

Abstract
Human cells are exposed to the electrophilic α,β-unsaturated aldehyde acrolein from a variety of sources. Reaction of acrolein with functionally critical protein thiol residues can yield important biological consequences. Protein tyrosine phosphatases (PTPs) are an important class of cysteine-dependent enzymes whose reactivity with acrolein previously has not been well characterized. These enzymes catalyze the dephosphorylation of phosphotyrosine residues on proteins via a phosphocysteine intermediate. PTPs work in tandem with protein tyrosine kinases to regulate a number of critically important mammalian signal transduction pathways. We find that acrolein is a potent time-dependent inactivator of the enzyme PTP1B (kinact = 0.02 ± 0.005 s−1, KI = 2.3 ± 0.6 × 10−4 M). Enzyme activity does not return upon gel filtration of the inactivated enzyme and addition of the competitive phosphatase inhibitor vanadate slows inactivation of PTP1B by acrolein. Together these observations suggest that acrolein covalently modifies the active site of PTP1B. Mass spectrometric analysis reveals that acrolein modifies the catalytic cysteine residue at the active site of the enzyme. Aliphatic aldehydes such as glyoxal, acetaldehyde, and propanal are relatively weak inactivators of PTP1B under the conditions employed here. Similarly, unsaturated aldehydes such as crotonaldehyde and 3-methyl-2-butenal bearing substitution at the alkene terminus are poor inactivators of the enzyme. Overall, the data suggest that enzyme inactivation occurs via conjugate addition of the catalytic cysteine residue to the carbon-carbon double bond of acrolein. The results indicate that inactivation of PTPs should be considered as a possible contributor to the diverse biological activities of acrolein and structurally-related α,β-unsaturated aldehydes.
Keywords: acrolein, protein tyrosine phosphatase
Introduction
Human cells are exposed to the electrophilic α,β-unsaturated aldehyde acrolein from a variety of sources (1, 2). Acrolein can be generated in the cell by endogenous lipid peroxidation (3–5) and oxidation of amino acids (6) or introduced via dietary sources (1), cigarette smoke (7), and the metabolism of drugs (8–11) or other xenobiotics (12, 13). Acrolein displays a wide range of biological activities (1, 2). One important chemical reaction underlying the biological action of acrolein involves covalent modification of cysteine thiol residues on proteins. Specifically, reactions of acrolein with proteins that contain functionally critical thiol side chains such as cysteine proteases (caspase-3) (14–16), some ion channels (TRPA1) (17). and some transcription factors (NF-κB, Keap1/Nrf2) (18, 19) have the potential to yield significant biological effects.
Protein tyrosine phosphatases (PTPs) are an important class of cysteine-dependent enzymes whose reactivity with acrolein has not been well characterized. These enzymes catalyze the dephosphorylation of phosphotyrosine residues on proteins via a phosphocysteine intermediate as shown in Scheme 1 (20–25). PTPs work in tandem with protein tyrosine kinases to regulate a number of critically important mammalian signal transduction pathways (20–26). Not surprisingly, inhibition or inactivation of PTPs has the potential to yield profound biological consequences (27–32).
Scheme 1.

In the work reported here, we examined the kinetics and mechanism of PTP inactivation by acrolein. We find that acrolein is a potent irreversible inactivator of the enzyme PTP1B. The data further suggest that the inactivation reaction occurs via conjugate addition of the active site cysteine-215 to the carbon-carbon double bond of acrolein.
Materials and Methods
Chemicals and Reagents
Reagents were purchased from the following suppliers: Buffers salts, p-nitrophenyl phosphate (pNPP), thiols, trifluoroacetic acid (TFA), sodium orthovanadate, acetaldehyde, 3-methyl-2-butenal, and crotonaldehyde were obtained from Sigma-Aldrich (St. Louis, MO). Catalase (catalog number 106810) and SOD (catalog number 837113) were obtained from Roche Bioscience (Palo Alto, CA). Acrolein and glyoxal were obtained from Acros Organics. Sequencing grade modified trypsin (catalog number V5111) was obtained from Promega. Ammonium bicarbonate was obtained from Fisher Scientific. Recombinant PTP1B (a.a. 1-322) was prepared in our laboratory as reported previously (33). The concentration of active PTP1B in the samples was determined as described previously (33).
Time-Dependent Inactivation of PTP1B
Inactivation assays were performed using modifications of existing literature protocols (34–36). Free thiols were removed from a stock solution of purified PTP1B using Zeba mini centrifugal buffer exchange columns (Pierce, catalog number 89882) according to manufacturer’s protocol. The exchange buffer contained 100 mM TRIS-HCl, 10 mM DTPA, 0.05% NP-40, pH 7.4. In the inactivation reactions, acrolein was added as a stock solution in water to a mixture containing PTP1B in TRIS-HCl (100 mM), DTPA (10 mM), and NP-40 (0.05% v/v) at 30 °C (final concentrations of acrolein 20–60 μM, TRIS-HCl (50 mM), DTPA (5 mM), and NP-40 (0.025% v/v), and PTP1B 75 nM). Aliquots (10 μL) were removed at various time points (1 min, 2 min, and 5 min) and placed in 490 μL of PTP1B assay buffer consisting of bis-Tris (50 mM, pH 6.0), NaCl (100 mM), DTPA (10 mM), and pNPP (20 mM) at 30 °C for 10 min. The enzymatic reaction was quenched by addition of NaOH (500 μL of a 2 N solution in water) and the amount of p-nitrophenol released during the assay determined at 25 °C by measuring the absorbance at 410 nm using a UV-vis spectrometer.
Gel Filtration of Acrolein-Inactivated PTP1B
A solution of acrolein in water (10 μL of 5 mM) was added to PTP1B (40 μL of a 4 μM solution in TRIS-HCl (100 mM), DTPA (10 mM), and NP-40 (0.05% v/v)). After 10 min, an aliquot (10 μL) was removed and tested for activity to confirm that the enzyme was completely inactivated. An aliquot of the remaining solution (12 μL) was gel filtered through a Zeba micro spin column according to the manufacturer’s protocol. The exchange buffer contained 100 mM TRIS-HCl, 10 mM NaCl, 10 mM DTPA, 0.05% NP-40, pH 7.4. Following buffer exchange, a 10 μL aliquot was tested for PTP1B activity as described above. No measurable return of PTP1B activity was observed versus a control sample treated in an identical manner except without inactivation by acrolein.
Time-Dependent Inactivation in the Presence of a Competitive PTP1B Inhibitor or Superoxide Dismutase or Catalase
An aliquot of thiol-free enzyme (20 μL of a 4 μM solution in TRIS-HCl (100 mM), DTPA (10 mM), and NP-40 (0.05% v/v)) was combined with an aqueous solution of acrolein and orthovanadate (20 μL of a mixture containing 500 μM acrolein and 1 mM orthovanadate in water) at 25 °C (final concentrations: PTP1B, 2 μM; acrolein, 250 μM; orthovanadate, 500 μM; TRIS-HCl, 50 mM; DTPA, 5 mM; and NP-40, 0.025% v/v). After 5 min, a 10 μL aliquot was removed and added to a cuvette containing a three component PTP1B assay buffer (990 μL) consisting of sodium acetate (100 mM), bis-Tris (50 mM), TRIS (50 mM), pNPP (10 mM), at pH 7.0 (final volume of 1 mL). Immediately following addition of enzyme to the cuvette, the assay was mixed by repeated inversion, and enzyme catalyzed release of p-nitrophenol monitored at 25 °C by measuring the increase in absorbance at 410 nm. Data points were taken every 2 seconds. In the assays designed to probe the potential role of reactive oxygen species in this inactivation process, acrolein (10 μL of a 100 mM stock solution) was added to a cuvette containing the assay buffer (985 μL of a mixture containing sodium acetate, 100 mM; bis-Tris, 50 mM; TRIS, 50 mM; pNPP, 10 mM, and either SOD, 10 μg/mL or catalase, 0.5 μg/mL, at pH 7.0), followed by thiol-free PTP1B (5 μL of a 4 μM solution in TRIS-HCl, 100 mM; DTPA, 10 mM; and NP-40, 0.05% v/v). PTP1B activity was then monitored as described above. The presence of catalase or superoxide dismutase exerted no measurable effect on the inactivation reaction.
MALDI TOF/TOF MS and Nanospray MS/MS Analysis of Acrolein-Modified PTP1B
A solution of acrolein (5 μL, 18 mM in water) was added to a solution containing thiol free PTP1B (45 μL of an approximately 14 pmol/μL solution in TRIS-HCl, 100 mM; NaCl, 10 mM; DTPA, 10 mM; and NP-40 0.05%, at pH 7.4) and the resulting mixture incubated 30 min at 25 °C. A control sample containing no acrolein was also prepared. After incubation, both solutions were passed through a Zeba mini centrifugal buffer exchange column. To the resulting mixtures, sequence grade modified trypsin (50 μL of a 1 μg/50 μL solution in 50 mM ammonium bicarbonate) was added. The digestion was incubated at 37 °C for 18 h then quenched with TFA (5 μL of a 10% aqueous solution). An aliquot (20 μL) of each solution was transferred to a microcentrifuge tube, frozen in liquid nitrogen, and lyophilized to dryness. The residue was resuspended in water (10 μL) and again lyophilized. The final dried sample was reconstituted in acetonitrile/water/88% formic acid (5 μL of 700/290/10 v/v/v). For MALDI TOF/TOF MS analysis, a 0.6 μL portion of diluted sample was mixed with an equal volume of alpha-cyano-4-hydroxycinnamic acid (CHCA) matrix solution (5 mg CHCA/mL in 500/455/20/25 (v/v/v/v) acetronitrile/water/10% TFA/400 mM aqueous ammonium dihydrogen phosphate). Aliquots (0.4 μL) of the mixture were deposited on a polished stainless steel target. Crystallization of the mixture proceeded under ambient conditions. Mass spectra were acquired on an Applied Biosystems Inc. 4700 MALDI TOF/TOF mass spectrometer with a 355 nm Nd:YAG laser (200 Hz) in the positive ion delayed extraction reflector MS mode. The MS spectra (2000 laser shots summed/averaged) were acquired over the mass range 700–4000 Da. Each MS spectrum was re-calibrated internally using the masses (monoisotopic [M+H]+) for the PTP1B tryptic fragments observed at 1366.675 Da ([13–24]) and 2508.288 Da ([293–314]). For nanospray MS/MS of the active site peptide 200–221, Ziptip (Millipore, standard C18) cleanup of sample with fractions of 50/940/10, 100/890/10, and 200/790/10 (v/v/v) acetonitrile/water/88% formic acid (AWF) were pooled then lyophilized to dryness. Sample was reconstituted in 4 μL 700/290/10 AWF for nanospray QqTOF MS analysis on an Applied Biosystems/MDS Sciex (Foster City, CA, USA) QStar/Pulsar/I instrument fitted with a Proxeon (Odense, Denmark) nanospray source. A pulsar frequency of 6.99 KHz was used for MS/MS mass range 50–2000 Da. MS/MS spectra were obtained with N2 collision gas. The MS/MS of the 3+ ion at 744.37 Da for the peptide of interest was obtained with a collision energy of 35 V. The instrument software was Analyst QS and the data analysis software was BioAnalyst 1.1.
Results
Kinetics of PTP1B Inactivation by Acrolein
We utilized the catalytic subunit of human PTP1B (a.a. 1–322) as an archetypal member of the PTP family of enzymes (20–25). We find that acrolein is a potent time-dependent inactivator of PTP1B (Figure 1A). A Kitz-Wilson replot of the inactivation data (Figure 1B) reveals that the maximum rate of inactivation at saturating concentrations of acrolein, kinact, is 0.02 ± 0.005 s−1 while the concentration of acrolein required to achieve half-maximal rate of inactivation, KI, is 2.3 ± 0.6 × 10−4 M. It is worth noting that the apparent second-order rate constant for inactivation of the enzyme by acrolein (kinact/KI = 87 M−1 s−1) is comparable to that for hydrogen peroxide (10 M−1 s−1) (37, 38), a known endogenous regulator of PTP1B activity (39, 40). Inactivation of PTP1B by acrolein is not reversed by gel filtration or dialysis to remove excess inactivator. This observation, along with the time-dependent nature of the reaction, indicates that the inactivation of PTP1B by acrolein involves covalent modification of the enzyme. The inactivation process is slowed by addition of the competitive PTP1B inhibitor ortho vanadate (35) (Figure 2), thereby providing evidence that the reaction is active-site directed.
Figure 1.

Semi-log plot of time courses for the inactivation of PTP1B by various concentrations of acrolein (Panel A) and Kitz-Wilson replot of the inactivation data (Panel B).
Figure 2.

Inactivation of PTP1B by acrolein is slowed by the presence of the competitive inhibitor orthovanadate.
Inactivation of PTP1B by Acrolein Does Not Involve Reactive Oxygen Species
PTPs can be inactivated by reactive oxygen species such as superoxide radical and hydrogen peroxide (33, 37, 38). These agents inactivate PTP1B via oxidation of the active site cysteine-215 to the sulfenic acid oxidation state (38, 41–43). In the context of the current study, it is important to note that spontaneous autooxidation of aldehydes in aerobic solution has the potential to generate superoxide radical and hydrogen peroxide (44). The possible involvement of reactive oxygen species in the inactivation of PTP1B by acrolein was ruled out by the observation that the presence of the superoxide- and hydrogen peroxide-destroying enzymes superoxide dismutase (SOD) and catalase (45) has no effect on the rate of the inactivation reaction (data not shown).
Mass Spectrometry of Acrolein-Inactivated PTP1B
Acrolein can covalently modify a number amino acid residues in proteins, including histidine, lysine, and cysteine (3, 14–17, 46–50). Importantly, the active site of PTP1B contains both a cysteine (Cys-215) and a histidine (His-216) residue that are required for catalytic activity (51). Mass spectrometry was employed to shed light on the site(s) at which acrolein covalently modifies the active site of PTP1B. Toward this end, acrolein-inactivated PTP1B was subjected to tryptic digestion and the resulting fragments analyzed by MALDI-TOF-TOF mass spectrometry. Signals for many of the expected (unmodified) tryptic fragments of PTP1B were observed in the mass spectrum. Importantly, a major signal was seen at m/z 2231.09 corresponding to the acrolein-modified active site peptide a.a. 200-221. A very weak signal was observed for the unmodified active site peptide (m/z 2175.17). In addition, weak signals were detected consistent with acrolein modification of four other cysteine-containing tryptic fragments corresponding to a.a. 25-33 (m/z 1117.47), 80-103 (m/z 2920.38), 121-128 (m/z 1079.66), and 222-237 (m/z 1824.80). Nanospray MS/MS-TOF was employed to further characterize the site(s) of acrolein modification in the active site peptide a.a 200–221 (m/z 2231.09 [M+H]+). The masses for the b12–b15 ions are those expected for the unmodified peptides (Figure 3). On the other hand, the masses of the b16–b17 ions are increased by +56 Da consistent with modification of these fragment ions by acrolein. Similarly, the masses for the y5 and y6 ions are consistent with those expected for the unmodified peptide fragments, while the masses for y7–y13 ions are increased by +56 Da. Overall the data indicates that acrolein inactivates PTP1B primarily via reaction with the active site cysteine-215. Weak signals corresponding to the acrolein-modified b15 and unmodified y7 ions are observed, suggesting that some modification also occurs at histidine-214 of the active site peptide.
Figure 3.

ESI+-MS/MS analysis of the acrolein-modified PTP1B active site tryptic fragment, E200SGSLSPEHGPVVVHCSAGIGR221. Cysteine 215 is the active site nucleophile of PTP1B. Acrolein-modified fragment ions are denoted by “*”.
Structure-Activity Studies with Related Aldehydes Suggest That Acrolein Inactivates PTP1B via a Conjugate Addition Mechanism
In aqueous solution, simple thiols react with acrolein via initial conjugate addition to the double bond of the α,β-unsaturated aldehyde, rather than by addition to the aldehyde residue (52). Thus, we anticipated that the inactivation of PTP1B by acrolein may proceed via the conjugate addition mechanism shown in Scheme 2. Consistent with this expectation, we find that aliphatic aldehydes such as glyoxal, acetaldehyde, and propanal, lacking the unsaturation found in acrolein, are relatively poor inactivators of the enzyme (Table 1). These results are in line with previous work showing that simple aldehydes such as acetaldehyde do not effectively inactivate PTP1B (53). It is worth noting that more functionalized peptidyl aldehydes that capture noncovalent binding interactions with the enzyme active site are slow-binding, reversible covalent inhibitors of protein tyrosine phosphatases (54, 55). Also consistent with our favored conjugate addition mechanism for the inactivation of PTP1B by acrolein, we find that analogues such as crotonaldehyde and 3-methyl-2-butenal, possessing steric bulk at the terminal position of the double bond, are relatively weak inactivators of the enzyme (Table 1).
Scheme 2.

Table 1.
Inactivation of PTP1B by Various Aldehydes.
| Compound | % Remaining Enzyme Activitya |
|---|---|
|
Aerolein |
4 ± 3% |
|
Crotonaldehyde |
85 ± 2% |
|
Glyoxal |
93 ± 2% |
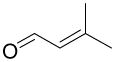 3-Methyl-2-butenal |
95 ± 3% |
|
Propanal |
93 ± 4% |
|
Acetaldehyde |
> 95% |
A solution containing PTP1B (225 nM) in sodium acetate (100 mM), bis-Tris (50 mM), Tris (50 mM), DETAPAC (5 mM) and DMF (5% by volume), pH 7 was incubated with various aldehydes (500 μM) at 25 °C. After 10 min, an aliquot (10 μL) was removed from the solution and the fraction of remaining enzyme activity (versus a control sample containing no aldehyde) was determined as described in the Materials and Methods section.
Discussion
Acrolein has previously been shown to inactivate a variety of enzymes including DNA methyltransferase (46), protein disulfide isomerase (49), glutathione reductase (47), caspase-3 (14–16), and carbonic anhydrase (48). In addition, modification of ion channels (17) and transcription factors (18, 19) by acrolein can alter their functional properties. Here we provide evidence that acrolein is a potent irreversible inactivator of the enzyme PTP1B (kinact = 0.02 ± 0.005 s−1, KI = 2.3 ± 0.6 × 10−4 M). The apparent second-order rate constant for the inactivation of PTP1B by acrolein (87 M−1 s−1) is comparable to that reported previously for hydrogen peroxide (10 M−1 s−1), a known endogenous regulator of the enzyme (37–40). In general, our findings mesh with previous reports that acrolein and structurally-related lipid peroxidation products inhibit PTP activity though, in the earlier work, neither the kinetics or mechanism of PTP inhibition by acrolein were elucidated (53, 56, 57). In our study, mass spectrometry of the inactivated enzyme indicates that the active site cysteine-215 residue is modified by acrolein. Examination of a series of structurally-related aldehydes further shows that the double bond found in acrolein is central to its properties as a phosphatase inactivator. This fact is revealed by the observation that simple aliphatic aldehydes do not inactivate PTP1B under the conditions employed for these studies. Furthermore, steric bulk at the alkene terminus diminishes the PTP1B-inactivating properties of compounds in this series. Together, these observations support an inactivation mechanism involving conjugate addition of the active site cysteine-215 to the carbon-carbon double bond of acrolein (Scheme 2).
In conclusion, inactivation of PTPs can yield profound biological consequences arising from the disruption of cellular signaling pathways (27–32). The results reported here indicate that inactivation of PTPs can be considered as a possible contributor to the diverse biological activities (1, 2) of acrolein and structurally-related α,β-unsaturated aldehydes.
Acknowledgments
We thank Beverly DaGue for mass spectrometry and the National Institutes of Health for partial support of this work (CA 83925 and CA 119131).
References
- 1.O’Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity, and possible effects on human health. Crit Rev Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- 2.Beauchamp RO, Jr, Andjelkovich DA, Kligerman AD, Morgan KT, Heck H. A critical review of the literature on acrolein toxicity. Crit Rev Toxicol. 1985;14:309–380. doi: 10.3109/10408448509037461. [DOI] [PubMed] [Google Scholar]
- 3.Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proc Nat Acad Sci USA. 1998;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. LIpid peroxidation in aging brain and Alzheimer’s. Free Rad Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 5.Arlt S, Beisiegel U, Kontush A. Lipid peroxidation in neurodegeneration: new insights into Alzheimer’s disease. Curr Opin Lipidology. 2002;13:289–294. doi: 10.1097/00041433-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. J Clin Investig. 1997;99:424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hecht SS. Smoking and lung cancer - a new role for an old toxicant? Proc Nat Acad Sci USA. 2006;103:15725–15726. doi: 10.1073/pnas.0607811103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramu K, Fraiser LH, Mamiya B, Ahmed T, Kehrer JP. Acrolein mercapturates: synthesis, characterization, and assessment of their role in the bladder toxicity of cyclophosphamide. Chem Res Toxicol. 1995;8:515–524. doi: 10.1021/tx00046a005. [DOI] [PubMed] [Google Scholar]
- 9.Patel JM. Metabolism and pulmonary toxicity of cyclophosphamide. Pharm Ther. 1990;47:137–146. doi: 10.1016/0163-7258(90)90049-8. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Tian Q, Zhou SF. Clinical pharmacology of cyclophosphamide and ifosfamide. Curr Drug Ther. 2006;1:55–84. [Google Scholar]
- 11.Kaijser GP, Korst A, Beijnen JH, Bult A, Underberg WJM. The analysis of ifosfamide and its metabolites (review) Anticancer Res. 1993;13:1311–24. [PubMed] [Google Scholar]
- 12.Hashmi M, Vamvakas S, Anders MW. Bioactivation mechanism of S-(3-oxopropyl)-N-acetyl-L-cysteine, the mercapturic acid of acrolein. Chem Res Toxicol. 1992;5:360–365. doi: 10.1021/tx00027a007. [DOI] [PubMed] [Google Scholar]
- 13.Atzori L, Dore M, Congiu L. Aspects of allyl alcohol toxicity. Drug Metab Drug Interact. 1989;7:295–319. [PubMed] [Google Scholar]
- 14.Finkelstein EI, Ruben J, Koot CW, Hristova M, van der Vliet A. Regulation of constitutive neutrophil apoptosis by the α,β-unsaturated aldehydes acrolein and 4-hydroxynonenal. Am J Physiol. 2005;289:L1019–L1028. doi: 10.1152/ajplung.00227.2005. [DOI] [PubMed] [Google Scholar]
- 15.Kern JC, Kehrer JP. Acrolein-induced cell death: a caspase-influenced decision between apoptosis and oncosis/necrosis. Chem Biol Interact. 2002;139:79–95. doi: 10.1016/s0009-2797(01)00295-2. [DOI] [PubMed] [Google Scholar]
- 16.Tanel A, Averill-Bates DA. The aldehyde acrolein induces apoptosis via activation of the mitochondrial pathway. Biochim Biophys Acta. 2005;1743(3):255–267. 255–267. doi: 10.1016/j.bbamcr.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 18.Horton ND, Biswal SS, Corrigan LL, Bratta J, Kehrer JP. Acrolein causes inhibitor kB-independent decreases in nuclear factor kB activation in human lung adenocarcinoma (A549) cells. J Biol Chem. 1999;274:9200–9206. doi: 10.1074/jbc.274.14.9200. [DOI] [PubMed] [Google Scholar]
- 19.Tirumalai R, Rajesh Kumar T, Mai KH, Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol Lett. 2002;132:27–36. doi: 10.1016/s0378-4274(02)00055-3. [DOI] [PubMed] [Google Scholar]
- 20.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Ostermann A, Godzik A, Hunter T, Dixon JE, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Barford D. Protein phosphatases. Curr Opin Struct Biol. 1995;5:728–734. doi: 10.1016/0959-440x(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 22.Jackson MD, Denu JM. Molecular reactions of protein phosphatases -insights from structure and chemistry. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 23.Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 24.Stone RL, Dixon JE. Protein-tyrosine phosphatases. J Biol Chem. 1994;269:31323–31326. [PubMed] [Google Scholar]
- 25.Zhang ZY. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc Chem Res. 2003;36:385–392. doi: 10.1021/ar020122r. [DOI] [PubMed] [Google Scholar]
- 26.Hunter T. Signaling - 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 27.Bialy L, Waldmann H. Inhibitors of protein tyrosine phosphatases: next generation drugs? Angew Chem Int Ed Eng. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 28.Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nature Rev Drug Discov. 2002;1:696–709. doi: 10.1038/nrd895. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman BT, Nelson MR, Burdick K, Baxter SM. Protein tyrosine phosphatases: strategies for distinguishing proteins in a family containing multiple drug targets and anti-targets. Curr Pharm Des. 2004;10:1161–1181. doi: 10.2174/1381612043452659. [DOI] [PubMed] [Google Scholar]
- 30.Bridges AJ. Therapeutic challenges of kinase and phosphatase inhibition and use in anti-diabetic strategy. Biochem Soc Trans. 2005;33:343–345. doi: 10.1042/BST0330343. [DOI] [PubMed] [Google Scholar]
- 31.Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, Kennedy BP, Muller WJ, Tremblay ML. Protein tyrosine phosphatase 1B deficiency or inhibition delays Erb2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genetics. 2007;39:338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 32.Tonks NK, Muthuswamy SK. A brake becomes an accelerator: PTP1B - A new therapeutic target for breast cancer. Cancer Cell. 2007;11:214–215. doi: 10.1016/j.ccr.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 33.LaButti JN, Chowdhury G, Reilly TJ, Gates KS. Redox regulation of protein tyrosine phosphatase 1B by peroxymonophosphate. J Am Chem Soc. 2007;129:5320–5321. doi: 10.1021/ja070194j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arabaci G, Guo XC, Beebe KD, Coggeshall KM, Pei D. alpha-Haloacetophenone derivatives as photoreversible covalent inhibitors of protein tyrosine phosphatases. J Am Chem Soc. 1999;121:5085–5086. [Google Scholar]
- 35.Montalibet J, Skorey KI, Kennedy BP. Protein tyrosine phosphatase: enzymatic assays. Methods. 2005;35:2–8. doi: 10.1016/j.ymeth.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. I. CRC Press; Boca Raton: 1988. [Google Scholar]
- 37.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 38.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 39.Mahedev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1B in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 40.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 41.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for the redox regulation of protein tyrosine phosphatase 1B (PTP1B) J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 42.van Montfort RLM, Congreeve M, Tisi D, Carr R, Jhoti H. Nature. Vol. 423. 2003. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B; pp. 773–777. [DOI] [PubMed] [Google Scholar]
- 43.Salmeen A, Anderson JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 44.Thornalley P, Wolff S, Crabbe J, Stern A. The autooxidation of glyceraldehyde and other simple monosaccharides under physiological conditions catalysed by buffer ions. Biochim Biophys Acta. 1984;797:276–287. doi: 10.1016/0304-4165(84)90131-4. [DOI] [PubMed] [Google Scholar]
- 45.Halliwell B, Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 46.Cox R, Goorha S, Irving CC. Inhibition of DNA methylase activity by acrolein. Carcinogenesis. 1988;9:463–465. doi: 10.1093/carcin/9.3.463. [DOI] [PubMed] [Google Scholar]
- 47.Vander Jagt DL, Hunsaker LA, Vander Jagt TJ, Gomez MS, Gonzales DM, Deck LM, Royer RE. Inactivation of glutathione reductase by 4-hydroxynonenal and other endogenous aldehydes. Biochem Pharmacol. 1997;53:1133–1140. doi: 10.1016/s0006-2952(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 48.Pocker Y, Janjic N. Differential modification of specificity in carbonic anhydrase catalysis. J Biol Chem. 1988;263:6169–6176. [PubMed] [Google Scholar]
- 49.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 50.Szapacs ME, Riggins JN, Zimmerman LJ, Liebler DC. Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: kinetic analysis of competing alkylation reactions. Biochemistry. 2007;45:10521–10528. doi: 10.1021/bi060535q. [DOI] [PubMed] [Google Scholar]
- 51.Zhang ZY. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Ann Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 52.Esterbauer H, Ertl A, Scholz N. The reaction of cysteine with a,b-unsaturated aldehydes. Tetrahedron. 1976;32:285–289. [Google Scholar]
- 53.Hernandez-Hernandez A, Sanchez-Yague J, Martin-Valmaseda EM, Llanillo M. Oxidative inactivation of human and sheep platelet membrane-associated phosphotyrosine phosphatase activity. Free Rad Biol Med. 1999;26:1218–1230. doi: 10.1016/s0891-5849(98)00306-2. [DOI] [PubMed] [Google Scholar]
- 54.Fu H, Park J, Pei D. Peptidyl adehydes as reversible covalent inhibitors of protein tyrosine phosphatases. Biochemistry. 2002;41:10700–10709. doi: 10.1021/bi0258748. [DOI] [PubMed] [Google Scholar]
- 55.Park J, Fu H, Pei D. Peptidyl aldehydes as reversible covalent inhibitors of Src homology 2 domains. Biochemistry. 2003;42:5169–5167. doi: 10.1021/bi034076u. [DOI] [PubMed] [Google Scholar]
- 56.Hernandez-Hernandez A, Garabatos MN, Rodriguez MC, Vidal ML, Lopez-Revuelta A, Sanchez-Gallego JI, Llanillo M, Sanchez-Yaguee J. Structural characteristics of a lipid peroxidation product, trans-2-nonenal, that favour inhibition of membrane-associated phosphotyrosine phosphatase activity. Biochim Biophys Acta 1726. 2005:317–325. doi: 10.1016/j.bbagen.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 57.Salsman SJ, Hensley K, Floyd RA. Sensitivity of protein tyrosine phosphase activity to the redox environment, cytochrome c, and microperoxidase. Antioxidants Redox Signaling. 2005;7:1078–1088. doi: 10.1089/ars.2005.7.1078. [DOI] [PubMed] [Google Scholar]