Cancer Center High Speed Sorter Core Facility for performing cell sorting segments of our experiments. The Siteman Cancer Center is supported in part by NCI Cancer Center Support Grant # P30 CA91842. This work was supported by National Institutes of Diabetes and Digestive and Kidney Disease (DK066408) and the O'brien Center Grant (5P30DK07933). SB was supported by institutional training grant (5T32DK007126).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. Notch1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2007;283:6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- Besseyrias V, Fiorini E, Strobl LJ, Zimber-Strobl U, Dumortier A, Koch U, Arcangeli ML, Ezine S, Macdonald HR, Radtke F. Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J Exp Med. 2007;204:331–343. doi: 10.1084/jem.20061442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, de Caestecker M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol. 2008;313:234–245. doi: 10.1016/j.ydbio.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Al-Awqati Q. Segmental expression of Notch and Hairy genes in nephrogenesis. Am J Physiol Renal Physiol. 2005;288:F939–952. doi: 10.1152/ajprenal.00369.2004. [DOI] [PubMed] [Google Scholar]
- Cheng H, Miner J, Lin M, Tansey MG, Roth KA, Kopan R. g-Secretase Activity is Dispensable for the Mesenchyme-to-Epithelium Transition but Required for Proximal Tubule Formation in Developing Mouse Kidney. Development. 2003;130:5031–5041. doi: 10.1242/dev.00697. [DOI] [PubMed] [Google Scholar]
- Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134:801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bianco C, Aster JC, Blacklow SC. Mutational and energetic studies of Notch 1 transcription complexes. J Mol Biol. 2008;376:131–140. doi: 10.1016/j.jmb.2007.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demehri S, Turkoz A, Kopan R. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol. 2006;22:509–529. doi: 10.1146/annurev.cellbio.22.010305.104340. [DOI] [PubMed] [Google Scholar]
- Friedmann DR, Wilson JJ, Kovall RA. RAM-induced allostery facilitates assembly of a notch pathway active transcription complex. J Biol Chem. 2008;283:14781–14791. doi: 10.1074/jbc.M709501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;21:180–184. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48:607–616. doi: 10.1002/hep.22381. [DOI] [PubMed] [Google Scholar]
- Givogri MI, Costa RM, Schonmann V, Silva AJ, Campagnoni AT, Bongarzone ER. Central nervous system myelination in mice with deficient expression of Notch1 receptor. J Neurosci Res. 2002;67:309–320. doi: 10.1002/jnr.10128. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Cheng HT, Surendran K. Molecular Insights into Segmentation along the Proximal-Distal Axis of the Nephron. J Am Soc Nephrol. 2007;18:2014–2020. doi: 10.1681/ASN.2007040453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Xue YZ, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes & Development. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Kuroda K, Han H, Tani S, Tanigaki K, Tun T, Furukawa T, Taniguchi Y, Kurooka H, Hamada Y, Toyokuni S, Honjo T. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 2003;18:301–312. doi: 10.1016/s1074-7613(03)00029-3. [DOI] [PubMed] [Google Scholar]
- Levine J, Kueh HY, Mirny L. Intrinsic fluctuations, robustness, and tunability in signaling cycles. Biophys J. 2007;92:4473–4481. doi: 10.1529/biophysj.106.088856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Morrissette JJ, Spinner NB. Conditional JAG1 Mutation Shows the Developing Heart Is More Sensitive Than Developing Liver to JAG1 Dosage. Am J Hum Genet. 2003;72:1065–1070. doi: 10.1086/374386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman OY, Ilagan MX, Kopan R, Barrick D. Quantitative Dissection of the Notch:CSL Interaction: Insights into the Notch-mediated Transcriptional Switch. J Mol Biol. 2007;365:577–589. doi: 10.1016/j.jmb.2006.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075–1082. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugford JW, Yu J, Kobayashi A, McMahon AP. High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev Biol. 2009;333:312–323. doi: 10.1016/j.ydbio.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry EP, Latifi T, Towler DA. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry. 1999;38:10678–10690. doi: 10.1021/bi990967j. [DOI] [PubMed] [Google Scholar]
- Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, Ludwig L, Wilda M, Hameister H, Knochel W, Liptay S, Schmid RM. SHARP is a novel component of the Notch/RBP-J signalling pathway. Embo J. 2002;21:5417–5426. doi: 10.1093/emboj/cdf549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Valerius MT, McMahon AP. Wnt/ß -catenin signaling regulates nephron induction during mouse kidney development. Development. 2007;134:2533–2539. doi: 10.1242/dev.006155. [DOI] [PubMed] [Google Scholar]
- Piccoli DA, Spinner NB. Alagille syndrome and the Jagged1 gene [Review] Seminars in Liver Disease. 2001;21:525–534. doi: 10.1055/s-2001-19036. [DOI] [PubMed] [Google Scholar]
- Piscione TD, Wu MY, Quaggin SE. Expression of Hairy/Enhancer of Split genes, Hes1 and Hes5, during murine nephron morphogenesis. Gene Expr Patterns. 2004;4:707–711. doi: 10.1016/j.modgep.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, Aster JC, Krishna S, Metzgers D, Chambon P, Miele L, Aguet M, Radtke F, Dotto GP. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO Journal. 2001;20:3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler RD, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. Embo J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratman JL, Barnes WM, Simon TC. Universal PCR genotyping assay that achieves single copy sensitivity with any primer pair. Transgenic Res. 2003;12:521–522. doi: 10.1023/a:1024225408961. [DOI] [PubMed] [Google Scholar]
- Tanigaki K, Han H, Yamamoto N, Tashiro K, Ikegawa M, Kuroda K, Suzuki A, Nakano T, Honjo T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat Immunol. 2002;3:443–450. doi: 10.1038/ni793. [DOI] [PubMed] [Google Scholar]
- Tanigaki K, Honjo T. Regulation of lymphocyte development by Notch signaling. Nat Immunol. 2007;8:451–456. doi: 10.1038/ni1453. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Shinkura R, Kuroda K, Yabe D, Honjo T. Msx2-interacting nuclear target protein (Mint) deficiency reveals negative regulation of early thymocyte differentiation by Notch/RBP-J signaling. Proc Natl Acad Sci U S A. 2007;104:1610–1615. doi: 10.1073/pnas.0610520104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadkar P, Kraman M, Despres D, Ma G, Lozier J, McCright B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev Dyn. 2008;237:1144–1152. doi: 10.1002/dvdy.21502. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Ong CT, Hadland B, Huppert S, Liu Z, Korving J, van den Born M, Stappenbeck T, Wu Y, Clevers H, Kopan R. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development. 2007;134:535–544. doi: 10.1242/dev.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Pereira FA, Beasley D, Zheng H. Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development. 2003;130:5019–5029. doi: 10.1242/dev.00682. [DOI] [PubMed] [Google Scholar]
- Yabe D, Fukuda H, Aoki M, Yamada S, Takebayashi S, Shinkura R, Yamamoto N, Honjo T. Generation of a conditional knockout allele for mammalian Spen protein Mint/SHARP. Genesis. 2007;45:300–306. doi: 10.1002/dvg.20296. [DOI] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.