

Abstract
The last 20 years have witnessed a tremendous explosion in the number of antiepileptic drugs (AEDs) as well as the introduction of AEDS developed for specific epilepsy syndromes. The study of the efficacy and side effect profile of AEDs for unique epilepsy syndromes has allowed neurologists to utilize evidence-based medicine when treating patients. In late 2008, the Food and Drug Administration approved rufinamide for adjunctive use in the treatment of seizures associated with Lennox–Gastaut syndrome. This unique chemical compound is also the first new AED to reach the market in the United States having a pediatric indication prior to approval for adults. Rufinamide appears to have a broad spectrum of efficacy, is well tolerated, and may be rapidly initiated—properties that will likely extend its use outside of Lennox–Gastaut syndrome.
Rufinamide's chemical name is: 1-[(2,6-difluorophenyl) methyl]-1H-1,2,3-triazole-4 carboxamide (see Figure 1); it is a triazole derivative structurally unrelated to any currently marketed antiepileptic drug (AED) (1). Rufinamide was granted orphan drug status for adjunctive treatment of patients with Lennox–Gastaut syndrome in October 2004, received its marketing authorization in Europe in January 2007, and was approved by the FDA in December in 2008 for adjunctive treatment of seizures associated with Lennox–Gastaut syndrome for children 4 years or older and for adults. The purposes of this paper are to present the significant parameters for the use of rufinamide in clinical practice and to summarize the results of phases II and III clinical trials.
FIGURE 1.

Chemical structure of rufinamide.
Pharmacology
The precise mechanisms by which rufinamide exerts its antiepileptic effect are unknown. In vitro studies suggest that a principal mechanism of action is the modulation of activity in sodium channels, particularly prolongation of the inactive state. In cultured cortical neurons from immature rats, rufinamide significantly slowed sodium channel recovery from inactivation after a prolonged prepulse and limited the sustained repetitive firing of sodium-dependant action potentials (1,2). Rufinamide has no effect on benzodiazepine or GABA receptors or on adenosine uptake; it also has no significant interactions with glutamate, adrenergic, tryptophan, histamine, and muscarinic cholinergic receptors.
The antiepileptic effect of rufinamide has been assessed in several animal models of generalized and partial seizures. For instance, oral rufinamide exhibited acute anticonvulsive activity in mice and rat models, suppressing maximal electroshock-induced tonic–clonic seizures in both species and pentylenetetrazol-induced clonic seizures in mice (2). In the maximal electroshock test conducted in mice, the effective dose required for a 50% response against induced seizures (i.e., ED50) was 23.9 mg/kg for rufinamide compared to values of 9.0, 20.1, 664.8, and >2,000 mg/kg for the established AEDs phenytoin, phenobarbital, valproate, and ethosuximide, respectively. In mouse pentylenetetrazol tests, the ED50 values were lower for rufinamide (45.8 mg/kg) than for ethosuximide (192.7 mg/kg), phenytoin (>300 mg/kg), and valproate (388.3 mg/kg). Similarly, the behavioral toxicity of rufinamide was equivalent or much better than the four AEDs tested in this study. Intraperitoneal rufinamide suppressed pentylenetetrazol-, bicuculline- and picrotoxin-induced clonus in mice. Efficacy in all seizure models suggests that rufinamide is likely to be of value in a broad spectrum of seizure types, although results in animal models may not translate to humans.
Pharmacokinetics
Rufinamide is well absorbed after oral administration. The extent of absorption decreases slightly as the dose is increased, however the effect is negligible at most clinical doses (3). Rufinamide absorption is enhanced by food, probably by improved solubility. This enhancement results in over a 50% increase in the peak exposure (Cmax) and approximately a one-third increase in overall absorption. Patients will need to be advised to take rufinamide each time in the same temporal relation to their meals to maintain steady concentrations from one dose to the next. Rufinamide has low protein binding (about 34%), suggesting that competition for protein binding would not be a source of drug interaction, and its volume of distribution after an oral dose approximates total body water (i.e., 50–80 L) (Table 1).
TABLE 1.
Pharmacokinetics of Rufinamide
| Bioavailability | Fed—85% |
| Tmax | 4 to 6 hours |
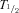 |
6 to 10 hours |
| Protein binding | 26 to 34% |
| Volume of distribution (Vd/F) | 50 to 80 L (0.8–1.2 L/kg) |
| Serum levels | 5 to 55 mcg/mL |
The elimination of rufinamide occurs via hepatic metabolism with the primary metabolite, resulting from carboxylesterase-mediated enzymatic hydrolysis of the carboxylamide moiety, to form an inactive carboxylic acid derivative (CGP 47,292) (1,3). The metabolite has no known pharmacologic activity, is excreted in the urine, and the metabolic route is not cytochrome P450 dependant. Rufinamide is a weak inducer of CYP3A4 enzymes and is susceptible to induction by other AEDs, with the resulting effect of a decrease in rufinamide serum levels in their presence. Rufinamide pharmacokinetics are not affected by impaired renal function. The renal excretion of unchanged rufinamide is less than 2% of the total dose. The half-life of rufinamide is approximately 6 to 10 hours and does not change with renal impairment. Dose adjustment is likely necessary for patients undergoing hemodialysis, as the drug's low protein binding would result in the free drug being removed during dialysis. There is no autoinduction of rufinamide metabolism. The effect of hepatic impairment has not been studied.
Clinical trials have shown no significant differences in the pharmacokinetic parameters as a function of age within the range tested (i.e., age 4 years to elderly subjects). However, applying the parameters derived from the pooled population pharmacokinetic analysis, one would predict rufinamide clearance at a full dose (45 mg/kg/day) to be 50% higher in a 4-year-old child than in an adult. Serum rufinamide levels can help guide the clinical decision making for a given patient, as variability in the rate and extent of absorption, comedications, and individual differences in drug clearance may impact the serum level and clinical efficacy. In addition, the significant relationship between therapeutic and adverse effects and plasma rufinamide concentrations suggests that measurement of rufinamide levels will be of value in clinical practice. Identifying the concentration at which a patient shows a good response provides a reference when evaluating the cause of a subsequent change in clinical status (4,5). Population pharmacokinetic studies reveal a positive correlation between reduction in seizure numbers and plasma rufinamide concentrations. Rufinamide reduced partial seizures and seizures associated with Lennox–Gastaut syndrome in a concentration-dependant manner. The mean plasma rufinamide concentration to reduce seizure frequency by 25% or 50% was predicted to be 15 and 30 mcg/mL, respectively (3).
Drug Interactions
Rufinamide does not have significant pharmacokinetic interactions with benzodiazepines, carbamazepine, lamotrigine, phenytoin, phenobarbital, valproate, topiramate, vigabatrin, oxcarbazepine, or primidone (3). However, cytochrome P450 enzyme inducers, such as phenobarbital, primidone, phenytoin, and carbamazepine, increase the clearance of rufinamide, which likely is secondary to induction of carboxylesterases activity. The coadministration of these enzyme-inducing AEDs with rufinamide leads to dramatically decreased rufinamide levels and potentially decreased efficacy (6). These patients may require a higher rufinamide dose. In contrast, valproate administration may lead to elevated levels of rufinamide; the effect was most dramatic in children, for whom rufinamide concentrations can increase by 60 to 70 percent (1,3). The highest serum levels of rufinamide are noted in patients with high serum valproate levels and who are concurrently taking high doses of rufinamide. The exact mechanism for this interaction is unclear, but valproate is known to inhibit a number of drug-metabolizing enzymes.
Clinical studies have shown that rufinamide can increase the clearance of oral contraceptives, specifically ethinyl estradiol and norethindrone. The clinical significance of this mild interaction is not known. The extent of the decreased plasma concentrations caused by rufinamide is much less than that caused by phenytoin, carbamazepine, and phenobarbital. The finding is consistent with the weak induction of the P450 3A4 enzyme by rufinamide.
Efficacy Demonstrated in Clinical Studies
Placebo-controlled studies for rufinamide that have efficacy data include studies involving: 1) patients with Lennox–Gastaut syndrome (see Table 2), 2) adult partial onset seizures (for both monotherapy and adjunctive therapy), 3) pediatric partial onset seizures as adjunctive therapy, and 4) patients with refractory generalized tonic–clonic seizures (7).
TABLE 2.
Summary of Clinical Studies with Rufinamide
| STUDY TYPE | SEIZURE TYPE | DAILY DOSE | AGE (YEARS) | OUTCOME* | REFERENCE |
|---|---|---|---|---|---|
| Adjunct | Lennox–Gastaut syndrome | 45 mg/kg (maximum 3,200 mg) or placebo | 4 to 30 | ↓Drop attacks ↓Total seizures ↓Seizure severity | 8 |
| Adjunct | Partial onset | 200, 400, 800, 1,600 or placebo | ≥15 | ↓Total seizures (+) Responder rate | 1 |
| Adjunct | Partial onset | 3,200 mg or placebo | ≥16 | ↓Total seizures (+) Responder rate | 1 |
| Monotherapy | Partial onset | 3,200 mg or placebo | ≥12 | Fewer seizures and longer time to first, second, and third seizure for rufinamide | 1 |
| Adjunct† | Primary GTC | 800 mg or placebo | ≥4 | No difference vs. placebo | 7 |
Abbreviations: GTC, generalized tonic–clonic.
All were significant (p < 0.05) except study Ref. 7.
The doses used did not provide patients with plasma rufinamide concentrations that are therapeutic for other seizure types, which could explain the lack of efficacy seen in this study.
Seizures Associated with Lennox–Gastaut Syndrome
The left end points evaluated were the percent of change in drop attacks (tonic–atonic seizures), total seizure frequency, and the seizure severity rating taken from a global evaluation of the patient's condition. Rufinamide-treated patients had a 42.5% median reduction in drop attacks per 28 days relative to the baseline compared to placebo-treated patients, who had a 1.4% median increase (p < 0.0001). The rufinamide-treated patients also had a significant decrease in the total seizure frequency per 28 days relative to the baseline (p= 0.0015: median reduction for rufinamide was 32.7% vs 11.7% for placebo). These results are comparable to the findings in other clinical trials involving topiramate, lamotrigine, and felbamate (see Figure 2). In addition, there was significant improvement on the seizure severity global evaluation for the rufinamide group compared with the placebo group (p < 0.005). Population pharmacokinetic modeling revealed that the reduction in atonic seizures, total seizures, and seizure severity was correlated with rufinamide serum concentrations. Patients who received rufinamide were approximately four times more likely to experience at least a 50% reduction in drop attacks, compared with those receiving placebo. The response to rufinamide could be seen as early as week 2. In the open label extension phase, patients who switched from double-blind rufinamide to open-label rufinamide continued responding to treatment (9). Figures 2 and 3 compare the clinical response to other trials involving patients with Lennox–Gastaut syndrome (10–14).
FIGURE 2.

Short-term, double-blind studies on Lennox–Gastaut syndrome. Abbreviations: FLB, felbamate; TPM, topiramate; LTG, lamotrigine; RFM, rufinamide; CLB, clobazam. References: 1, #10; 2, #11; 3, #12; 4, #8; 5, #15. Felbamate: approved for all ages. Lamotrigine and topiramate: approved for ages 2 years and older in Lennox–Gastaut syndrome. Clobazam: high dose is 1 mg/kg/day, max 40 mg/day, given BID; low dose is 0.25 mg/kg/day, max 10 mg/day. There was a significant 14% decrease over 4 weeks.
FIGURE 3.
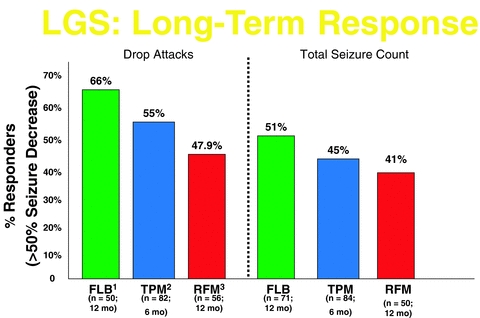
Long-term, open-label studies of AED efficacy for Lennox–Gastaut syndrome. Abbreviations: FLB, felbamate; TPM, topiramate; RFM, rufinamide. References: 1, #13; 2, #14; 3, #9. Lamotrigine: no long-term data reported.
An right, multicentered, double-blind, placebo-controlled, randomized, parallel-group study, performed between early 1998 and fall of 2000, enrolled 138 patients (ages 4–30 years) with a diagnosis of inadequately controlled seizures associated with Lennox–Gastaut syndrome (including both drop attacks and atypical absence seizures) and who were being treated with one to three AEDs (felbamate therapy was not allowed in this study) (8). Each patient was required to have had at least 90 seizures in the month prior to study entry. After a 4-week baseline phase, patients were randomized to receive either rufinamide or placebo during a 12-week double-blind phase. The double-blind phase consisted of a titration period (over 1–2 weeks) and a maintenance period (10 weeks). During the titration period, the dose was increased to approximately 45 mg/kg/day (maximum dose 3,200 mg/day); 77% of patients achieved their final dose level by the end of the first week, which was kept stable during the maintenance period. Doses were given on a twice-daily schedule.
Partial Onset Seizures
Two double-blind, placebo-controlled, randomized, parallel-group studies (n= 313 and 647) have been performed using rufinamide as adjunctive therapy for partial onset seizures. One was a fixed-dose study of adolescents and adults, 16 years or older, and the other was a dose-ranging study of adolescents and adults, ages 16 to 65 years (1). In both studies, patients had inadequately treated partial seizures and were on AED therapy. In the first study, the patients were required to have had at least one partial seizure in each 4-week period of a baseline phase and were then randomized to rufinamide or placebo during a 13-week double-blind phase (1). Titration of rufinamide occurred over 1 to 2 weeks. The initial dose of 800 mg/day was increased to a target dose of 3,200 mg/day, given as a twice-daily dose for an 11-week maintenance period. Rufinamide-treated patients experienced a significant, although modest, reduction (p= 0.0158) in partial seizure frequency per 28 days compared with placebo-treated patients (a 20.4% median decrease vs a 1.6% median increase). In addition, the responder rate (at least a 50% reduction in partial seizure frequency per 28 days) during the double-blind phase relative to the baseline phase was 28.2% for rufinamide compared with 18.6% for placebo (p= 0.0381).
In the second adjunctive trial for partial onset seizures, patients were required to have experienced nine or more seizures during the 12-week baseline phase (1). They were then randomized to one of five treatment groups (placebo or rufinamide at 200, 400, 800, or 1,600 mg/day); treatments were administered on a twice-daily schedule for the 3-month double-blind phase. Significant dose response was observed and pairwise comparisons between placebo and each rufinamide treatment group showed that the seizure frequency ratio was statistically significantly lower for the 400-, 800-, and 1,600-mg groups. In addition, a significant dose response was observed for the responder rate (p < 0.04).
A single monotherapy study has been performed—a double-blind, placebo controlled, randomized, parallel-group study (n= 104) involving inpatients, ages 12 years and older, with uncontrolled partial seizures, who had just completed an inpatient presurgical evaluation. The patients had a 48-hour baseline prospective phase and then were randomized to either to rufinamide, 2,400 mg/day on day 1 and 3,200 mg/day on days 2 to 10 (given three times per day), or to placebo. The primary efficacy variable was the mean time to meet the exit criteria. Outcome data favored rufinamide (p < 0.05) over placebo, with a median time to exit of 4.8 days compared with 2.4 days. Statistically significantly differences between treatments were observed for the time to first, second, and third partial seizures (p < 0.04), however the time to the fourth partial seizure failed to reach significance (p= 0.0509).
Long-Term Follow-Up
Both the Lennox–Gastaut study and the studies on partial seizures were followed by long-term, open-label extension studies. The patients who switched from double-blind placebo to open-label rufinamide quickly responded to treatment, with a marked decrease in seizure frequency. There was no evidence of tolerance to the anticonvulsant effect of rufinamide, during up to 3 years of follow-up (1).
Dosing, Tolerability, and Safety
Table 3 provides the authors suggestions for dosing in children and adults. The clinical trials were performed with administration of the drug with food (resulting in enhanced absorption), which is the recommended protocol.
TABLE 3.
Rufinamide Dosing
| LABEL (FDA) | AUTHORS’ RECOMMENDATIONS | |
|---|---|---|
| Children | Given BID: Begin 10 mg/kg/day, Increase by 10 mg/kg, every other day to 45 mg/kg/day or 3,200 mg/day (whichever is less) | Given BID or TID: Begin 15 mg/kg/day Increase by 15 mg/kg/day, every week to 45 mg/kg/day or 3,600 mg/day (whichever is less) |
| Adults | Given BID: Begin with 400 to 800 mg/day Increase by 400 to 800 mg every 2 days, up to a maximum of 3,200 mg/day | Given BID or TID: Begin with 1,200 mg/day Increase by 1,200 mg/day every week up to 3,600 mg/day |
Abbreviations: BID, twice daily dosing; TID, three times daily dosing.
Take with food. Supplied in 200- and 400-mg scored tablets (and 100 mg in Europe), which can be administered whole, in half tablets, or crushed.
Based on the clinical trials, rufinamide appears to be well tolerated. A small number of rufinamide-treated patients (9% vs 4% for placebo) discontinued treatment because of adverse effects (15). The adverse experiences most commonly associated with discontinuation of rufinamide (>1%) were similar in adults and children: dizziness (1.8%), fatigue (1.6%), and headache (1.1%). The majority of adverse events in the clinical trials were judged to be mild to moderate and often transient in nature, largely occurring during the titration phase. The most commonly observed adverse events (i.e., occurring in >10% and at a higher frequency than placebo-treated patients), pooled from all of the studies of patients with epilepsy, were headache, dizziness, fatigue, somnolence, and nausea. Adverse events were reported more often in adults than in children and with plasma rufinamide concentrations in the higher ranges. Only somnolence and vomiting were significantly more common in the rufinamide group of the Lennox–Gastaut syndrome trial. At the fixed titration dose of 45 mg/kg/day in all pediatric trials, only somnolence, vomiting, and headache were significantly more common with rufinamide than placebo (i.e., observed >5% more often). In doses up to 3,200 mg/day in all adult clinical trials, only dizziness, fatigue, and diplopia were significantly more common with rufinamide than placebo. Neuropsychiatric side effects were rare (all <5%) and were no more common in rufinamide than in placebo groups. The rufinamide side effect profile is similar to other drugs that have an effect on the sodium channel.
The overall tolerability of rufinamide is good. During the clinical trials, there were no cases of Stevens–Johnson syndrome, hepatic failure, agranulocytosis, or pancytopenia. The incidence of cognitive disorders in rufinamide-treated patients was higher than placebo-treated patients only because of the increased occurrence of somnolence. Psychiatric adverse events were similar between rufinamide and placebo patients.
AED hypersensitivity syndrome has occurred in association with rufinamide therapy. While the clinical symptoms varied, patients generally presented with fever and rash associated with other organ system involvement. In the clinical trials, this syndrome occurred in close temporal association (within the first 4 weeks) to the initiation of rufinamide therapy and was more likely in the pediatric population. If a serious rash related to rufinamide is suspected, rufinamide should be discontinued and alternative treatment started.
In the randomized trial, cognitive assessments were performed at baseline (before rufinamide treatment) and after 3 months of adjunctive therapy at doses of 200, 400, 800, and 1,600 mg/day for adolescents and adults (ages 15–64 years) with partial seizures (16). None of the cognitive tests for psychomotor speed and attention or for working memory demonstrated a significant worsening at any of the doses of rufinamide. In a placebo-controlled study of the QT interval, a higher percentage of subjects taking 2,400–4,800 mg of rufinamide per day had a QT shortening of greater than 20 milliseconds compared with placebo, but none had a reduction below 300 milliseconds. Patients with potassium channelopathy associated with familial short QT syndrome cannot be treated with rufinamide. Caution is advised when administering rufinamide with other drugs or disease states that shorten the QT interval (e.g., digoxin toxicity, hypercalcemia, hyperkalemia, and acidosis). There is no known clinical risk associated with the degree of QT shortening induced by rufinamide. No meaningful changes in laboratory data were observed, and rufinamide is designated pregnancy Category C. When assessing the risk of rufinamide or any new drug, it is important to remember that not all potential risks may have been identified, which is because only a relatively small number of patients have been exposed to the drug for a long period of time.
Conclusions
Rufinamide is a new broad-spectrum AED that is structurally unique. It offers various advantages: 1) the ability to rapidly escalate dosing and obtain a clinical response, 2) few drug interactions, and 3) a good cognitive and psychiatric adverse event profile. The CNS-related adverse events (primarily somnolence) largely occurred during the first 2 weeks of therapy, which may be related to the rapid fixed titration schedule used in the clinical trials. Slower titration helps minimize side effects. No laboratory monitoring is required, and plasma levels correlate with clinical efficacy. All of these characteristics will make it a commonly used drug in Lennox–Gastaut syndrome. Further trials are ongoing. Continuing clinical experience may elucidate whether rufinamide eventually will prove beneficial for a wider spectrum of patients.
References
- 1.Arroyo S. Rufinamide. Neurotherapeutics. 2007;4:155–162. doi: 10.1016/j.nurt.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White HS, Franklin MR, Kupferberg HJ, Schmutz M, Stables JP, Wolf HH. The anticonvulsant profile of rufinamide (CGP33101) in rodent seizure models. Epilepsia. 2008;49:1213–1220. doi: 10.1111/j.1528-1167.2008.01552.x. [DOI] [PubMed] [Google Scholar]
- 3.Perucca E, Cloyd J, Critchley D, Fuseau E. Rufinamide: Clinical Pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia. 2008;49:1123–1141. doi: 10.1111/j.1528-1167.2008.01665.x. [DOI] [PubMed] [Google Scholar]
- 4.Patsalos PN, Berry DJ, Bourgeois BF, Cloyd JC, Glauser TA, Johannessen SI, Leppick IE, Tomson T, Perucca E. Antiepileptic drugs–best practice guidelines for therapeutic drug monitoring: A position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia. 2008;49:1239–1276. doi: 10.1111/j.1528-1167.2008.01561.x. [DOI] [PubMed] [Google Scholar]
- 5.Perucca E. Is there a role for therapeutic drug monitoring of new anticonvulsants? Clin Pharmacokinet. 2000;38:191–204. doi: 10.2165/00003088-200038030-00001. [DOI] [PubMed] [Google Scholar]
- 6.Brodie M, Li H, Wang W, Narurkar M, Richardson S. Differential effects of rufinamide in adults with partial onset seizures as a function of concomitant anti-epileptic drug therapy: A post hoc analysis. Neurol. 2009;72(11, suppl 3):A228. [Google Scholar]
- 7.Biton V, Rosenfeld W, Schachter S, Perdomo C, Arroyo S. Multicenter, randomized, double-blind, placebo-controlled, parallel trial comparing the safety and efficacy of rufinamide. Ann Neurol. 2005;58(suppl 9):S114. [Google Scholar]
- 8.Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut Syndrome. Neurology. 2008;70:1950–1958. doi: 10.1212/01.wnl.0000303813.95800.0d. [DOI] [PubMed] [Google Scholar]
- 9.Glauser T, Kluger G, Krauss G, Perdomo C, Arroyo S. Short term and long term efficacy and safety of rufinamide as adjunctive therapy in patients with inadequately controlled Lennox-Gastaut Syndrome. Neurol. 2006;66(5, suppl 2):A36. [Google Scholar]
- 10.Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of Felbamate in Childhood Epileptic Encephalopathy (Lennox-Gastaut Syndrome) N Engl J Med. 1993;328:29–33. doi: 10.1056/NEJM199301073280105. [DOI] [PubMed] [Google Scholar]
- 11.Sachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double-blind, randomized trial of topiramate in Lennox-Gastaut Syndrome. Topiramate YL Study Group. Neurol. 1999;52:1882–1887. doi: 10.1212/wnl.52.9.1882. [DOI] [PubMed] [Google Scholar]
- 12.Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalized seizures associated with the Lennox-Gastaut Syndrome. Lamictal Lennox-Gastaut Study Group. N Engl J Med. 1997;337:1807–1812. doi: 10.1056/NEJM199712183372504. [DOI] [PubMed] [Google Scholar]
- 13.Dodson WE Felbamate in the treatment of Lennox-Gastaut Syndrome: Results of a 12-month open-label study following a randomized clinical trial. Epilepsia. 1993;34(suppl 7):S18–S24. doi: 10.1111/j.1528-1157.1993.tb04590.x. [DOI] [PubMed] [Google Scholar]
- 14.Glauser TA, Levisohn PM, Ritter F, Sachdeo RC, and the Topiramate YL Study Topiramate in Lennox-Gastaut Syndrome: Open-label treatment of patients completing a randomized controlled trial. Epilepsia. 2000;41(suppl 1):S86–S90. doi: 10.1111/j.1528-1157.2000.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 15.Wheless J, Conry J, Krauss G, Mann A, LoPresti A, Narurkar M. Safety and tolerability of rufinamide in children with epilepsy: A pooled analysis of seven clinical trials. J Child Neurol. 2009;50:1158–1166. doi: 10.1177/0883073809350508. [DOI] [PubMed] [Google Scholar]
- 16.Aldenkamp AP, Apherts WCJ. The effect of the new antiepileptic drug rufinamide on cognitive functions. Epilepsia. 2006;47:1153–1159. doi: 10.1111/j.1528-1167.2006.00589.x. [DOI] [PubMed] [Google Scholar]