

Abstract
It has long been assumed that the red cell membrane is highly permeable to gases because the molecules of gases are small, uncharged, and soluble in lipids, such as those of a bilayer. The disappearance of 12C18O16O from a red cell suspension as the 18O exchanges between labeled CO2 + HCO3− and unlabeled HOH provides a measure of the carbonic anhydrase (CA) activity (acceleration, or A) inside the cell and of the membrane self-exchange permeability to HCO3− (Pm,HCO−3). To test this technique, we added sufficient 4,4′-diisothiocyanato-stilbene-2,2′-disulfonate (DIDS) to inhibit all the HCO3−/Cl− transport protein (Band III or capnophorin) in a red cell suspension. We found that DIDS reduced Pm,HCO−3 as expected, but also appeared to reduce intracellular A, although separate experiments showed it has no effect on CA activity in homogenous solution. A decrease in Pm,CO2 would explain this finding. With a more advanced computational model, which solves for CA activity and membrane permeabilities to both CO2 and HCO3−, we found that DIDS inhibited both Pm,HCO−3 and Pm,CO2, whereas intracellular CA activity remained unchanged. The mechanism by which DIDS reduces CO2 permeability may not be through an action on the lipid bilayer itself, but rather on a membrane transport protein, implying that this is a normal route for at least part of red cell CO2 exchange.
18O exchange between C18O16O/HC18O16O2− and H216O in a red cell suspension at chemical equilibrium can be used in principle to measure the proportional intracellular acceleration (A) of CO2 hydration produced inside intact red blood cells by carbonic anhydrase (CA) compared with the uncatalyzed rate (1) and the self-exchange permeability of the membrane to HCO3 (Pm,HCO−3). As predicted, A was found to be the same in intact erythrocytes as in hemolysate, both under normal conditions and when exposed to varying concentrations of a membrane-permeable CA sulfonamide inhibitor (ethoxzolamide), whereas Pm,HCO−3 was not affected (2). Validation of the ability of the technique to differentiate between CA activity and HCO3− permeability in intact red cells was extended by exposing them to phlorizin (3), an established Band III inhibitor (1); this drug decreased Pm,HCO−3 but did not change A.
Recently we exposed red cells to a more specific inhibitor of Band III protein, 4,4′-diisothiocyanato-stilbene-2,2′-disulfonate (DIDS), to decrease Pm,HCO−3 without inhibiting CA. However, we found that whereas DIDS lowered HCO3− transport, it also lowered the membrane permeability to CO2.
METHODS
Two groups of experiments were carried out, one in Philadelphia and one in Hannover, Germany. Blood from healthy adults, freshly drawn into heparin, was washed three times with 145 mM NaCl at 4°C; the red cells were diluted to approximately 40% hematocrit and used the same day. Hemolysates were prepared by freezing and thawing the cell suspension.
The fractional water volume, v, of the red cells in the reaction suspension was calculated as the hematocrit of the stock cell suspension × water content of red cells [0.61 (1) for the Philadelphia experiments and 0.65 (4) for the Hannover experiments] × its dilution in the reaction mixture. In later experiments, v was calculated as 55.5 × [cyanmethemoglobin] in mM in the reactant mixture. NaHCO3 was prepared by incubating 2% and 5% 18O labeled HOH with unlabeled NaHCO3 at 150°C in a pressure bomb for several days, after which the water was removed by lyophylization. DIDS was obtained from Research Organics and phlorizin (phloridzin) was obtained from Sigma.
The technique of the 18O exchange method for measuring A and PHCO−3 has been described in detail (1, 5, 6), so only a brief summary will be given here. The reactions take place in an airtight 3-ml glass water-jacketed and stirred chamber connected to the ion source of a mass spectrometer through a thin 0.012-mm-thick Teflon membrane supported by a sintered glass disc, through which gases are dissolved in the solution diffuse. The chamber has a glass pH electrode and a removable stopper for the addition of reactants. For experiments done in Philadelphia, the mass spectrometer was at first a Model 21 620A (Consolidated Electrodynamics, Pasadena, CA) and later a Model CH7 Varian MAT, which recorded masses 32 (16O2), 44 (12C16O2), and 46 (12C18O16O) peak every 22 sec on a strip chart. For experiments done in Hannover, the mass spectrometer was a Europa 1 stable isotope detector (Europa Scientific, Crewe, U.K.), which had three separate detectors tuned to record masses 44 (12C16O2), 45 (13C16O2), and 46 (12C18O16O) simultaneously, and a computer program that calculated and recorded the ratio [12C18O16O]/[12C16O2] once per second. The response time of the whole system to a change in 12C18O16O2 in the reaction mixture was about 3 sec, including a time delay.
To initiate an experiment, sufficient dry 18O-labeled NaHCO3 to produce a 25-mM solution was added to 120 mM NaCl in the chamber. pH was adjusted within several minutes, and an aliquot of red cell suspension or of hemolysate was added sufficient to produce a fractional cell water volume, from about 0.000095 to 0.001.
The overall CO2 hydration velocity constant, k1, in homogeneous solution can be calculated from the linear semilogarithmic slope (n) of the uncatalyzed or catalyzed reaction of hemolysate (Figs. 1 and 2), by using the equation of Mills and Urey (7).
 |
1 |
where: Φ = {1 + [H+]/K′} ± ((1 + [H+]/K′)2 − 4/3 × [H+]/K′)1/2;
Figure 1.

A semilogarithmic graph of the disappearance of 12C18O16O, mass 46, with time, in the presence of intact human red blood cells at 37°C and pH 7.4, from the Philadelphia experiments. At time zero, dry NaHCO3 2% enriched with 18O to produce 25 mM was added to 125 mM saline and at the arrow, an aliquot of the red cell suspension producing a v of 0.0013 in the control (open circles) and 0.00121 in the DIDS (solid circles) experiment. For the control graph (experiment 25, run 8) and DIDS graph (experiment 25, run 3), the SRs were 3.1 and 3.2 and n’s for the regressions were 0.014 and 007 sec−1, respectively.
Figure 2.

A semilogarithmic graph of the disappearance of 12C18O16O under the same conditions as in Fig. 1, except lysate was added instead of red blood cells. At the arrow, hemolysate was added equivalent to a cell fractional water volume, v, of 0.001. In the DIDS experiment (solid circles), the solution contained 0.4 μM DIDS.
n = the exponential constant after hemolysate has been added. K′ = [HCO3] [H+]/[CO2], the Henderson–Hasselbalch constant, taken to be 10−6.1;
[H+] = 10−7.4;
ku, the uncatalyzed hydration reaction velocity constant = 0.18 sec−1 (1);
kcat = effective catalyzed reaction velocity constant, calculated by dividing the catalyzed reaction rate in M × sec−1 by [CO2];
k1 = ku + kcat, and acceleration (A) of the CO2 reaction by CA = k1/ku.
The diffusion and chemical processes taking place during the exchange of 18O in a red cell suspension are more complicated than in homogenous solution and are diagrammed in the cartoon of Fig. 3. The initial rapid phase is produced by 12C18O16O diffusing into the cells, where the 18O is exchanged with HOH at a rate accelerated some 14,000 times by intracellular CA. This rapid intracellular consumption of 12C18O16O in relation to its production at the uncatalyzed rate from extracellular HC18O16O3 lowers 12C18O16O in the extracellular solution. If the intracellular carbonic anhydrase were as accessible to HCO3− as to 12C16O2, there would be no step, that is, no initial rapid phase, as can be seen in Fig. 2, in the presence of hemolysate. Therefore, the lower the effective membrane permeability to HCO3−, the larger the step and the lower [12C18O16O] in the suspending fluid at the end of the first phase. Itada et al. (1) developed a double exponential equation, the first term of which describes the rapid initial fall in mass 46, and the second term describes the slower linear (exponential) phase as a function of the intracellular CA activity (A), the membrane permeability to HCO3− (Pm,HCO−3) and v the fractional cell water volume. With this equation, one can calculate the CA activity inside the cell (A) and HCO3− permeability (PHCO−3) from the experimental step ratio (SR) and n, on the assumption that CO2 is highly permeable through the cell membrane. The rate of the initial rapid drop was too fast for the Philadelphia instrumentation to follow accurately, so we measured instead the SR, defined as the reciprocal of the fractional drop in mass 46 peak during the first fast phase.
Figure 3.

A cartoon [Fig. 6 from (1)] of a red blood cell and the reversible reaction of CO2 and transmembrane exchanges taking place intra- and extracellularly during 18O exchange. The solid circles represent 18O and the open circles represent 16O. The fractions appended to the reaction arrows indicate the proportion of the total chemical reaction going to the designated product.
The relation between the exponential decay constant, n, of the second phase and the acceleration of CO2 hydration by carbonic anhydrase in the cells is as follows.
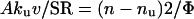 |
2 |
On the left-hand side of the equation, Aku is the catalyzed reaction velocity constant in the cells. This product is multiplied by v because the catalyzed reaction takes place only in this small fraction of the total volume and is reduced by 1/SR because the 18O abundance in 12C18O16O is only this fraction of its abundance in the extracellular H12C18O16O2−, which approximates that in 12C18O16O before the cells were added. On the right-hand side, the reaction velocity constant of the catalyzed process is calculated from the exponential decay constant, n. According to Eq. 1, nu is the velocity constant for uncatalyzed exchange that is seen in Figs. 1 and 2 before cells or lysate are added. This constant can also be calculated from Eq. 2 and the normal value of ku, 0.18 second. From Eq. 2, Av should be proportional to (n − nu)SR, the other factors remaining constant. Although CA-catalyzed reactions follow Michealis–Menten kinetics, because the system is at chemical equilibrium we can approximate it as a simple monomolecular reaction with the velocity constant, kcat.
The mass spectrometer in Hannover was able to record the exponential decay constant of the rapid phase as well as its magnitude and the exponential constant of the slow phase. Therefore, we solved the set of six linear differential equations describing the change with time of the intra- and extracellular concentrations of the labeled species CO2, HCO3− and HOH (8). Each of these equations contains a term for the chemical reactions and the transmembrane flux affecting the species concentration. Solution of this system of differential equations was achieved with the software package matlab (Mathworks, Natick, MA). Parameter values were obtained from a fitting procedure generating an optimal fit of the numerical solution and the experimental data (such as shown in Fig. 4). All data points (one data point per second) of curves like those in Fig. 4 were included in the fitting procedure. This meant that the time course not only of the second slow phase but also of the first fast phase was used for parameter estimation. The continuous recording made this latter datum available, whereas previously we have been able to use only the magnitude of the rapid drop in 12C18O16O, the SR, but not its time course. The water permeability of the red cell membrane was taken from the literature to be 0.002 cm sec−1 (9). The fitting procedure yielded best estimates of A, Pm,HCO−3, and PCO2.
Figure 4.

A graph of the disappearance of 12C18O16O, mass 46, with time, in the presence of intact human red blood cells at 37°C and pH 7.4, from the Hannover experiments. At time zero, dry NaHCO3 2% enriched with 18O to produce 25 mM was added to 125 mM saline and at the first arrow, an aliquot of red cell suspension sufficient to produce a v of 0.0000953 in the control (open circles) and in the DIDS (solid circles) experiment. For the control experiment, the intracellular acceleration (A) was 24,700, Pm,HCO−3 was 0.00255 cm × sec−1, and Pm,CO2 was 2.19 cm × sec−1. For the experiment in which red cells were preincubated in 0.1 M DIDS, A was 26,740, Pm,HCO−3 0.00121 cm × sec−1, and Pm,CO2 0.0664 cm × sec−1. At the second arrow, CA was added to accelerate the exchange and to provide a measure of the 46 peak height at final equilibrium. Curves calculated from the Hannover theoretical model are also plotted, but lie so closely on the experimental curves that they cannot be distinguished.
RESULTS
The results of typical experiments are shown in Figs. 1 and 2. Immediately after the labeled NaHCO3 was added, the uncatalyzed reactions caused the mass 46 peak to decrease slowly as 18O exchanged with 16O in water. When red cells were added (Fig. 1), the mass 46 peak fell rapidly and then resumed a slower linear decrease when plotted semilogarithmically. In the control experiment, SR was 3.0 and the rate constant, n, was 0.017 sec−1. In the DIDS experiments, the procedure was the same except that the red cells were preincubated in 4 μM DIDS for 45 minutes. In the experiment in Fig. 1, the SR was 3.2, approximately the same as in the control experiment, but n decreased to 0.01 sec−1 so that, according to Eq. 2, intracellular CO2 hydration velocity was inhibited.
We repeated these experiments in fractional cell volumes from 0.00008 to 0.0056 in 13 different samples of fresh human red cells in 52 experiments in the presence of from 0.004 to 6.5 μM DIDS, compared with 42 controls in the absence of DIDS. There was considerable scatter in the data at the low red cell volumes because of reduced sensitivity. As expected, DIDS decreased Pm,HCO−3 in 51 of 52 preparations. Unexpectedly, pooling all experiments, including those at low red cell volumes, A was decreased from an average of 16,200 (SEM ± 1032) to 12,700 (±591) by DIDS, which was significant (P < 0.01). We also graphed (n × SR) against v, where the slope is proportional to A according to Eq. 2. The regression for the experiments with DIDS had a significantly lower (P < 0.01) slope, again indicating that the drug appears to inhibit CA in the cell.
To determine whether DIDS inhibited CA, we carried out a series of experiments in which red cell lysate was added to 120 mM NaCl reaction solution (control) and to the same solution containing 10 μM DIDS. The time course of 12C18O16O2 disappearance in homogeneous solution was much simpler, as shown in Fig. 2. There was no rapid initial step because the CO2 and HCO3 were equally accessible to the active site of the carbonic anhydrase. The slope, n, was 0.042 sec−1 for the control experiment and 0.043 sec−1 for the DIDS experiment, which is not a significant difference. We did similar comparisons on three human hemolysates, one human CA III, and one bovine CA II preparation, a total of seven experiments without DIDS and twelve with DIDS concentrations sufficient to bind all Band III proteins in the membranes.
DIDS does not inhibit CA activity in human hemolysate or in solutions of human CA III or bovine CA. Because the intracellular CO2 reaction velocity was decreased by DIDS and the enzyme activity was unaffected by the drug, we propose that the intracellular [12C18O16O] was decreased by inhibition of CO2 transport through the membrane. This inhibition would impede intracellular 12C18O16O exchange and reduce both n and the SR, the latter opposing the increase in SR produced by the inhibition of HCO3− transport. Because our original mathematical model of 18O exchange in cell suspensions (1) assumes membrane CO2 permeability is infinite, we could not use this model to analyze the data further.
Therefore, a second group of experiments on human red cells in the presence and absence of 100 μM DIDS and of an equivalent concentration of hemolysate was carried out in Hannover. The mass spectrometer recorded masses 44 and 46 and computed the abundance of 18O continuously (every second), as shown in Fig. 4. One of us (M.W.) solved the set of six linear differential equations, including the reversible chemical reactions, intra- and extracellular, of CO2 and HCO3− and their diffusion across the red cell membrane along with that of HOH (see Fig. 3), with the software package matlab. The experimental data used were the exponential constant for the slow phase and for the fast phase, instead of the magnitude of the rapid drop (SR) previously chosen, because the continuous recording made this datum available. The permeability of the membrane to water was considered infinitely high.
Typical experimental records and calculated time courses are shown in Fig. 4 for a control experiment and for an experiment with DIDS. Note that the rapid phase is noticeably slower in the presence of DIDS, indicating a decreased Pm,CO2. Because the rate of 18O exchange in the fast phase is at least an order of magnitude greater than in the slow phase and depends mainly on CA activity and Pm,CO2, and because there is initially no gradient of labeled HCO3− across the cell so that no anion exchange occurs, the effect of a decrease in Pm,CO2 is more noticeable. The results of the new calculations on eight experiments analogous to those in Fig. 4 are summarized in Table 1. DIDS (100 μM) did not alter the calculated CA activity in the intact red cells, but did lower Pm,HCO−3 to about half. The interesting finding is that membrane CO2 permeability, from a value of 1 cm × sec−1 or higher in the control experiments, was lowered by the drug to a value of 0.1 cm × sec−1.
Table 1.
Effect of DIDS on acceleration (A) and membrane permeability to HCO3− (Pm,HCO−3) and CO2 (Pm,CO2)
| Intact red cells | Hemolysate | Intact red cells + DIDS | |
|---|---|---|---|
| Acceleration (A) | 17,220 ± 1,120 | 17,780 ± 4,410 | 17,710 ± 3,080 |
| Pm,HCO−3, cm/sec | 0.00164 ± 0.00015 | 0.00080* ± 0.00010 | |
| Pm,CO2, cm/sec | >1 | 0.09* ± 0.04 |
n = 8, at 37°C and pH 7.4. Fractional red cell water volume (v) in reaction mixture averaged 0.000095.
DIDS = 10−4 M. Acceleration (A) is the intracellular CO2 hydration velocity constant divided by the uncatalyzed constant at 37°C (0.18 sec−1). Values with ∗ were significantly reduced; all other values were not.
DISCUSSION
The permeability of the red cell membrane to gases is so great that it has been technically impossible to determine. Forty years ago, Roughton and colleagues (10) investigated the diffusion resistance of the red cell membrane to CO, O2, and NO by comparing: (i) the calculated initial rate of gas uptake by a membraneless layer of hemoglobin, analogous to a red cell, using measurements of reaction velocities and diffusion coefficients in homogenous solution with (ii) experimental measurements of gas uptake by red cell suspensions using a continuous-flow rapid-mixing apparatus. The calculated rate, assuming there was no membrane resistance, divided by the experimental rate was from 1.5 to 5.5, suggesting that the membrane posed a significant resistance to gas transport. There were many approximations in the calculations and the discrepancy did not increase consistently with the velocity of the ligand–hemoglobin reactions, as would be expected. Therefore, the results have remained unconvincing.
Constantine et al. (11) measured the initial rate of CO2 uptake by red cells with a continuous-flow rapid-mixing apparatus and a PCO2 electrode and found it only 60% of the theoretical expected value predicted from the reaction velocity constants for the hydration of CO2 in hemolysate, again suggesting that there is a significant resistance in the membrane. This result suggests that a drop in [CO2] across the cell wall might be lowering the intracellular CO2 reaction rate, particularly as Barinov et al. (2) and Itada et al. (1) had found the extra- and intracellular enzyme rates to be the same. However, the red cell CO2 uptake becomes limited by the accumulation of H+ and HCO3− so quickly (12) that it was unlikely that Constantine et al. were able to measure the actual enzyme catalyzed reaction velocity in intact cells.
Silverman and colleagues reported studies on 18O exchange of rat red cell suspensions in a series of stimulating papers from 1974 to 1981 (5, 13–15). Their final conclusion was that diffusion resistance across the red cell membrane, rather than the intracellular CA activity rate, limited CO2 exchange. The value of Pm,CO2 that they obtained in rat red cells was 0.0075 cm × sec−1 (15), at least two orders of magnitude less than the approximate value for normal human red cells in Table 1. This value of Pm,HCO−3 also appears to be too small in comparison with the value for an artificial lipid bilayer of 0.35 cm/sec obtained by Gutknecht, Bisson, and Tosteson (16). In their 1981 report, Silverman, Tu, and Roessler (15) found that methazolamide did not inhibit CA in intact red cells to the same extent as in hemolysate, supporting their conclusion that the rate of 18O exchange was limited more by the permeability of the membrane than by the effective catalyzed reaction velocity constant inside the cell. However, Barinov et al. (2) measured 18O exchange at low enrichment (about 2%), calculating A from their data with the equations of Itada et al. (1), and found the same fractional inhibition by ethozxolamide in intact human red cells as in lysate; the Ki was the same for both, 2.6 × 10−9 M. A significant Δ [12C18O16O] across the membrane is incompatible with these results. Barinov et al. calculated, allowing for a 10% error in their data, that 1.5 cm × sec−1 was a minimal value for Pm,CO2. We have no explanation for the differences between our findings and those of Silverman et al. They carried out pertinent experiments in the presence of 0.08 M methazolamdie to inhibit about half the intracellular CA activity in lysate to improve the accuracy of the measurements, but this should not have produced the difference between their results and ours. The properties of rat red cells also would not be expected to differ that much from those of man. We point out that the equations used to calculate the results of Table 1, developed by Wunder et al. (8), represent the first complete mathematical description of 12C18O16O exchange in cell suspensions and that all previous approaches, including those by Silverman et al. (14) and Itada et al. (1), contain various simplifications.
Gros and Bartag (17) compared the rate of diffusion of CO2 through 1-mm-thick layers of packed intact red cells vs. similar layers of membrane-free hemoglobin solution of equivalent concentration and found that the presence of a cell membrane lowered CO2 transport by a significant 6.5%. A comparison of nearly hemoglobin-free ghosts and an equivalent hemoglobin solution (1.1 g per 100 ml) gave similar results. From these data, they estimated a membrane CO2 permeability of 1.6 to 3.4 cm sec−1.
The failure of DIDS to increase the SR might be a technical artifact caused by a delay in its binding to the cell. There are comments in the literature (18) that DIDS reacts with Band III slowly and that it may dissociate. Incubating the red cells in DIDS (n = 26) before an experiment did not lower Pm,HCO−3 to a greater extent than did adding the cells directly to a reaction solution containing DIDS, which exposed them to the drug only a few seconds before effects were seen (see Fig. 1). We conclude that the apparent decrease in CA activity caused by DIDS was not caused by any delay in the action of the drug.
Diffusion resistance in a stagnant layer around the cells in the membrane proper and within the cytoplasm could be interpreted as resistance in the bilayer. The first of these possible diffusion resistances is considered next. Although the cell suspension in the reaction chamber is stirred continuously, the vigor is in nowise comparable to the mixing in a rapid mixing apparatus, and even in these instruments there is a stagnant layer around each cell (19). However, 24 mM bicarbonate should act as a transport facilitator and, by dehydration, should supply 12C18O16O at the very surface of the membrane, as explained by Gutknecht et al. (16). Unfortunately, CA cannot be added to the suspending solution to amplify this facilitation as they did. If an unstirred layer were important, it would produce an underestimate of Pm,CO2. Because the CA activity in intact cells is the same as in lysate using the 18O exchange method (2), a stagnant layer does not appear to decrease CO2 transport significantly. DIDS should have no effect on the stagnant layer in any case.
Simultaneous chemical reaction and diffusion within the cell can also slow 18O exchange. As 12C18O16O2 diffuses into the red cell, its 18O exchanges with the unlabeled water so the diffusion gradient is reduced, reducing the overall rate of exchange in the cell. This consumption of 18O makes the average [12C18O16O2] in the cell less than that at the surface, with the same effect as diffusion resistance in the membrane. Constantine et al. (11) computed the magnitude of this effect in a semiinfinite layer of 0.8-micron-thick hemoglobin on the rate of uptake of CO2 and found the average intracellular [CO2] was 0.76 of that outside the cell. The equation used, developed by Roughton et al. (20), was
 |
3 |
where ω = b(k/D)1/2 and b is the half-thickness of the layer in cm, k is the hydration velocity constant in sec−1, and D is the diffusion coefficient in cm2 × sec−1. In the present case of C18O16 exchange, the thickness assumed for the cell is the same, D for CO 2 is taken as 6.5 × 10−6 × cm2 × sec−1, and the exchange velocity constant as 44.8 sec−1. This value is calculated from the Mills and Urey relationship (Eq. 1), a CA acceleration of 14,000, an uncatalyzed hydration velocity constant of 0.18 sec−1, and a pH of 7.4. The average intracellular [CO2]/extracellular [CO2] is 1, so that the rate of 18O exchange should be the same in cells and in lysate, as Barinov et al. found (2).
Silverman (15) made a similar calculation and found that the average intracellular [CO2] would be significantly reduced. However, he used a sphere of equal volume to a red cell as a model that increased the diffusion distance from the surface almost 3-fold. We believe the semiinfinite layer in which gases can diffuse from either surface is a better model of the discoid red cell. Silverman also appeared to use a hydration reaction velocity constant instead of the exchange velocity constant. The difference in these values accounts for his different conclusion. We believe simultaneous chemical reaction and diffusion do not contribute to a ‘membrane’ resistance.
The rate of O2 uptake by red cells is about 1/30th of that in hemolysate, owing to the effect of simultaneous chemical reaction and diffusion, in distinct contrast to CO2, a fact that deserves explanation. This difference results simply because the velocity constant k in Eq. 3 for O2 is 3,500 mM−1 × sec−1 × 18 mM hemoglobin, equal to 63,000 sec−1, 1,400 times larger than the exchange velocity constant for 18O in CO2, making average cell [O2] about 1/30 of the surface concentration according to Eq. 3.
To measure PCO2 in red cells, it is necessary to produce a flux across the membrane high enough that the transmembrane concentration can be detected. The flux of labeled CO2 into the red cell equals (n − nu)[12C18O16O]21. The number 21 is ([CO2] + [HCO3]), the total pool of 18O that is exchanging 18O with water divided by [CO2]. nu is the uncatalyzed exchange rate in the extracellular fluid. The 12C18O16O diffusion into the cells equals Pm,CO2 20,000 v Δ[12C18O16O], where 20,000 is the red cell surface area/volume in cm−1 (21) and v is the red cell volume in the reaction mixture. Therefore, setting these two statements equal to each other, one obtains
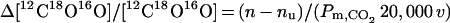 |
4 |
By using the data for the control experiment in Fig. 1, n = 0.014 sec−1, nu = 0.003 sec−1, and v = 0.0013, the right-hand side of Eq. 4 becomes 0.009/Pm,CO2. This means that if Pm,CO2 is as large as 3 cm × sec−1, the Δ[[12C18O16 ] across the membrane would be only 0.3% of the [12C18O16O], producing an equivalent small decrease in flux of 12C18O16O. However, this decrease in flux into the cells would raise [12C18O16O], decreasing the SR by a greater proportion. If Pm,CO2 is decreased to 0.09 cm × sec−1, as in Table 1, the change in uptake is dropped 10%, a measurable value. It is the effect of DIDS that makes the CO2 permeability detectable by using presently available techniques.
In an analogous calculation for O2 uptake by red cells, the flux of ligand gas into the red cell is the bimolecular reaction velocity constant for the combination of O2 with hemoglobin in intact cells, 92 mM−1 sec−1 (20) times the intracellular hemoglobin concentration, 18 mM. Following the same arguments as for CO2, Δ[O2]/[O2 ] = 0.097/Pm,O2. Thus a Pm,O2 of 3 cm × sec−1 should reduce O2 uptake by 3%, which might be detected. Unfortunately, Roughton et al. (10) had to compare experimental gas uptake by cells with computed values, whose error was probably much greater.
To summarize, our conclusions are that the CO2 permeability of the red cell membrane (Pm,CO2) is normally so great that it is difficult to measure, but that exposure to DIDS lowers it enough that it can be measured precisely with the 18O exchange method. Two possible explanations for this action of DIDS are a decrease in the permeability of the phospholipid bilayer itself and inhibition of a transport protein. Classically, like other gases, CO2 has been assumed to traverse a cell membrane by the first mechanism, that is by dissolving in the phospholipid bilayer and diffusing through its structure (22). The flux through a given surface area and membrane thickness should be proportional to the solubility of CO2 in the lipid membrane over that in plasma (saline), the partition coefficient times the diffusion coefficient of CO2 in the membrane. The permeability of cell membranes to a solute has long been known to be linearly related to the oil/water partition coefficient of that solute (22). However, the oil/water partition of CO2 is near unity and can be half that in egg lecithin liposome bilayers. The oils often used as models for membrane investigations of membrane permeability have a greater CO2 solubility than do lipid bilayers (23). The CO2 permeability of an artificial lipid black membrane studied by Gutknecht et al. (16) was high, 0.3 cm × sec−1, but this property could be much lower if the lipid composition were different. The isothiocyanate groups in DIDS might react with amine groups in the phospholipids of membrane, changing its structure and decreasing its permeability.
For CO2 to permeate by means of a membrane protein is not unreasonable and has been suggested before (15, 24). The human red cell membrane is at least 50% protein (25). CO2 would have to pass through a channel or pore rather than by a saturating process, which would be too slow. The self-exchange of HCO3− in red cells through Band III (capnophorin) is extremely rapid, and the cells have an effective permeability under physiological conditions of 4 × 10−4 cm × sec−1 (1), with about 2.5 million proteins per cell (26), but this is about 1/10,000 of our minimal estimate of CO2 permeability. Two obvious candidates for a CO2 transport protein are Band III and aquaporin. Considering the red cell area/volume as 20,000 cm−1 (20), there was on the average 540 times this concentration of DIDS in the experiments in Philadelphia and 31,000 times in the experiments in Hannover. Thus there was enough free DIDS in the cell suspension to react with all the capnophorin and possibly with other components of the membrane to change its solubility and/or diffusion coefficient. However, Itada et al. (3) measured CA activity and Pm,HCO−3 by the 18O exchange method in the absence (n = 3) and presence (n = 22) of 1 and 2 mM phlorizin, an inhibitor of Band III (v was 0.00017 to 0.0017). Pm,HCO−3 was reduced by 50%, as expected, but the experimental value of intracellular carbonic anhydrase activity was not decreased, so there was no indication of a decreased Pm,CO2. This result makes it less likely that the action of DIDS is to inhibit Band III. However, Band III might have a transport channel for CO2, a linear uncharged unpolarized molecule, which is different from that for HCO3/Cl exchange and, while inhibited by DIDS, is not inhibited by phlorizin. The other protein candidate is aquaporin, which is present in the red cell membrane in high concentration. Boron and Cooper (27) have reported in an abstract that DIDS inhibits the CO2 flux through the aquaporin proteins inserted into Xenopus ova membranes, but does not impede water flux through these channels.
These results lead us to favor an inhibitory action by DIDS on a transport protein in the red cell membrane to explain its effect on CO2 permeability. If borne out, this means that contrary to classical theory, a significant portion of CO2 fluxes across cell membranes in gas exchange is carried by a membrane protein(s), altering our understanding of respiratory gas exchange and raising the possibility of unsuspected dysfunction in disease.
Acknowledgments
We thank Mrs. Sisko Bauer for her expert technical help in the Hannover experiments. We also acknowledge the helpful criticisms of Dr. Bayard T. Storey and Dr. Robert Post. This research was supported in part by a grant from National Institutes of Health HL 47815 and in part by a grant from the Deutsche Forschungsgemeinschaft (SFB 28).
ABBREVIATIONS
- CA
carbonic anhydrase (EC 4.2.1.1)
- DIDS
4,4′-diisothiocyanato-stilbene-2,2′-disulfonate
- k1
CO2 hydration velocity constant
- A
proportional acceleration of CO2 hydration by carbonic anhydrase
- the effective velocity constant for the reaction of CO2 hydration
k1 divided by the uncatalyzed reaction velocity constant ku
- Pm
HCO−3, membrane permeability of HCO3− in cm × sec−1
- Pm
CO2, membrane permeability of CO2 in cm × sec−1
- SR
step ratio
References
- 1.Itada N, Forster R E. J Biol Chem. 1977;252:3881–3890. [PubMed] [Google Scholar]
- 2.Barinov I, Dodgson S J, Forster R E, Itada N, Lin L. J Physiol. 1985;371:159. [Google Scholar]
- 3.Itada N, Chow E, Forster R E. Fed Proc. 1976;35:750. (abstr.). [Google Scholar]
- 4.Diem K, editor. Scientific Tables. 6th Ed. Ardsley, New York: Geigy Pharmaceuticals; 1962. p. 576. [Google Scholar]
- 5.Tu C, Wynn G C, McMurray R E, Silverman D N. J Biol Chem. 1978;253:8178–8184. [PubMed] [Google Scholar]
- 6.Dodgson S J, Gros G, Krawiec J, Lin L, Bitterman N, Forster R E. J Appl Physiol. 1990;68:2443–2450. doi: 10.1152/jappl.1990.68.6.2443. [DOI] [PubMed] [Google Scholar]
- 7.Mills C A, Urey H C. J Am Chem Soc. 1940;62:1019–1026. [Google Scholar]
- 8.Wunder M, Bòllert P, Gros G. Isotopes Environ Health Stud. 1997;33:197–205. doi: 10.1080/10256019808234064. [DOI] [PubMed] [Google Scholar]
- 9.Forster R E. In: Current Topics in Membranes and Transport. Bronner F, Kleinzeller A, editors. New York: Academic; 1971. , 2, 41–97. [Google Scholar]
- 10.Roughton F J W. In: Handbook of Physiology. Respiration I. Fenn W O, Rahn H, editors. Washington, DC: Am. Physiol. Soc.; 1964. pp. 767–825. [Google Scholar]
- 11.Constantine H, P, Craw M R, Forster R E. Am J Physiol. 1965;208:801–811. doi: 10.1152/ajplegacy.1965.208.4.801. [DOI] [PubMed] [Google Scholar]
- 12.Forster R E, Crandall E D. J Appl Physiol. 1975;38:710–718. doi: 10.1152/jappl.1975.38.4.710. [DOI] [PubMed] [Google Scholar]
- 13.Silverman D N. Mol Pharmacol. 1974;10:820–836. [Google Scholar]
- 14.Silverman D N, Tu C, Wynn G C. J Biol Chem. 1976;251:4428–4435. [PubMed] [Google Scholar]
- 15.Silverman D N, Tu C K, Roessler N. Respir Physiol. 1981;44:285–298. doi: 10.1016/0034-5687(81)90024-4. [DOI] [PubMed] [Google Scholar]
- 16.Gutknecht J, Bisson M A, Tosteson D C. J Gen Physiol. 1977;69:779–794. doi: 10.1085/jgp.69.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gros B, Bartag I. Pflügers Arch. 1979;382:83. (R21). [Google Scholar]
- 18.Knauf P. In: Current Topics in Membranes and Transport. Bronner F, Kleinzeller A, editors. New York: Academic; 1978. , 12, 249–363. [Google Scholar]
- 19.Coin J T, Olson J S. J Biol Chem. 1979;254:1178–1190. [PubMed] [Google Scholar]
- 20.Roughton F J W, Forster R E. J Appl Physiol. 1957;11:290–302. doi: 10.1152/jappl.1957.11.2.290. [DOI] [PubMed] [Google Scholar]
- 21.Ponder E. Hemolysis and Related Phenomena. New York: Grune and Stratton; 1948. p. 14. [Google Scholar]
- 22.Collander R. Physiol Plant. 1949;2:300–311. [Google Scholar]
- 23.Simon S A, Gutknecht J. Biochim Biophys Acta. 1980;596:352–358. doi: 10.1016/0005-2736(80)90122-4. [DOI] [PubMed] [Google Scholar]
- 24.Forster R E. In: CO2: Chemical, Biochemical and Physiological Aspects. Forster R E, Edsall J T, Otis A B, Roughton F J W, editors. NASA; 1969. pp. 53–59. [Google Scholar]
- 25.Guidotti G. In: Membrane Physiology. Andreoli T E, Hoffman J F, Fanestil D D, editors. New York: Plenum; 1978. pp. 49–60. [Google Scholar]
- 26.Jennings M L, Solomon A K. J Gen Physiol. 1976;67:381–397. doi: 10.1085/jgp.67.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boron W F, Cooper G J. Biophys J. 1998;74:A374. [Google Scholar]