

Alzheimer’s disease (AD) is now the most common form of dementia, affecting up to 15 million people worldwide. A common pathogenic event that occurs in all forms of AD is the progressive accumulation of amyloid-β peptide (Aβ) in brain loci responsible for cognition[1]. Genetic and epidemiological data support a role for altered cholesterol homeostasis in the pathogenesis of AD[2] with polymorphisms of a number of cholesterol-related genes being linked to elevated central levels of Aβ[3]. Recent studies suggest that intracellular levels of cholesteryl esters (CEs) correlate closely with Aβ production and secretion[4–6], and that the cellular enzyme responsible for CE production, acyl-CoA: cholesterol acyltransferase (ACAT) may play a central role in the regulation of Aβ processing. Kovacs and co-workers[5, 6] have recently shown that the ACAT inhibitors, CP-113,818 and Dup 128, reduce Aβ secretion in CHO cells stably transfected with human amyloid precursor protein (APP751) (Fig. 1). Dorsal implantation of CP-113,818, in a transgenic murine model of familial AD, resulted in the reduction of brain Aβ plaque load[4].
Figure 1.
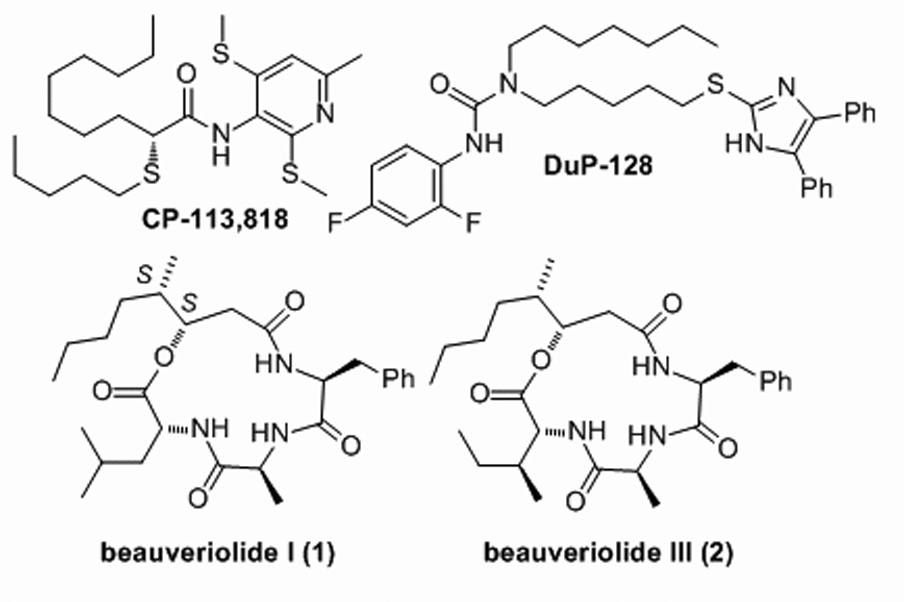
ACAT inhibitors
The natural product depsipeptides, beauveriolide I (1) and III (2) were originally isolated from the fungal strain Beauveria sp. FO-6979 during a screening program for inhibitors of lipid droplet accumulation in murine macrophages[7] (Fig. 1). The assignment of the absolute stereochemistry of the 3-hydroxy-4-methyloctanoic acid (HMA) as (3S, 4S) and the first formal syntheses of all stereiosiomers of 1 and 2 have been reported recently[8]. Depsipeptides 1 and 2 prevent lipid droplet accumulation in primary mouse peritoneal macrophages via ACAT inhibition and importantly, they are orally active and reduce atherogenic lesions in mouse models of atherosclerosis[9]. Currently, these depsipeptides are being studied as a new approach for the treatment of atherosclerosis[9, 10].
Given our ongoing interest in the pathogenesis of atherosclerosis[11] and AD[12] and our expertise in synthetic approaches to cyclodepsipeptides[13] we wondered whether 1 and 2 may be a valid chemical scaffold that would, for the first time, link orally active ACAT inhibition to therapeutically-relevant neuronal Aβ-reduction. Towards this goal, herein we report a new flexible homochiral synthesis of the fungal depsipeptide natural products, beauveriolide I (1) and III (2) and reveal that these cyclodepsipeptides are potent inhibitors of cellular Aβ40 and Aβ42 secretion in vitro.
Our synthesis of 1 and 2 commences with a cis-crotylation of PMB-protected aldehyde 3 that proceeds with high diastereo- (> 98:2) and enantio-selectivity (> 95:5), to give cis-alcohol 4 (Scheme 1). Homologation of 4 proceeds via initial TBS protection of the secondary alcohol, hydroboration of the terminal double bond, TPAP oxidation of the resultant primary alcohol and Wittig olefination to give TBS-protected cis-alkene 5a (> 85 % Z as determined by 1H NMR).
Scheme 1.
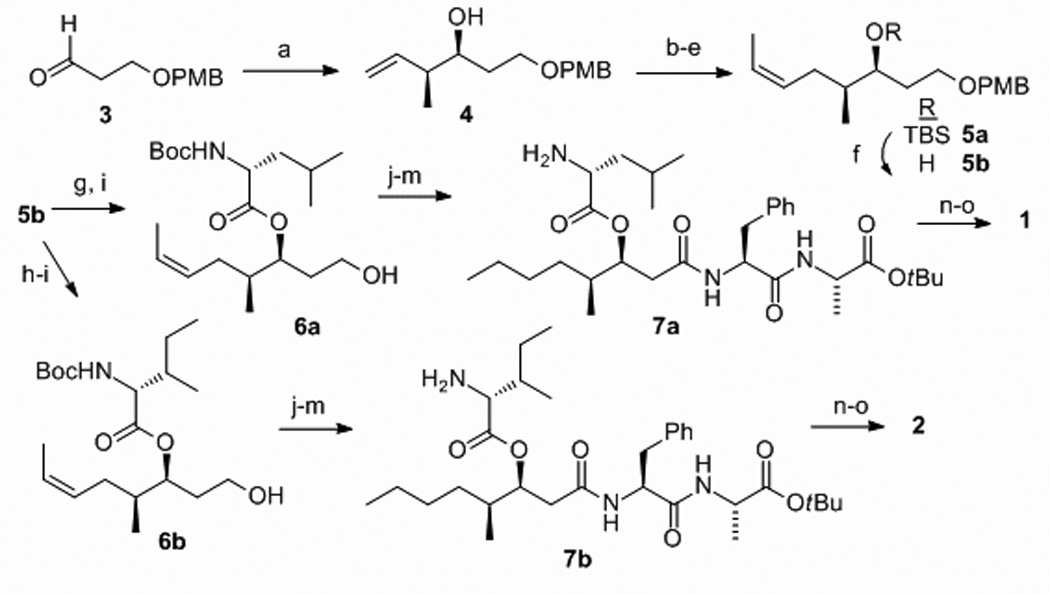
Synthesis of 1 and 2 (a) cis-2-butene, t-BuOK, n-BuLi, −78°C to −50°C; (+)-B-methoxydiisopinocamphorylborane; BF3.OEt2; 78% (b) TBSOTf, 2,6-lutidine; 95% (c) 9-BBN, THF; H2O2, NaOH; 82% (d) TPAP, NMO; 96% (e) EtPPh3, NaHMDS; 83% (f) TBAF; 97%; (g) DIC (2.4 eq.), DMAP (2.0), N-Boc-D-Leu (2.0); Sc(OTf)3 (0.4); 87%; (h) DIC (2.4 eq.), DMAP (2.0), N-Boc-D-allo-Ile (2.0) ; Sc(OTf)3 (0.4); 79% (i) DDQ, CH2Cl2/H2O; (j) Dess-Martin, CH2Cl2; (k) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O; (l) EDC, HOBt, H2N-l-Phe-l-Ala-COO-t-Bu, CH2Cl2; 55% four steps from i–l; (m) H2, Pd/C (5%), EtOAc (quant.); (n) TFA/CH2Cl2; (o) HATU, HOAt, collidine, DMF; (1) 74% (2) 71 %.
A key aspect of the synthesis of depsipeptides is the strategy for macrocyle formation, either lactonization or lactamization. Our synthesis, that prepares the key (3S,4S)-HMA in a masked form (5a), is amenable to both approaches. However, for brevity only the macrolactamization approach is described in Scheme 1. Thus, the TBS group of 5a is removed and the secondary alcohol 5b is esterified with either N-Boc-D-Leu-OH (for 1), or N-Boc-d-allo-Ile-OH (for 2) with Sc(OTf)3 catalysis; conditions that result in no measurable racemization of the amino acid esters 6a or 6b. The completion of the syntheses of 1 and 2 are then entirely parallel and involve PMB removal, primary alcohol oxidation, coupling of H2N-l-Phe-l-Ala-CO-tBu, alkene reduction and protecting group removal to give amino acids 7a or 7b. The critical macrolacatamization reaction was achieved with HATU/HOAt activation and proceeded in > 70 % yield with both 7a and 7b to give 1 and 2 respectively.
With synthetic 1 and 2 in hand, we studied the ability of these depsipeptides to reduce cholesterol esterification in the stable transgenic chinese hamster ovary (CHO) cell line (7WD10) expressing human APP751[14]. Cultured 7WD10 cells incubated with depsipeptide 1 or 2 (5 µM) in media and co-solvent (DMSO, 0.1 % v/v) exhibit reduced cellular cholesteryl ester (CE) levels within 12 h. This reduction in CE levels reaches a maximum level (~ 5 % CEs relative to vehicle treated cells) after 24–48 h of incubation with 1 and 2 (data not shown). Depsipeptides 1 and 2 exhibit a dose-dependent reduction in CE levels in the 7WD10 CHO cells, with measured IC50 values after 96 h incubation of 0.08 ± 0.02 µM and 0.17 ± 0.08 µM respectively (Fig 2a). These values are similar to the IC50 values of 1 and 2 in primary murine macrophages[9]. Importantly, the cellular total cholesterol (TC), that is the sum of free cholesterol (FC) and CE remains essentially unchanged in the CHO cells throughout incubation with 1 and 2 (data not shown). This null effect of the beauveriolides on TC causes a shift in cellular cholesterol from esterified to unesterified and is in-line with what has been observed for other ACAT inhibitors, such as CP113,818 and DUP-128, on other cell-lines[6]. This shift supports the notion that the reduction in CE levels by 1 and 2 is due to ACAT inhibition alone and not a result of either reduced endogenous cholesterol biosynthesis, via inhibition of HMG-CoA-reductase, or reduced uptake of cholesterol from media. This lowering of cellular CEs is expressed macroscopically as a reduced number of cytoplasmic lipid droplets. Using fluorescence microscopy, and cellular lipid droplet staining with oil-red-O dye, we observe this macroscopic effect as is an almost complete lack of lipid droplets in 1 and 2-treated 7WD10 CHO cells relative to control cells (Figs. 2b–d).
Figure 2.

Depsipeptides 1 and 2 reduce cholesteryl ester (CE) levels and fat droplets in the stable transgenic human APP751 expressing CHO cell-line 7WD10. a Graph of cellular CE levels as a percent of vehicle treated cells after incubation with 1 ( ) or 2 (
) or 2 ( ) for 96 h. Values are plotted as the mean value ± SEM of at least triplicate experiments. (b–d) Fluorescence microscopy of fixed live 7WD10 CHO cells stained with oil red O (red, that stains lipid droplets marked with white arrows) and lysenin (green that stains membrane sphingomyelin) after 48 h culture with b vehicle (0.1 % DMSO in media), c 1 (1 µM) or d 2 (1 µM). Bar = 10 µm.
) for 96 h. Values are plotted as the mean value ± SEM of at least triplicate experiments. (b–d) Fluorescence microscopy of fixed live 7WD10 CHO cells stained with oil red O (red, that stains lipid droplets marked with white arrows) and lysenin (green that stains membrane sphingomyelin) after 48 h culture with b vehicle (0.1 % DMSO in media), c 1 (1 µM) or d 2 (1 µM). Bar = 10 µm.
We next investigated whether beauveriolide treatment has any impact on Aβ secretion from this cell line using an ELISA-based method that can quantify both Aβ40 and Aβ42 (Fig. 3). Incubation of the 7WD10 cells 1 and 2 (1 µM) for up to 4 d causes a decrease in Aβ40 and Aβ42 secretion, the intensity and time profile of which, is compound-dependent (Fig. 3a upper). The time-dependence of reduction in Aβ secretion induced by 1 (~ 25 and 10 % reduction in Aβ40 and Aβ42 levels after 4 d respectively) reaches a maximum effect after 1–2 d, paralleling this depsipeptide’s effect on CE lowering vide supra. Beauveriolide III (2), is more effective at lowering Aβ secretion (~ 45 and 65 % reduction in Aβ40 and Aβ42 levels after 4 d respectively) than 1 and the time-dependence in Aβ reduction of 2 does not parallel the CE effect as closely as 1, with reductions in Aβ40 and Aβ42 still increasing after 4 d incubation (Fig. 3a upper).
Figure 3.

Depsipeptides 1 and 2 reduce Aβ secretion from the CHO cell-line 7WD10 and do not perturb trafficking of Aβ. UPPER a Graph of time-dependent changes in Aβ40 (▲) and Aβ42 (■) secretion by 7WD10 cells when incubated with either 1 ( ) or 2 (
) or 2 ( ) (1 µM) as determined by ELISA analysis of Aβ40 and Aβ42 in conditioned media standardized to total protein. b Concentration-dependence of 1 (red bars) and 2 (green bars) on Aβtotal secretion from the 7WD10 cell line. c Concentration-dependence of 1 (red bars) and 2 (green bars) on Aβ42 secretion from the 7WD10 cell line. Data in a–c is expressed as the mean value ± SEM of at least triplicate separate experiments and is recorded as a percent of vehicle treated cells (0.1 % DMSO v/v). Statistical analysis was performed using a student t test and significance *P < 0.05 and **P < 0.01. LOWER Confocal microscopy of 7WD10 cells incubated with either vehicle (0.1 % DMSO, v/v) (a–c) or beauveriolide III (1 µM) (d–f) for 48 h. Cells were fixed with paraformaldehyde and immunolabeled with either a rabbit anti-Rab5 antibody with an Alexa-fluor488 (green)-labeled 20 antibody (a, d), or the murine anti-Aβ antibody 4G8, with an Alexafluor 543-labeled (red) 20 antibody (b, e). Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Images were recorded with a Zeiss LSM510 confocal microscope at 63 × and image analysis and merges were performed with LSM image examiner software (4.2). Note: yellow color in the merged images (c, f) corresponds to co-localized Rab5 and Aβ; Bar = 10 µm.
) (1 µM) as determined by ELISA analysis of Aβ40 and Aβ42 in conditioned media standardized to total protein. b Concentration-dependence of 1 (red bars) and 2 (green bars) on Aβtotal secretion from the 7WD10 cell line. c Concentration-dependence of 1 (red bars) and 2 (green bars) on Aβ42 secretion from the 7WD10 cell line. Data in a–c is expressed as the mean value ± SEM of at least triplicate separate experiments and is recorded as a percent of vehicle treated cells (0.1 % DMSO v/v). Statistical analysis was performed using a student t test and significance *P < 0.05 and **P < 0.01. LOWER Confocal microscopy of 7WD10 cells incubated with either vehicle (0.1 % DMSO, v/v) (a–c) or beauveriolide III (1 µM) (d–f) for 48 h. Cells were fixed with paraformaldehyde and immunolabeled with either a rabbit anti-Rab5 antibody with an Alexa-fluor488 (green)-labeled 20 antibody (a, d), or the murine anti-Aβ antibody 4G8, with an Alexafluor 543-labeled (red) 20 antibody (b, e). Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Images were recorded with a Zeiss LSM510 confocal microscope at 63 × and image analysis and merges were performed with LSM image examiner software (4.2). Note: yellow color in the merged images (c, f) corresponds to co-localized Rab5 and Aβ; Bar = 10 µm.
The time-dependant effects of the beauveriolides 1 and 2 on Aβ-secretion were then validated in a concentration-effect study (1 µM and 5 µM)(Figs. 3b–c upper). Significant reductions in Aβtotal (Aβ40 + Aβ42) levels, with respect to vehicle treated cells (0.1 % DMSO v/v), were observed after a 96 h incubation with either beauveriolide I (1) or beauveriolide III (2) (at 1 and 5 µM). The most effective compound at reducing Aβ secretion is depsipeptide 2, which induces a mean reduction in Aβtotal of 39 ± 10 % (P < 0.05) and 57 ± 14 %(P < 0.05) (Fig. 3b upper). Depsipeptide 2 also had a more profound effect on reduction of Aβ42 secretion than 1, causing levels to be reduced by 58 ± 12 % (P < 0.05) and 59 ± 14 % (P < 0.05) respectively (Fig. 3c upper).
To assess whether the reduction in Aβ secretion induced by treatment with 2 corresponds with measurable changes in intracellular Aβ localization, immunolabeling was performed with the anti-Aβ antibody 4G8 and an anti-Rab5 antibody (Figs. 3a–f lower). This microcscopy study revealed that beauveriolide III treatment does not alter the known early endosomal localization and trafficking of Aβ as merged images of vehicle-treated (0.1 % DMSO, v/v) and depsipeptide 2-treated cells show the same degree of co-localization of Aβ and Rab5 (the early endosome-localized Rab GTPase)[15] (Figs. 3c,f).
The nature of the link between ACAT inhibition, CE homeostasis and the biogenesis of Aβ is still an ongoing area for study and has been linked to either intracellular Aβ-accumulation and mistrafficking[16] or a change in APP enzymatic processing[17]. Our microscopy studies of 7WD10 cells treated with the ACAT inhibitor beauveriolide III reveal no indication of either altered localization or accumulation of Aβ (Fig. 3f) and no mistrafficking of normal lipids, such as sphingomyelin (Fig. 2d). These observations are in-line with a mechanism of decreased biogenesis of Aβ recently proposed by Kovacs and co-workers[17] that involves enzymatic N-terminal processing of APP and APP degradation dependent upon cellular CE levels.
In conclusion, we have developed a flexible homochiral synthesis of the fungal-derived beauveriolide natural products, and shown them to be extremely effective at lowering cellular Aβ secretion in the transgenic CHO cell line 7WD10 that expresses human APP751. What makes this study of particular significance is the potency and rapid onset with which these depsipeptides, especially beauveriolide III (2), reduce the cellular secretion of Aβ42. While the major peptide generated by APP processing is Aβ40, the more hydrophobic Aβ42, is the predominant peptide found in senile AD-plaques[18] and therefore reducing central levels of this peptide is seen as a clear therapeutic unmet need. A incubation with 2 (1 µM) reduces Aβ42 secretion by ~ 58 % after 4 d and ~ 30 % after 2 d relative to vehicle treated cells. Previous studies with the ACAT inhibitor CP113,818 on cellular Aβ42 secretion (using a CHO cell line expressing human APP751) revealed that no measurable reduction in Aβ42 secretion was observed with a 1 mM incubation for 4 d. In fact, a 10 fold higher concentration of CP113,818 (10 µM) was required to induce the same reduction in Aβ42 secretion as depsipeptide 2.
As mentioned above, existing ACAT inhibitors such as CP-113,818 and DUP-128, are limited by low bioavailability[19]. They are absorbed poorly from the gut and are rapidly metabolized in blood or tissue. This is the case for the lipidic pyridylamide CP-113,818, which for an in vivo study to assess Aβ in mice had to be prepared in a 60 day release pellet formulation that was implanted under the skin[4]. In contrast, beauveriolide III has been shown to be orally active in both apoE-knockout and LDL-receptor knockout mouse models of atherosclerosis[9]. Therefore, if our discovery that these compounds are excellent at reducing Aβ secretion in vitro can be translated to an in vivo setting and is then coupled with the known pharmacokinetic properties of the beauveriolides, these depsipeptides may well be excellent new candidates for reduction of senile plaque burden in AD.
Supplementary Material
Acknowledgements
This work was supported by a grant from The Scripps Research Institute (PW), The Skaggs Institute for Chemical Biology (PW) and the NIH (AG032549).
Footnotes
Attacking Alzheimer’s via ACAT. The aggregation of β-amyloid peptides, especially Aβ42, into senile plaques is a hallmark of Alzheimer’s disease (AD). We show that the fungal natural products beauveriolides I and III are potent at reducing Aβ secretion from cells expressing human APP offering a potential new scaffold for the development of compounds with proven bioavailability for the treatment of AD.
References
- 1.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Hartmann T. Trends Neurosci. 2001;24:S45–S48. doi: 10.1016/s0166-2236(00)01990-1. [DOI] [PubMed] [Google Scholar]; Burns M, Duff K. Ann N Y Acad Sci. 2002;977:367–375. doi: 10.1111/j.1749-6632.2002.tb04839.x. [DOI] [PubMed] [Google Scholar]; Puglielli L, Tanzi RE, Kovacs DM. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 3.Wolozin B, Brown J, 3rd, Theisler C, Silberman S. CNS Drug Rev. 2004;10:127–146. doi: 10.1111/j.1527-3458.2004.tb00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, Kovacs DM. Neuron. 2004;44:227–238. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 5.Huttunen HJ, Greco C, Kovacs DM. FEBS. 2007;581:1688–1692. doi: 10.1016/j.febslet.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puglielli L, Konopka G, Pack-Chung E, Ingano LAM, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM. Nat Cell Biol. 2001;3:905–912. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki K, Ohmori K, Tamura H, Shizuri Y, Nishiyama S, Miyoshi E, Yamamura S. Bull. Chem. Soc. Jpn. 1993;66:3041–3046. [Google Scholar]; Namatame I, Tomoda H, Arai H, Inoue K, Omura S. J. Biochem. 1999;125:319–327. doi: 10.1093/oxfordjournals.jbchem.a022289. [DOI] [PubMed] [Google Scholar]; Namatame I, Tomoda H, Si S, Yamaguchi Y, Masuma R, Omura S. J. Antibiot. 1999;52:1–6. doi: 10.7164/antibiotics.52.1. [DOI] [PubMed] [Google Scholar]
- 8.Oshiro T, Namatame I, Nagai K, Sekiguchi T, Doi T, Takahashi T, Akasaka K, Rudel LL, Tomoda H, Omura S. J. Org. Chem. 2006;71:7643–7649. doi: 10.1021/jo0611667. [DOI] [PubMed] [Google Scholar]
- 9.Namatame I, Tomoda H, Ishibashi S, Omura S. Proc. Natl. Acad. Sci. U. S. A. 2004;101:737–742. doi: 10.1073/pnas.0307757100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomoda H, Doi T. Acc. Chem. Res. 2008;41:32–39. doi: 10.1021/ar700117b. [DOI] [PubMed] [Google Scholar]
- 11.Wentworth P, Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, DeLaria GA, Saven A, Babior BM, Janda KD, Eschenmoser A, Lerner RA. Science. 2003;302:1053–1056. doi: 10.1126/science.1089525. [DOI] [PubMed] [Google Scholar]
- 12.Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, Jr, Lerner RA, Kelly JW. Nat. Chem. Biol. 2006;2:249–253. doi: 10.1038/nchembio782. [DOI] [PubMed] [Google Scholar]; Zhang Q, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Jr, Lerner RA, Kelly JW. Proc. Natl. Acad. Sci. USA. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen YP, Gambs C, Abe Y, Wentworth P, Janda KD. J. Org. Chem. 2003;68:8902–8905. doi: 10.1021/jo034765b. [DOI] [PubMed] [Google Scholar]
- 14.Koo EH, Squazzo SL. J. Biol. Chem. 1994;275:17468–17475. [Google Scholar]; Yamazaki T, Chang T-Y, Haass C, Ihara Y. J. Biol. Chem. 2001;276:4454–4460. doi: 10.1074/jbc.M009598200. [DOI] [PubMed] [Google Scholar]
- 15.Colicelli J. Sci. STKE. 2004;250:1–31. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin LW, Shie FS, Maezawa I, Vincent I, Bird T. Am. J. Pathol. 2004;164:975. doi: 10.1016/s0002-9440(10)63185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huttunen HJ, Puglielli L, Ellis BC, Mackenzie Ingano LA, Kovacs DM. J. Mol. Neurosci. 2009;37:6–15. doi: 10.1007/s12031-008-9088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.