

Abstract
Over the last 10 years, promising data has emerged from both animal and human studies that both active immunization with amyloid-β (Aβ) as well as passive immunization with anti-Aβ antibodies offer promise as therapies for Alzheimer’s disease (AD). Data from animal models suggests that antibodies to Aβ through several mechanisms can decrease Aβ deposition, decrease Aβ-associated damage such as dystrophic neurite formation, and improve behavioral performance. Data from human studies suggests that active immunization can result in plaque clearance and that passive immunotherapy might result in slowing of cognitive decline. Despite this, a recent analysis from a phase I trial that involved active immunization with Aβ42, while not powered to determine efficacy, suggested no large effect of active immunization despite the fact that plaque clearance was very prominent in some subjects. An important issue to consider is when active or passive immunization targeting Aβ has the chance to be most effective. Clinicopathological and biomarker studies have shown that in terms of the time course of AD, Aβ deposition probably begins about 10–15 years prior to symptom onset (preclinical AD) and that tau aggregation in tangles and in neurites does not begin to accelerate and build up in larger amounts in the neocortex until just prior to symptom onset. By the time the earliest clinical signs of AD emerge, Aβ deposition may be close to reaching its peak and tangle formation and neuronal cell loss is substantial though still not at its maximal extent. Since immunization targeting Aβ does not appear to have major effects on tangle pathology, for immunization to have the most chance for success, performing clinical trials in individuals who are cognitively only very mildly impaired or even in those with preclinical AD would likely offer a much better chance for success. Current work with AD biomarkers suggests that such individuals can now be identified and it seems likely that targeting this population with immunization strategies targeting Aβ would offer the best chance of success.
Keywords: Alzheimer’s disease, immunization, biomarkers, amyloid-β, tau, imaging, antecedent biomarkers, plaques
INTRODUCTION
The aggregation, buildup, and deposition of the 37–43 amino acid peptide amyloid-β (Aβ) predominantly in the extracellular space of the brain in diffuse and neuritic plaques as well as in cerebral arterioles in the form of cerebral amyloid angiopathy (CAA) is one of the main pathological features of Alzheimer’s disease (AD). The premise underlying the amyloid hypothesis is that Aβ accumulation and toxicity is a primary upstream driving event in AD pathogenesis. This idea which is based on genetic, biochemical, and animal model data, has provided a theoretical background for many attempts to develop disease -modifying therapies for AD, aiming to decrease Aβ production, inhibit its aggregation, or promote its clearance from the brain. Immunotherapeutic approaches targeting Aβ gained major international interest after the publication of promising results of studies in transgenic mouse models that develop Aβ accumulation and other changes in the brain. Despite some positive results that have come from the first human trial of Aβ immunotherapy on amyloid plaque clearance, long-term follow up from the first phase I study showed no major effect on survival or disease progression. While there are many issues and caveats to consider in regard to the AN1792 trial, several lines of evidence suggest that many of the pathological processes that characterize the early stages of AD may become self-propagating elements in cell destruction and degeneration in the advanced pathological stages of AD, independent of the presence of amyloid deposition. Importantly, the advanced pathological stages of AD are almost always present by the time that AD patients have reached the clinical stages that are termed mild to moderate dementia. Based on these results, it remains unclear whether targeting amyloid pathology in the presence of significant tau pathology, synaptic dysfunction, or neuronal loss which are present by the mild to moderate clinical stages of AD, will be effective. These results have highlighted the importance of early disease detection to maximize the efficacy of disease-modifying therapy targeting Aβ and amyloid-associated pathology. This has translated into significant research efforts in search of biological or radiological biomarkers that can correlate with the presence of AD-like pathology that may be detectable prior to the onset of significant cognitive impairment, especially if such changes can be used to predict clinical outcome. The utilization of such antecedent biomarkers for AD has the potential to be critical in the design of future clinical trials for passive and active immunotherapy approaches targeting Aβ, and for the identification of subjects who are most likely to benefit from such disease-modifying therapies. Moreover, such information has the potential to greatly influence decisions in clinical trial enrollment, initiation of treatment, and assessment of treatment outcomes.
PATHOLOGICAL HALLMARKS OF AD
The characteristic pathological features of AD include Aβ-containing amyloid plaques, dystrophic neurites, neurofibrillary tangles (NFT), and neuropil threads [1]. Plaques consist predominantly of abnormal insoluble extracellular aggregates of the Aβ peptide; a normally soluble 4 kDa peptide of 37–43 amino acids in length, which is derived from the proteolytic cleavage and processing of a larger protein, amyloid precursor protein (APP) by β and γ secretases [2]. “Neuritic” plaques are mainly composed of extracellular deposits of Aβ in a β-pleated sheet conformation (i.e. they bind to dyes such as Congo red and thioflavin-S) as well as cellular components that include degenerating neuritic processes (referred to as dystrophic neurites), astrocytes, and microglia [3]. “Diffuse” plaques consist of Aβ deposits that contain little to no fibrillar Aβ and lack prominent neuritic or glial changes [4]. Approximately 80–90 % of patients with AD also have evidence of amyloid deposition in the walls of small-to-medium blood vessels in the meninges and cerebral parenchyma, referred to as CAA [5, 6].
Tau is a cytoplasmic, microtubule-associated protein that plays a major role in the assembly and stabilization of microtubules. In normal circumstances, tau is soluble and there is equilibrium between the phosphorylation and dephosphorylation of tau. Hyperphosphorylation and conversion of tau to insoluble forms inside the neuronal cytoplasm is thought to be one of the earliest changes in AD, and impairs the ability of tau to bind to and stabilize microtubules [7, 8]. Paired helical filaments are generated by the self-aggregation of tau, which subsequently assemble in the neuronal perikarya leading to the formation of neurofibrillary tangles or in the neuronal dendrites leading to the formation of neuropil threads [8, 9].
TEMPORAL PATTERN OF THE PATHOLOGICAL PROCESSES IN AD
There has been a large amount of evidence, from both animal and human studies, supporting a crucial role for Aβ in the pathogenesis of AD. Based on genetic and biochemical studies, it is strongly believed that the deposition of insoluble forms of Aβ in the form of both oligomers and amyloid plaques is a pivotal step in the pathogenesis of AD, later culminating in neuronal loss, synaptic loss, and synaptic dysfunction. In addition, while tau aggregation may initiate as an independent process, there is evidence that Aβ accumulation exacerbates tau accumulation [10–12]. The “amyloid hypothesis” of AD [13] represents an attempt to explain the neurodegenerative substrates of AD as direct consequences of accumulation, and increased deposition, of insoluble extracellular, and perhaps, intracellular Aβ [14] with neurotoxic properties. This idea was first introduced after the isolation of Aβ from AD plaques in 1984 and has since been supported by multiple lines of evidence. Aβ is produced by the proteolytic cleavage of the full length amyloid precursor protein (APP) by the action of β and γ secretases [2, 15]. Point mutations in APP, presenilin -1 (PS-1), or presenilin-2 (PS-2) can cause familial forms of AD, most likely due to a common mechanism of increased Aβ production (particularly of the more aggregation -prone form of Aβ, Aβ42) leading to early onset of Aβ aggregation [16, 17]. There are some mutations in APP that appear not to increase Aβ production, but rather alter its propensity to aggregate and/or slow its clearance from the brain [15]. Trisomy 21 or Down syndrome is associated with early onset of AD pathology and dementia in affected individuals due to an extra copy of the APP gene [18] which leads to increased levels of APP and Aβ that probably promote early onset of Aβ aggregation in the brain [19]. In addition, duplication of the APP gene leads to CAA and AD [20].
The amyloid hypothesis has provided the first link to the cascade of destructive events that characterize this disease, and has stimulated interest in understanding the temporal pattern and relative contribution of pathological events implicated in the disease process. Multiple studies of molecular and pathological indicators of disease progression have proposed models for the temporal pattern of biochemical and pathological changes in AD. Data from these studies, studies of familial cases of AD, and mouse models of AD support the notion that amyloid deposition is an early occurrence in AD that begins many years prior to the appearance of clinical signs of cognitive decline that characterize AD. In fact, amyloid deposition has been estimated to begin 10–15 years prior to any clinically detectable signs of dementia [21], progress with time, and reach what many refer to as a “ceiling” effect. There is evidence to suggest that Aβ accumulation achieves a high steady state by the early clinically evident stage of disease, with only little increase afterwards. In other words, amyloid deposition has already reached or is close to reaching its peak by the time there is evidence of even very mild dementia, as sometimes termed mild cognitive impairment (MCI) or in terms of staging of disease, a clinical dementia rating (CDR) of 0.5, very mildly impaired [21, 22].
Results from neuropathological studies of non-demented elderly, individuals with mild cognitive impairment (MCI), and early stage Alzheimer’s disease have shown that individuals with MCI or very mild dementia (CDR 0.5) have profuse numbers of senile plaques in all neocortical regions [21]. In addition, such individuals have increased NFT densities in the hippocampus and entorhinal cortex. These studies suggest an important notion: by the time dementia is minimally apparent clinically, the histopathological changes of AD have already been established with substantial numbers of both diffuse and neuritic plaques distributed widely throughout the cerebral cortex. In fact, even cases with the mildest stages of dementia (CDR 0.5) have sufficient plaques and tangles that usually meet pathological criteria for a diagnosis of AD [23]. Since these lesions are believed to accumulate relatively slowly, it has been proposed that the disease process must begin at an even earlier stage. These studies have changed our thinking of the pathological time course of AD, and along with other studies in this field, have introduced the concept of the preclinical stage of AD (i.e. neuropathological evidence in the absence of clinical signs) [24]. Such stages have long been described in Down syndrome [25].
The identification of significant AD neuropathology in individuals with even the mildest stages of the AD dementia syndrome has raised interest in identifying the pathological correlates of cognitively intact elderly, with the hope of detecting preclinical stages of pathology. Interestingly, these studies have demonstrated the presence of significant AD pathology in a subset of cognitively intact elderly, with quantitative and qualitative differences from what can be attributed to normal aging [21]. Although previous reports have suggested that the neuropathological distinction between aging and AD is based on higher densities of senile plaques in the neocortex of AD [26], there also appear to be qualitative differences in the type of plaques in AD compared to aging. While a few diffuse plaques can be seen in the neocortex of healthy elderly, the neuritic plaques are generally not part of ‘healthy aging”. Neuropathological reports from a subset of non-demented elderly have indicated the presence of diffuse and to a lesser extent neuritic plaques distributed extensively across the neocortex. Furthermore, there was evidence of predominantly neuritic plaques in the limbic structures, particularly the entorhinal and perirhinal cortex, resembling those changes seen in MCI/very mild AD (CDR-0.5). Based on these results, it has been suggested that previous reports attributing the presence of neuritic plaques in non-demented individuals to normal aging were very likely contaminated by cases of unrecognized pre-clinical AD [21].
NFT formation differs both spatially and temporally from plaque pathology. At least a few NFTs can be seen in the brains of virtually all non-demented elderly with or without plaques, including vulnerable brain regions such as the entorhinal cortex and the hippocampus. Moreover, there appears to be an exponential increase in the rate of NFT formation with normal aging [21]. Some studies have suggested that tau pathology occurs very early, even preceding any signs of amyloid deposition by decades [27]. However, in the absence of plaques, neurofibrillary changes are relatively slow and appear to be confined to the medial temporal lobe structures. On the other hand, their more extensive buildup in the neocortex appears to occur later than Aβ deposition, and in fact, there is correlative evidence that it may be driven by the presence of extensive Aβ pathology in preclinical AD. The presence of amyloid plaques is correlated with a significantly larger amount of NFT formation compared to elderly with few or no plaques. In other words, while early and slowly progressive NFT formation can be seen in the limbic structures with normal aging, independent of amyloid pathology, this is thought to occur at a too indolent rate to produce AD. It appears that early Aβ deposition is a key process that promotes NFT formation at higher rates and results in the formation of dystrophic neurites, and subsequently neuritic plaques. This is supported by recent genetic data in which single nucleotide polymorphisms in the tau gene are associated with both tau levels in the cerebrospinal fluid (CSF) as well as earlier onset of dementia of the Alzheimer’s type, but only in individuals who have evidence of brain amyloid deposition [28]. This presumed role for Aβ in inducing tau accumulation is supported by the observation that tau-containing dystrophic neurites tend to form around Aβ plaques [29]. In addition, animal studies show that Aβ accumulation appears to exacerbate tau accumulation [11, 12]. Further accumulation of neuritic plaques and tangle pathology progresses over time, and in the absence of sufficient neuronal, axonal, and synaptic damage to cause clinically detectable dementia, marks the preclinical stages of AD. Supportive of the fact that Aβ accumulation generally drives early events in AD, we have recently found that reduction in CSF Aβ42, that reflects Aβ aggregation in the brain, is associated with brain atrophy in the pre-clinical phase of AD [30]. In contrast, CSF tau levels were not correlated with brain atrophy in cognitively normal elderly but the levels were negatively correlated with brain atrophy in individuals with very mild and mild dementia of the Alzheimer’s type [30]. This suggests that increases in CSF tau (and ptau181), probably reflecting overall levels of neurofibrillary- linked neurodegeneration, are later events that correlate with further structural damage and occur with clinical onset and progression of disease [30].
The earliest clinical signs of dementia are thought to occur when a threshold of neuronal loss, axonal loss, and loss of synaptic integrity is reached in several areas of the brain such as the hippocampus, entorhinal cortex, and specific neocortical regions. This is suggested by studies comparing tissue volumes and neuronal numbers in the entorhinal cortex and hippocampal field CA1 in healthy brain aging, preclinical AD, and very mild AD (MCI) [31]. These results indicated that measures of tissue volume and neuronal numbers in cases with preclinical AD were comparable to those of the healthy elderly. On the other hand, both of these measures decrease substantially in cases with MCI/very mild AD. These results suggest that a threshold of neuronal loss and synaptic dysfunction must occur prior to the appearance of the first clinical manifestations of AD (CDR-0.5), despite the presence of amyloid plaques, and to a lesser degree NFT, in the preclinical stage.
Following the first clinical signs of cognitive impairment, further disease progression is associated with progressive neuronal loss, synaptic loss, and progressive increase in NFT pathology, on a background of maximally elevated Aβ [29]. A decrease in synapse density, altered synaptic signaling, and synaptic composition are seen in the early stages of AD, even prior to the occurrence of synaptic degeneration [32]. Although these changes may be driven by amyloid deposition in the early stages, there is evidence to suggest that these processes may have a more immediate effect on the severity and progression of dementia. This has been supported by many studies suggesting that clinical progression correlates well with the extent of neuronal loss [33, 34], with decreasing levels of synaptophysin (a synaptic marker), and with increasing numbers of NFTs in the hippocampus, entorhinal, and association cortices [29].
Other mechanisms implicated in the pathogenesis of AD include oxidative stress, calcium-mediated toxicity, microglial activation and neuroinflammation (Interleukin-1 [IL-1], Interleukin-10 [IL-10], and Tumor Necrosis Factor [TNF-α]) [7]. Inflammation observed in the brains of patients with AD is thought to be secondary to the presence of amyloid plaques as well as neurofibrillary changes. The inflammatory reaction in AD includes the activation and recruitment of microglia and astrocytes, and upregulation of immune mediators such as IL-1, IL-10, and TNF-α. Astrocytosis, evidenced by increased glial fibrillary acidic protein levels (GFAP), is also commonly observed in pathological studies of patients with AD, and seems to correlate with longer disease duration [29]. As the numbers of plaque-associated microglia and cytokines vary with plaque evolution [35], gliosis is thought to reflect a plaque and tangle-associated event to some extent.
In summary, preclinical AD is characterized by significant Aβ deposition and lesser degrees of tau aggregation, with only minimal synaptic and neuronal loss. MCI or very mild dementia that marks the first clinically detectable stage of AD occurs when neuronal, axonal, and synaptic loss and dysfunction has reached a threshold, and is associated with further increases in tau and Aβ42 aggregation. By the time patients have mild or moderate dementia (e.g. CDR-1 or --2), Aβ deposition has likely already peaked while tau aggregation, neuronal and synaptic loss, and inflammation continue through the more advanced stages of disease (Fig. 1).
Fig. 1.
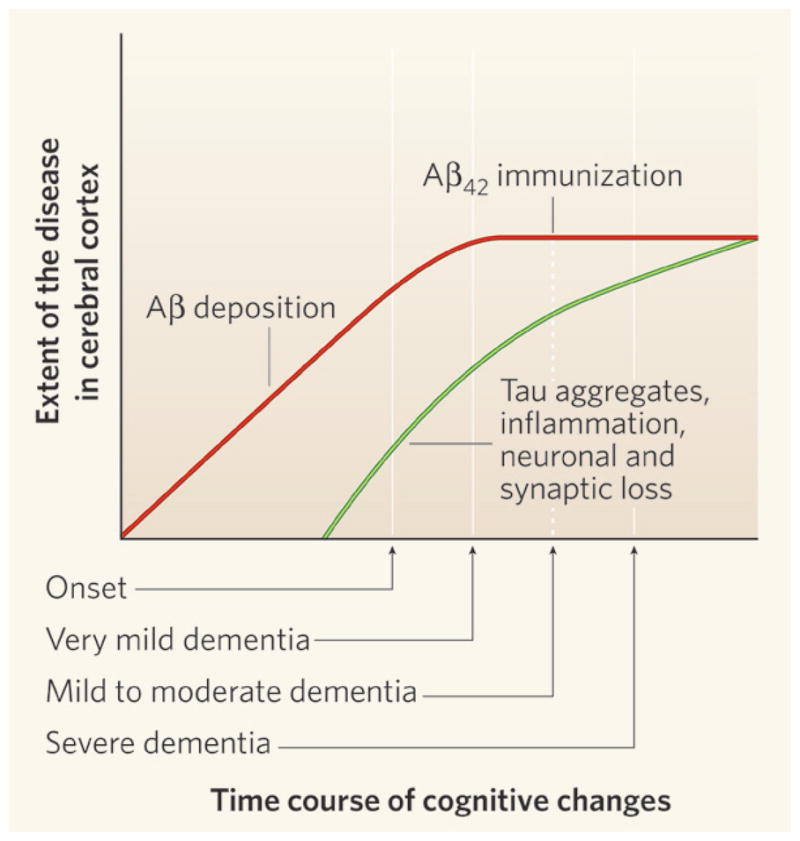
Time course of the development of Alzheimer’s-type pathology in the brain. Approximately 10–20 years prior to the onset of cognitive decline that is recognized as dementia due to Alzheimer’s disease (AD), deposition of the amyloid-β (Aβ) protein begins in the cerebral cortex. It is likely that the accumulation of Aβ in the brain peaks by the stage of very mild dementia due to AD. High levels of tau protein deposition in the cerebral cortex (in the form of neurofibrillary tangles), inflammatory changes, and both neuronal and synaptic loss, also likely begin several years prior to the onset of cognitive decline due to AD. These pathologies then probably increase throughout the course of the disease. The Phase I clinical trial of active immunization with Aβ42, called AN1792, was performed on individuals with mild to moderate dementia due to AD. Reprinted with permission from Nature, 2008, 454, 418–420.
SUMMARY OF THE IMMUNOTHERAPY TRIALS IN HUMANS
Immunotherapy, including both active and passive immunization targeting Aβ, has been extensively studied as a potential target for AD therapy. The promising results of the animal experiments with immunotherapy led to the launching of the first human trial with active immunization with Aβ42 in 2000 (Elan/Wyeth). The phase I component of this trial was designed to assess the safety of active immunization with multiple doses of Aβ42 in adjuvant (AN1792 and QS-21) in 80 patients with mild to moderate dementia. A significant percentage of patients developed antibodies to Aβ (53%) and no adverse events were reported. Therefore, a subsequent larger phase IIa trial of 372 patients was started in 2001 to assess efficacy. This trial was halted prematurely when 18/298 patients (6%) developed a subacute encephalopathy due to meningoencephalitis [36]. Subsequent postmortem pathological evaluation of patients enrolled in this trial showed a remarkable reduction in Aβ deposition in some patients [37, 38], similar to what might have been expected based on the animal models [39]. There was a significant reduction and clearance of plaques from different cortical regions, and residual plaques were surrounded by microglia and demonstrated a moth eaten appearance probably due to Aβ phagocytosis. Evidence of microglial activation and localization of Aβ inside microglia was observed [37]. Tau -containing plaque-associated dystrophic neurites were removed from areas where plaques had been removed [37]. Long term follow up of some of these patients for 6 years after initial immunization showed that immunization was associated with a long-term reduction in Aβ load in some immunized individuals. The effect of immunization on other pathological correlates of AD was less evident; there were no significant changes in quantitative measures of tau immunoreactive tangles, neuropil threads, or CAA even in areas where amyloid plaques had been removed [37].
Despite the premature termination of the trial and the lack of significant effects on other forms of pathology, this human trial provided a proof of principle for what animal studies had shown regarding the efficacy of this approach in reducing plaque load. However, data from different studies are conflicting as to whether active immunotherapy actually translated into functional benefits in cognitive and behavioral terms. Previous reports suggested that there was no evidence of statistically significant improvement in cognitive function on several measures or survival even in patients who had almost complete plaque removal at autopsy [40]. No differences were observed in five individual cognitive tests (including the mini-mental state examination [MMSE] and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale [ADAS-cog]). However, it is important to consider that this study was not powered to detect small differences in the rate of progression and that many patients were lost to follow up. It is also important to consider that only about half of the patients produced antibodies and in variable titers [41]. On the other hand, analysis of the z-score of the Neurological Test Battery that focuses on tests of episodic memory showed less progression and even some improvement in certain domains. Also, a few subgroup analyses of the initial trials suggest there may be some beneficial effect [40, 42], and the results remained significant even after patients with meningoencephalitis were excluded.
More recently, a six-year follow up study of 80 subjects from the phase I of Aβ42 immunization trial by Holmes et al showed evidence of persistently raised Aβ antibodies in some subjects and significant reduction in the brain amyloid plaques in a subset of subjects even 5 years after the last injection [43]. Despite plaque reduction in some individuals, the study reported no significant difference in survival or the time to severe dementia in the AN1792 and the placebo groups, even after correction for age and cognitive stage at the time of enrollment [43]. Moreover, there was no difference in the rate of decline over the 6 year follow up period between the immunized and placebo groups.
Why this analysis did not show a slowing of progression is not clear. These findings may be at least partially explained by the possibility that this study was not designed to detect small but significant changes in disease progression between the immunization and placebo groups as this was a phase I study. In addition, while Aβ immunization reduces insoluble amyloid plaques in some people, its effect on other putatively toxic soluble forms of Aβ, such as oligomers, remains unclear. In addition to these considerations, results from this trial have potentially very important implications regarding the lack of obvious benefit of amyloid reduction on delaying disease progression. All study subjects enrolled in this study had mild to moderate dementia believed to be due to AD at the start of the study. As mentioned earlier, amyloid deposition probably begins to occur 10–15 years prior to the first clinical signs of dementia. While active immunization may be able to clear existing amyloid deposits, it seems unlikely that it will result in reversing or even halting NFT, synaptic loss, inflammation and neuronal loss which have already been set to motion and have reached substantial levels.
Passive immunization has also been the subject of several human trials. Passive immunization with a humanized recombinant Aβ monoclonal antibody directed against the N terminus of Aβ (AAB-001 or Bapineuzumab) has recently entered phase III clinical trials [http://clinicaltrials.gov/ct2/show/NCT00574132?term=Bapineuzumab&rank=1]. Again, it is important to note that this large phase III trial is being carried out in individuals with mild to moderate dementia of the Alzheimer’s type. As the AAB-001 antibody is a humanized version of mouse monoclonal antibody m3D6 directed against the first 8 amino acids at the N-terminus of Aβ that has been shown to be able to decrease amyloid plaques in mouse models of AD [44], it is hoped that the beneficial effects of reducing amyloid burden and potentially improving cognition will be reproduced. In a phase II trial of AAB-001, two-hundred-thirty-four (234) patients were randomized to receive one of four doses of bapineuzumab or placebo by intravenous infusion every 13 weeks. Findings were reported for 229 patients in a modified intent-to-treat (MITT) analysis. The pre-specified primary efficacy endpoints were change from baseline in Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-cog) and Disability Assessment Scale for Dementia (DAD) in the treatment versus placebo groups. Preliminary findings of the phase II trial were reported at the 2008 International Conference on Alzheimer’s Disease. While the primary end-point was not met, post-hoc analysis suggested a beneficial effect in the ApoE4 non-carrier group. In the ApoE4 non-carrier patients, statistically significant differences from baseline to week 78 were observed in favor of bapineuzumab -treated patients on both cognitive and functional efficacy endpoints. Additionally, MRI results showed significantly less brain volume reduction compared to placebo in the non-carrier patients. On the other hand, no statistically significant changes were observed in any of the cognitive or functional efficacy endpoints in the ApoE4 carrier patients.
There are now several trials of passive immunization targeting Aβ including a trial of LY2062430 (Eli Lilly and Co.), which involves passive immunization with an Aβ central domain antibody. This compound just completed phase II testing (http://clinicaltrials.gov/ct2/show/NCT00329082?term=LY2062430&rank=1). A very similar monoclonal antibody in mice (m266) has been shown to improve behavior [45] and reduce amyloid plaques [46] in an APP transgenic mouse model. Other ongoing trials for passive immunization with humanized monoclonal antibodies directed toward Aβ include RN-1219 (PF-04360365) (http://www.clinicaltrials.gov) and R-1450 (http://www.centerwatch.com). Results from these trials have not yet been published.
It is interesting to note that studies of passive immunization in mouse models have also highlighted the importance of early immunization. Passive immunization of 3xTg-mice that develop Aβ and tau-accumulation with the monoclonal antibodies 4G8 and 1560 not only resulted in the clearance of intracellular and extracellular amyloid, but also of early tau aggregates in the somatodendritic compartment. On the other hand, there was no effect on hyperphosphorylated tau or neurofibrillary tangles, which are indicative of more advanced stages of tau pathology [10]. These findings support the notion that while Aβ may drive tau pathology in the early stages, the neutralization of Aβ does not seem to affect tau deposition as tau pathology advances [10].
The recognition of the neuropathological processes that characterize preclinical AD has important implications for future therapies in AD. This becomes relevant in designing trials and developing potential disease modifying therapies for AD. Since most attempts at therapeutic strategies for AD target different steps of the amyloid cascade, it is critically important to understand that amyloid deposition is an early process in the pathogenesis of AD that may then prime or trigger a cascade of secondary events (such as inflammation, tau deposition, production of free radicals, alteration of calcium metabolism, and cell death) that may contribute significantly to further damage and cognitive decline despite the clearance of amyloid. By the time these secondary changes become prominent and there is significant synaptic and neuronal loss, reducing amyloid burden is unlikely to be of any benefit.
Our improved understanding of the spectrum and temporal pattern of the neuropathological changes that characterize AD highlights the importance of early detection of disease, prior to the appearance of any clinical signs. Intervention at an early stage should improve our ability to target disease pathology, such as amyloid deposition, at a relatively reversible stage, and prior to the occurrence of irreversible neuronal and synaptic loss. The success of current and future trials of disease-modifying therapies for AD will be significantly influenced by improving preclinical detection. This requires the identification of surrogate markers of disease pathology including CSF and blood biomarkers, as well as imaging markers. In other words, using these “antecedent” markers offers the possibility of preclinical intervention of AD.
EARLY DETECTION OF AMYLOID PATHOLOGY: BLOOD AND CSF BIOMARK-ERS
There has been vast expansion in our knowledge of many CSF markers that might aid in the detection, early diagnosis, and prediction of disease progression of AD [47]. A large variety of potential markers in the blood and CSF have been studied [48, 49], including proteins involved in inflammation, oxidative stress, apolipoproteins, and markers of neurodegeneration, with conflicting results [50]. Plasma levels of Aβ40 and 42 have been an area of interest; baseline levels or the ratio of Aβ40/42 appear to be related to the subsequent risk of developing AD [51–54], though the levels are low and difficult to measure and the methods have not been standardized across studies. However, other studies have shown that plasma Aβ40 and 42 levels are not altered in AD vs control subjects [55, 56]. Further search for plasma markers is underway with the exploitation of genomics, proteomics, and metabolomics [57, 58]; categories such as cytoskeletal maintenance, cellular trafficking, cellular stress response, redox homeostasis, transcription, and DNA repair show unique gene expression in patients with AD compared to age matched controls [59].
Much more promising results have been achieved to date with studies of CSF biomarkers, as the CSF appears to be more reflective of underlying pathological processes occurring in the brain. The CSF biomarkers with the highest diagnostic potential are CSF tau, phosphorylated forms of tau (p-tau), and Aβ42. This is based on the finding of marked elevations of tau in AD CSF, and its ability to discriminate AD from normal aging in ~80% of the cases [49]. More importantly, tau elevation seems to occur at the early stages of dementia, suggesting its utility in early detection. CSF tau seems to be an indicator of the intensity of neurodegeneration, phosphorylated tau (p-tau) has been hypothesized as a marker of tangle formation, and CSF Aβ42 is persistently decreased as it is deposited in AD plaques [49]. The use of the Aβ42/37 ratio [60] has been shown to differentiate AD from non-demented controls and from other forms of dementia. More recently, CSF biomarkers of neuronal injury such as visinin-like protein 1 (VLP-1) have gained some interest. In one study, VLP-1 was higher in AD compared to age matched controls, and correlated with CSF tau levels as well with MMSE scores [61].
Despite these major advancements in the area of CSF biomarkers for the differential diagnosis and prognosis of dementia, until recently, little was known about the ability of these markers to predict conversion to very mild dementia or MCI in asymptomatic elderly or predict progression from MCI to AD. Our improved understanding of the pathological changes that characterize MCI and preclinical AD has highlighted the importance of early detection of AD at preclinical stages, and has translated into significant research efforts in search of biological or radiological surrogate markers that can detect it prior to the onset of cognitive impairment. Since only 15% of MCI or very mild dementia patients progress to mild to moderate dementia per year and not all people with MCI have AD or clinically progress, biomarkers or radiological markers are needed to accurately determine the presence or absence of underlying AD pathology and predict rate of progression of disease. These patients are the most likely to benefit from disease- modifying therapies, as intervention is applied prior to the development of significant degrees of irreversible neuronal and synaptic damage. The utility of CSF markers in predicting progression has gained even more attention with the publication of promising results from the first longitudinal studies in this field. Not surprisingly, these studies have focused on patients with the earliest stages of cognitive impairment (MCI) and patients with identifiable risk factors for AD (pre-senilin or APP mutations).
CSF BIOMARKERS PREDICT THE PROGRESSION FROM MCI TO AD
A few studies have addressed the utility of biomarkers in predicting the conversion from MCI (very mild dementia) to AD with mild to moderate dementia. High CSF tau was found to predict progression from MCI to AD with 90% sensitivity and 100% specificity in one study [62]. These results were later reproduced in another study that showed that CSF tau was markedly increased in the CSF of MCI patients who later progressed to AD compared to those MCI cases with no evidence of progression [63]. Similar results were obtained with the combination of CSF tau and Aβ42: increased tau and low Aβ42 was found in 90% of cases that later progressed to AD compared to only 10% in stable MCI patients [64]. These findings have now been confirmed in a large longitudinal study of MCI patients, who were followed for 4–6 years [65]. This study demonstrated that the relative risk of progression from MCI to clear-cut dementia typical of Alzheimer’s disease was substantially increased in patients with MCI who had elevated concentrations of tau, phosphorylated forms of tau, and low Aβ42 at baseline. The association between pathological CSF and progression to Alzheimer’s disease was much stronger than, and independent of, established risk factors including age, sex, education, ApoE genotype, and plasma homocysteine. The combination of total tau and Aβ42/P-tau181 ratio yielded closely similar results in the same study. The ratio of CSF tau/Aβ42 has also been shown to predict progression from mild cognitive impairment (MCI, CDR-0.5) to AD [65]. By 5 years, of those with MCI with a normal tau/Aβ42 ratio, less than 10% progressed to dementia of the Alzheimer’s type while greater than 90% progressed to mild to moderate AD if there was a high tau/Aβ42 ratio.
CSF BIOMARKERS IN PREDICTING PROGRESSION FROM COGNITIVELY NORMAL TO VERY MILD DEMENTIA/MCI
While MCI/very mild dementia patients can be identified clinically, the detection of preclinical cases poses challenging questions as to how can we identify patients at risk for developing the clinical signs and symptoms characteristic of AD. Clinical diagnostic evaluations, including neuropsychological tests, have not been useful in predicting future progression from normal to MCI/very mild dementia in cognitively intact individuals. With the recognition of the importance of detecting preclinical changes to potentially improve therapeutic efficacy, many studies have recently focused on the utility of CSF biomarkers for detection of pre-symptomatic or so called “pre-clinical” AD. The ability to identify such individuals should have a major impact on the identification of appropriate candidates for disease modifying therapies and better characterization of subjects one might enroll in disease-modifying clinical trials such as immunization targeting Aβ.
The increased ratio of tau/Aβ42 and p-tau/Aβ42 in cognitively normal individuals was reported to be associated with an increased risk for conversion from normal to mild cognitive impairment/very mild dementia (CDR-0.5) in 2 recent studies [56, 66]. Li et al. reported that over the follow up period of 42 months, while no conversions to MCI occurred in the normal CSF tau/Aβ42 subgroup [56, 66], all subjects who converted to MCI had elevated tau/Aβ42 ratios. In the Fagan et al. study, using a tau/Aβ42 cutoff, ~70% of those with a high ratio converted from normal to very mild dementia over a 3 year period while only ~10% of those with a normal ratio converted to very mild dementia. Since the high tau/Aβ42 subgroup of non-demented elderly is clinically indistinguishable from the subgroup of non-demented elderly with normal tau/Aβ42 ratios, it has been suggested that the subgroup with abnormal ratio represents a subgroup that has developed Aβ accumulation as well as tau aggregation in the brain. If such results are replicated across large sample sets, it may be possible to design clinical trials involving immunization in such individuals who are at very high risk to convert from normal to dementia over a relatively short period of time.
RADIOLOGICAL MARKERS FOR IN VIVO AMYLOID IMAGING
The introduction of positron emission tomography (PET) imaging using compounds such as 11C labeled Pittsburgh compound B (11C-PIB) ligand has revolutionized imaging in dementia and opened new avenues for advanced diagnostic tools in AD. 11C-PIB is a Carbon 11 derivative of the thioflavin-T amyloid dye which binds with high affinity and high specificity to neuritic Aβ plaques [67]. Human 11C-PIB studies have shown robust binding in several brain regions in cases of AD, with a significant 1.5–2 fold increase of 11C-PIB binding compared to controls [68]. 11C-PIB binding correlates with the regional distribution of amyloid in the frontal, parietal and temporal cortex in postmortem cases [69]. These areas also correspond to areas with reduced FDG (2-fluoro-2-deoxy-D-glucose)-PET activity [70]. In contrast, no differences in 11C-PIB retention between AD and normal controls were observed in areas of the brain not typically affected in AD [68]. Some studies have suggested that the initial 11C-PIB load is indicative of disease progression in the next 2 years [67, 71]. However, no significant change in amyloid load was observed over the two year course of the study, in association with disease progression. This is not surprising based on the notion that amyloid deposition most likely has peaked and stabilized by the time cognitive deficits become clinically detectable.
Since amyloid deposition is known to precede clinical signs of dementia by many years, 11C-PIB imaging may allow early detection of amyloid during preclinical AD. Importantly, it has been shown that up to 30% of cognitively normal elderly by their mid ‘70s have substantial 11C-PIB retention in the cortex, similar in extent to the amounts observed in subjects who have mild to moderate dementia of the Alzheimer’s type [72]. Further longitudinal studies are needed to determine how well this imaging change predicts eventual progression to dementia. Since 11C-PIB does not detect tangles, it may be that 11C-PIB positivity will begin to occur many years prior to dementia and some measurements of other markers such as tau accumulation (as marked by CSF tau) will be needed to accurately predict dementia onset. One imaging marker, FDNNP (18F-2-(1-(6-[(2-fluoroethyl(methyl)amino]-2-naphthyl)ethylidene), appears to bind to both plaques and tangles and it will be interesting to determine its usefulness in predicting cognitive decline in cognitively normal elderly [73]. Global 18F-FDDNP retention differentiated patients with MCI from AD or non-demented controls in one study of 28 patients. Although it was superior to FDG-PET or MRI volumetry in differentiating AD, MCI, and normal individuals from each other, its use may be limited by its narrow range of binding [47, 73]. Studies have not yet been done correlating CSF biomarkers with FDNNP.
COMBINED USE OF CSF BIOMARKERS AND PIB
The utility of the combined assay of CSF Aβ42 with in vivo amyloid imaging (using 11C-PIB) in both demented and non-demented individuals as an antecedent marker for AD has recently been reported by Fagan et al. [56, 74]. Subjects fell into two non-overlapping groups: subjects with low Aβ42 levels and positive 11C-PIB retention, and subjects with high CSF Aβ42 and no 11C-PIB retention. Among the first group were three subjects who were cognitively intact (CDR-0), consistent with observations from pathological studies indicating the presence of amyloid deposition in a subset of cognitively normal elderly (as described previously). These non-demented 11C-PIB-positive subjects with low CSF Aβ42 levels performed comparably with the non-demented 11C-PIB -negative subjects on neuropsychological testing. In other words, the combination of Aβ42 and 11C-PIB allowed a detection of amyloid deposition in individuals, who appeared normal clinically and via neuropsychological testing. The combination of CSF Aβ42 and 11C-PIB imaging may be very useful in selecting individuals with preclinical AD or with very mild dementia for entry into future immunization trials. In addition, 11C-PIB or similar agents may be useful to determine if certain immunization strategies are removing amyloid over time.
STRUCTURAL AND FUNCTIONAL IMAGING
In addition to the exciting results of studies using novel markers for in vivo amyloid imaging, structural imaging such as volumetric MRI has also been shown to offer benefit in predicting progression to MCI or AD. Measures of hippocampal volume have recently gained interest as anatomical measurements useful in predicting the transition from normal to MCI and from MCI to AD [75]. These results have been seen in studies of hippocampal volume [76] and hippocampal perfusion [77]. In addition, qualitative estimates of hippocampal atrophy in patients with MCI have been shown to predict cognitive decline and progression to AD [78, 79]. In addition to hippocampal volume, the inclusion of the fusiform gyrus volume significantly improved the ability to discriminate subjects with MCI from AD [80]. Moreover, findings have suggested that the reduction in the size of the entorhinal cortex (EC) can discriminate MCI from cognitively intact elderly as well as predict progression from MCI to AD [76].
Functional imaging studies suggest that glucose metabolism in the temporal neocortex and posterior cingulate gyrus can predict conversion from MCI to AD. Promising results were also reported with the use of FDG-PET in predicting decline to MCI in “normal” patients; baseline reductions in glucose metabolism in the entorhinal cortex (EC) accurately predicted decline to MCI with a sensitivity and specificity of 83% and 85% respectively. Normal subjects who progressed to MCI had significant reductions in glucose metabolism in the hippocampus and temporal neocortex as compared with non declining normal elderly on follow up [75]. What is not clear is how well structural and functional imaging measures compare to CSF biomarkers in predicting cognitive decline and whether if one used a combination of structural imaging and CSF biomarkers, this would improve diagnosis and prognosis to an even greater extent. It is possible that results from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) as well as studies from individual groups will greatly assist in answering these questions.
CONCLUSION/FUTURE DIRECTIONS
Despite the success of the first human trial with active immunization with Aβ42 (AN1792) in reducing and even clearing amyloid plaques in AD, the long term effects on dementia progression have been less compelling to date. Many factors should be taken into consideration in the design of future trials in AD, such as choice of immunogen and adjuvant, routes of administration, and duration of the immune response. Importantly, the success of future trials of immunotherapy for AD will be greatly influenced by the selection of the study population. It seems likely the earlier the treatment starts, the more likely it will be to get a positive clinical effect. Consideration should be given to including subjects with preclinical AD as well as very mild dementia of the Alzheimer’s type (MCI). Clinical evaluation and neuropsychological testing cannot differentiate cognitively intact subjects who do have latent AD pathology from those who do not. While MCI is a description of a clinical syndrome, there is a lot of heterogeneity within this diagnostic category. For example, many of these individuals have very mild dementia due to AD while others do not have AD but have other diagnoses. This results in variable cognitive profiles and rates of progression. Prediction of what subgroup of very mild dementia/MCI will clinically progress to mild to moderate dementia on clinical evaluations alone is probably not possible. Use of CSF biomarkers as well as neuroimaging can greatly assist in picking out those people with MCI/very mild dementia who are highly likely to progress. Preclinical detection of AD will depend on the utilization of biomarkers and radiological markers for selection of patients likely to benefit from amyloid-targeting therapies. In addition to its role in therapeutic trial design, there is no doubt that preclinical detection of AD pathology will have major impacts on entry criteria into trials as well as assessment of the efficacy of current and future treatments.
Finally, with the knowledge that amyloid deposition has almost peaked by the time dementia is clinically detectable, it becomes important to address the other pathological substrates when evaluating the efficacy of an immunotherapeutic agent or designing future trials for AD. More recently, the effects of immunization strategies on downstream effects of Aβ on the geometry of neuronal processes, early tau pathology, loss of synaptophysin immunoreactivity, and neuritic dystrophy have become more active areas of interest. This mechanistic understanding of the pathology that underlies AD, combined with the ability to detect preclinical stages of disease with CSF and radiological markers will pave the way to the development of a safe and effective therapy to this disease, and give us hope to detect and prevent the progression of this devastating and multifaceted disease.
Acknowledgments
This work was supported by NIH grants AG13956, NS32636, AG03991, AG026276, and NS034467.
ABBREVIATIONS
- AD
Alzheimer’s disease
- ADAS-cog
Alzheimer’s Disease Assessment Scale-Cognitive Subscale
- APP
Amyloid precursor protein
- Aβ
Amyloid β
- CAA
Cerebral amyloid angiopathy
- CDR
Clinical Dementia Rating
- 11C-PIB
11C labeled Pittsburgh compound B
- CSF
Cerebrospinal fluid
- DAD
Disability Assessment Scale for Dementia
- EC
Entorhinal cortex
- FDG
2-Fluoro-2-deoxy-D-glucose
- FDNPP
18F-2-(1-(6-[(2-fluoroethyl(methyl) amino]-2-naphthyl)ethylidene
- GFAP
Glial fibrillary acidic protein
- IL-1
Interleukin-1
- IL-10
Interleukin-10
- MCI
Mild cognitive impairment
- MMSE
Mini-mental state examination
- NFT
Neurofibrillary tangles
- PET
Positron emission tomography
- PS-1
Presenilin-1
- PS-2
Presenilin-2
- TNF-α
Tumor necrosis factor-α
- VLP-1
Visinin-like protein 1
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991 ;82(4):239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75(6):1039–42. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- 3.Benzing WC, Ikonomovic MD, Brady DR, Mufson EJ, Armstrong DM. Evidence that transmitter-containing dystrophic neurites precede paired helical filament and Alz-50 formation within senile plaques in the amygdala of nondemented elderly and patients with Alzheimer’s disease. J Comp Neurol. 1993;334(2):176–91. doi: 10.1002/cne.903340203. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Ihara Y. A variety of cerebral amyloid deposits in the brains of the Alzheimer-type dementia demonstrated by beta protein immunostaining. Acta Neuropathol. 1988;76(6):541–9. doi: 10.1007/BF00689591. [DOI] [PubMed] [Google Scholar]
- 5.Price JL, McKeel DW, Jr, Morris JC. Synaptic loss and pathological change in older adults--aging versus disease? Neurobiol Aging. 2001;22(3):351–2. doi: 10.1016/s0197-4580(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg SM. Cerebral amyloid angiopathy and vessel dysfunction. Cerebrovasc Dis. 2002;13(suppl 2):42–7. doi: 10.1159/000049149. [DOI] [PubMed] [Google Scholar]
- 7.Maccioni RB, Munoz JP, Barbeito L. The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch Med Res. 2001;32(5):367–81. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 8.Ballatore C, Lee VM, Trojanowski JQ. Taumediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Braak E. Neuropil threads occur in dendrites of tangle-bearing nerve cells. Neuropathol Appl Neurobiol. 1988;14(1):39–44. doi: 10.1111/j.1365-2990.1988.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 10.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 12.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 14.Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T. Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005;19(2):255–7. doi: 10.1096/fj.04-2637fje. [DOI] [PubMed] [Google Scholar]
- 15.Golde TE, Eckman CB, Younkin SG. Biochemical detection of Ab isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:172–87. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 16.Beyreuther K, Masters CL. Alzheimer’s disease. The ins and outs of amyloid-beta. Nature. 1997;389(6652):677–8. doi: 10.1038/39479. [DOI] [PubMed] [Google Scholar]
- 17.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1(5):355–8. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 18.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3(1):16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 19.Heinrich G. Amyloid A4 protein and its precursors in Down’s syndrome and Alzheimer’s disease. N Engl J Med. 1989;320:1446–52. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- 20.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 21.Price JL, Morris JC. Tangles and plaques in non-demented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45(3):358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 22.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491–500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JC, McKeel DW, Jr, Storandt M, Rubin EH, Price JL, Grant EA, Ball MJ, Berg L. Very mild Alzheimer’s disease: informant-based clinical, psychometric, and pathologic distinction from normal aging. Neurology. 1991;41(4):469–78. doi: 10.1212/wnl.41.4.469. [DOI] [PubMed] [Google Scholar]
- 24.Morris JC, Price AL. Pathologic correlates of non-demented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17(2):101–18. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 25.Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17:278–82. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 26.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38(11):1682–7. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 27.Silverman W, Wisniewski HM, Bobinski M, Wegiel J. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18(4):377–9. doi: 10.1016/s0197-4580(97)00051-1. discussion 89–92. [DOI] [PubMed] [Google Scholar]
- 28.Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM, Holtzman DM, Goate AM. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA. 2008;105(23):8050–4. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 30.Fagan Aea. Decreased CSF Abeta-22 correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2008 doi: 10.1002/ana.21559. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58(9):1395–402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 32.Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41(1):17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 34.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 35.Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54(2):276–81. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 36.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 37.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65(11):1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 38.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 39.Boche D, Nicoll JA. The role of the immune system in clearance of Abeta from the brain. Brain Pathol. 2008;18(2):267–78. doi: 10.1111/j.1750-3639.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 41.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64(1):94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 42.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38(4):547–54. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 43.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 44.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 45.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 46.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98(15):8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeKosky ST. Taking the next steps in the diagnosis of Alzheimer’s disease: the use of biomarkers. CNS Spectr. 2008;13(3 Suppl 3):7–10. doi: 10.1017/s1092852900017193. [DOI] [PubMed] [Google Scholar]
- 48.Thal LJ, Kantarci K, Reiman EM, Klunk WE, Weiner MW, Zetterberg H, Galasko D, Praticò D, Griffin S, Schenk D, Siemers E. The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(1):6–15. doi: 10.1097/01.wad.0000191420.61260.a8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2(10):605–13. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 50.Racchi M, Uberti D, Govoni S, Memo M, Lanni C, Vasto S, Candore G, Caruso C, Romeo L, Scapagnini G. Alzheimer’s disease;new diagnostic and therapeutic tools. Immun Ageing. 2008;5(1):7. doi: 10.1186/1742-4933-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, Mehta PD. Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61(9):1185–90. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 52.Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, Ravetch J, Mayeux R. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci USA. 2008;105(37):14052–7. doi: 10.1073/pnas.0805902105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graff-Radford NR, Crook JE, Lucas J, Boeve BF, Knopman DS, Ivnik RJ, Smith GE, Younkin LH, Petersen RC, Younkin SG. Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007;64(3):354–62. doi: 10.1001/archneur.64.3.354. [DOI] [PubMed] [Google Scholar]
- 54.van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5(8):655–60. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 55.Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP. Amyloid beta protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci, Lett. 2001;304(1–2):102–6. doi: 10.1016/s0304-3940(01)01754-2. [DOI] [PubMed] [Google Scholar]
- 56.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64(3):343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 57.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 58.Burczynski ME, Dorner AJ. Transcriptional profiling of peripheral blood cells in clinical pharmacogenomic studies. Pharmacogenomics. 2006;7(2):187–202. doi: 10.2217/14622416.7.2.187. [DOI] [PubMed] [Google Scholar]
- 59.Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM. Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging. 2007;28(12):1795–809. doi: 10.1016/j.neurobiolaging.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Bibl M, Mollenhauer B, Esselmann H, Lewczuk P, Klafki HW, Sparbier K, Smirnov A, Cepek L, Trenkwalder C, Rüther E, Kornhuber J, Otto M, Wiltfang J. CSF amyloid-beta-peptides in Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease dementia. Brain. 2006;129(Pt 5):1177–87. doi: 10.1093/brain/awl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JM, Blennow K, Andreasen N, Laterza O, Modur V, Olander J, Gao F, Ohlendorf M, Ladenson JH. The Brain Injury Biomarker VLP-1 Is Increased in the Cerebrospinal Fluid of Alzheimer’s Disease Patients. Clin Chem. 2008 ;54(10):1617–23. doi: 10.1373/clinchem.2008.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arai HNT, Kosaka Y. Elevated cerebrospinal fluid tau protein level as a predictor of dementia in memory-impaired patients. Alzheimer’s Res. 1997;3:211–3. [Google Scholar]
- 63.Buerger K, Teipel SJ, Zinkowski R, Blennow K, Arai H, Engel R, Hofmann-Kiefer K, McCulloch C, Ptok U, Heun R, Andreasen N, DeBernardis J, Kerkman D, Moeller H, Davies P, Hampel H. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology. 2002;59(4):627–9. doi: 10.1212/wnl.59.4.627. [DOI] [PubMed] [Google Scholar]
- 64.Riemenschneider M, Lautenschlager N, Wagenpfeil S, Diehl J, Drzezga A, Kurz A. Cerebrospinal fluid tau and beta-amyloid 42 proteins identify Alzheimer disease in subjects with mild cognitive impairment. Arch Neurol. 2002;59(11):1729–34. doi: 10.1001/archneur.59.11.1729. [DOI] [PubMed] [Google Scholar]
- 65.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 66.Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, Kaye JA, Raskind MA, Zhang J, Peskind ER, Montine TJ. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69(7):631–9. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 67.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 68.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 69.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C] PIB and [18F] FDG PET study. Neurology. 2007;68(7):501–8. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 71.Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Långström B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129(Pt 11):2856–66. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 72.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 73.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355(25):2652–63. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 74.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 75.de Leon MJ, Mosconi L, Blennow K, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Tsui W, Saint Louis LA, Sobanska L, Brys M, Li Y, Rich K, Rinne J, Rusinek H. Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann NY Acad Sci. 2007;1097:114–45. doi: 10.1196/annals.1379.012. [DOI] [PubMed] [Google Scholar]
- 76.Jack CR, Jr, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52(7):1397–403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson KA, Jones K, Holman BL, Becker JA, Spiers PA, Satlin A, Albert MS. Preclinical prediction of Alzheimer’s disease using SPECT. Neurology. 1998;50(6):1563–71. doi: 10.1212/wnl.50.6.1563. [DOI] [PubMed] [Google Scholar]
- 78.de Leon MJ, George AE, Stylopoulos LA, Smith G, Miller DC. Early marker for Alzheimer’s disease: the atrophic hippocampus. Lancet. 1989;2(8664):672–3. doi: 10.1016/s0140-6736(89)90911-2. [DOI] [PubMed] [Google Scholar]
- 79.de Leon MJ, Golomb J, George AE, Convit A, Tarshish CY, McRae T, De Santi S, Smith G, Ferris SH, Noz M. The radiologic prediction of Alzheimer disease: the atrophic hippocampal formation. AJNR Am J Neuroradiol. 1993;14(4):897–906. [PMC free article] [PubMed] [Google Scholar]
- 80.Convit A, de Asis J, de Leon MJ, Tarshish CY, De Santi S, Rusinek H. Atrophy of the medial occipitotemporal, inferior, and middle temporal gyri in non-demented elderly predict decline to Alzheimer’s disease. Neurobiol Aging. 2000;21(1):19–26. doi: 10.1016/s0197-4580(99)00107-4. [DOI] [PubMed] [Google Scholar]