

Abstract
Janus kinase 2 (JAK2) plays a crucial role in the pathomechanism of myeloproliferative disorders and hematologic malignancies. A somatic mutation of JAK2 (Val617Phe) was previously shown to occur in 98% of patients with polycythemia vera and 50% of patients with essential thrombocythemia and primary myelofibrosis. Thus, effective JAK2 kinase inhibitors may be of significant therapeutic importance. Here, we applied a structure-based virtual screen to identify novel JAK2 inhibitors. One JAK2 inhibitor in particular, G6, demonstrated remarkable potency as well as specificity, which makes it as a potential lead candidate against diseases related to elevated JAK2 tyrosine kinase activity.
Keywords: Janus kinase 2, Virtual screening, Inhibitor, Pharmacological evaluation
Janus kinases (JAKs) are cytoplasmic protein kinases that play a crucial role in cytokine-mediated signal transduction. In the absence of ligand, JAK kinases are constitutively bound to the cytoplasmic tails of cytokine receptors. The binding of ligand to the extracellular portion of the receptor results in a conformational change of the receptor itself and subsequent activation of the JAKs. Activated JAKs tyrosine autophosphorylate and phosphorylate specific tyrosine residues on the C-terminal end of the receptors. This results in the recruitment and activation of the Signal Transducers and Activators of Transcription (STAT) proteins. Activated STATs subsequently translocate into the nucleus where they alter specific gene transcription patterns.1 JAKs contain seven conserved Janus homology (JH)2 domains: a tyrosine kinase domain at the C-terminus (JH1), a pseudokinase domain (JH2) and five further domains that are believed to interact with regulatory proteins or cytokine receptors (JH3–JH7).3,4 The mammalian JAK kinase family comprises four members: JAK1, JAK2, JAK3 and TYK2.5 JAKs are expressed ubiquitously, except JAK3 which is primarily expressed in hematopoietic cells.1 Their constitutive or enhanced activity is usually associated with abnormal cell proliferation in a series of hematologic malignancies including lymphoid and myeloid leukemias, Hodgkin's lymphoma and various B-cell non-Hodgkin's lymphomas.6,7 Constant stimulation of JAKs by interleukin (IL)-6 may be of critical importance in multiple myeloma cell growth.8,9 Quite recently, a somatic mutation within JAK2 (Val617Phe) has been identified in patients with myeloproliferative disorders.10 This mutation occurs in the JH2 pseudokinase domain and leads to constitutive JAK2 activation. Substitution of Val617 to other large non-polar residues also results in increased JAK2 kinase activity.11 Previous work demonstrated that JAK2-deficiency is embryonically fatal in mice due to a lack of erythropoesis.12,13 As a consequence, no in vivo JAK2 knock out animal model exists, which makes the biological characterization of JAK2 more difficult. siRNA knock down models may be useful for this purpose,14,15 but for the time being, they cannot be considered as therapeutic agents. JAK2 antagonists, however, could be used efficiently for analyzing the possible therapeutic benefit of JAK2 inhibition in hematologic malignancies and myeloproliferative disorders. On the other hand, only a small number of JAK2 inhibitors have been reported in the literature so far. AG490 possesses significant JAK2 inhibition, for that reason it has been used extensively as a research tool. AG490 blocks leukemic cell growth significantly both in vitro and in vivo.16 On the other hand, this compound lacks sufficient target specificity, therefore the interpretation of results obtained with AG490 may not be limited to JAK2 inhibition.17–20 Several other non-specific JAK2 inhibitors have already been reported in the literature (LFM-A13,21 INCB20,22 WP106623 and SD-100824). Since off-target effects may cause serious immuno-modulative or proliferative side effects, specific JAK2 inhibition is highly desirable. Very recently, Wernig et al. reported that TG101348, a nanomolar JAK2 inhibitor, was highly specific for JAK2 as evidenced by a 300-fold selectivity over JAK3.25 Antonysamy et al. published a fragment-based JAK2 inhibitor having more than 35-fold selectivity versus JAK3.26 Our group has also identified potent JAK2 inhibitors showing selectivity over EGFR, TYK2 and c-Src.27,28
Structure-based drug design has been effectively used on kinases for decades identifying and optimizing novel ligands. Many successful studies have already proven the usefulness of structure-based applications, even in the field of JAKs.26–29 We previously reported the successful application of a JAK2 homology model (JH1 domain) based on the crystal structure of the tyrosine kinase domain of fibroblast growth factor receptor 1 (FGFR1) (PDB ID: 1FGI) for virtual screening.27,28 Meanwhile, the crystal structure of JAK3 (JH1 domain) in complex with a staurosporine analog (AFN941) was solved by Boggon and his co-workers (PDB ID: 1YVJ).30 Since the JH1 domain of JAK2 shows significantly higher sequence identity to that of JAK3 (62%) than to that of FGFR1 (38%), we decided to build a new JAK2 homology model using the JAK3 crystal structure as a template. Moreover, the JAK3 crystal structure and the sequence alignment of JAKs showed that these kinases contain an additional helix, which is not found in other tyrosine kinases.30 Therefore, a JAK2 homology model including this structural domain could be of higher quality. In this paper, we report a successful structure-based virtual screen conducted on our newly developed JAK2 model. We screened the compound library of the National Cancer Institute (NCI) in silico, and identified several novel JAK2 inhibitors by subsequent in vitro and ex vivo assays.
The crystal structure of the human JAK3 kinase domain was retrieved from the Protein Data Bank (PDB ID: 1YVJ). Sequence alignment of the human JAK3 and mouse JAK2 kinase domains was carried out by ClustalW (version 1.81).31 100 initial models of the mouse JAK2 kinase domain was generated by MODELLER (version 8.0)32 with default settings. The JAK3 crystal structure contains two phosphotyrosine residues (pTyr980 and pTyr981). The equivalent residues in JAK2 (pTyr1007 and pTyr1008) were replaced to tyrosine because of the lack of proper template in MODELLER. The best model was chosen according to the Modeller Objective Function. This model was validated by PROCHECK33 and WHATIF34 quality tests. All values were within acceptable limits and comparable to those of the template structure. Tyrosine residues (Tyr1007 and Tyr1008) of this model were phosphorylated back to phosphotyrosine in sybyl (version 7.0),35 and the 5 Å environment of the two residues were minimized (MMFF94 force field36 and charges, conjugated gradient method, 5000 step, distance dependent dielectric constant: 1, NB cutoff: 8).
After the development of our JAK2 homology model, the high-resolution structure of JAK2 in complex with a potent and selective pan-Janus inhibitor was reported by Lucet et al. (PDB ID: 2B7A).37 During the preparation of this manuscript further crystal structures of JAK2 in complex with small-molecule inhibitors were published by Antonysamy et al. (PDB IDs: 3E62, 3E63 and 3E64).26 Alignment of the Cα atoms of the binding site of our homology model to that of the available JAK2 crystal structures showed RMSD values of 0.79 Å (2B7A), 0.74 Å (3E62), 0.77 Å (3E63), 0.64 Å (3E64) and 0.83 Å (3FUP). It also has to be mentioned that alignment of the crystal structures showed a comparable level of RMSD values ranging from 0.17 Å to 0.47 Å. Moreover, the side chain orientations of the crucial residues at the binding site in our model were found to occupy a similar conformation to those in the crystal structures.
Next, we set up a virtual screen using flexx (version 1.13.2)38 which demonstrated its efficacy in a number of structure-based screens (e.g., see Ref. 39). flexx was used in combination with its own scoring function as well as with four other scoring functions available in the CSCORE package implemented in sybyl. The discriminating power of the scoring functions was investigated in two distinct phases of scoring (pose extraction and ranking) by enrichment tests. A subset of random decoys from the World Drug Index (WDI) was specifically designated to reduce artificial enrichment.40,41 The number of JAK2 inhibitors available at the beginning of this project was low. Lyne et al. previously showed that enrichment results obtained on a close analog of the target kinase are transferable.42 Accordingly, we performed an enrichment test using the JAK3 crystal structure (PDB ID: 1YVJ) and 51 known, diverse JAK3 ligands collected from the Prous Integrity database.43 Enrichment tests were carried out with and without pharmacophore constraints. Pharmacophore constraints were based on the interactions between the ligand and the protein as found in the crystal structure, that is, ligands had to form an H-bond with either the backbone oxygen of Glu930 or the backbone nitrogen of Leu932. Analysis of enrichment factors (EFs) calculated at the top 1% of the ranked database revealed that the application of pharmacophore constraints improved the results significantly (Fig. S1 in the Supplementary data).
The best scoring scheme was found to be FlexX-Score for pose extraction and ChemScore for ranking that was consequently applied in our virtual screen against JAK2. The active site of JAK2 was defined as a collection of residues within 6.5 Å of the bound inhibitor merged from JAK3 to the JAK2 homology model. The NCI subset of the ZINC database44 was screened against the JAK2 homology model. 26589 out of the 223481 compounds fulfilling the pharmacophore constraints were scored. We ranked the database according to their scores, and selected the top 1% of the whole database (2235 compounds). Next, we applied a diverse selection using Unity 2D fingerprints resulting in a set of 245 compounds with Tanimoto coefficients below 0.61. The best scored 60 compounds were ordered from NCI, and 39 arrived at our laboratory. These compounds were tested in a human erythroleukemia (HEL) cell growth inhibition assay at a final concentration of 25 μM. These cells contain the JAK2-Val617Phe mutation on both alleles, which confers their JAK2-dependent pathological growth. Five out of the 39 tested compounds showed inhibition higher than 50% (details of the assay and the results, Table S1, are provided in the Supplementary data).
The most potent compound, G6, inhibited HEL cell growth by greater than 99%. The structure of G6 is depicted on Figure 1.
Figure 1.
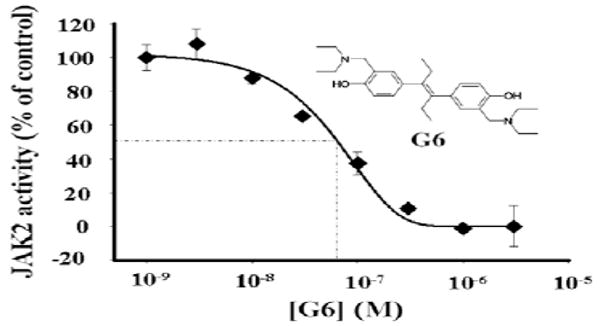
Inhibition of JAK2 Val617Phe kinase activity by G6 (IC50 = 60 ± 4.5 nM).
To determine whether G6 is a direct inhibitor of JAK2, per se, a cell free kinase assay was utilized whereby recombinant JAK2-Val617Phe protein (JH1+JH2) was incubated with increasing concentrations of G6. After completion of the kinetic reactions, relative kinase inhibition was determined by measuring the residual ATP levels in each reaction (further details of the assay are provided as Supplementary data). We found that G6 directly inhibited JAK2-Val617Phe kinase activity in a dose-dependent manner with an IC50 of 60 nM (Fig. 1).
Next, specifically to correlate G6 treatment with reduced levels of phospho-JAK2 protein within cultured cells, HEL cells were treated with G6 for varying concentrations and times. We found that G6 reduced the levels of phospho-JAK2 in both a dose- and time-dependent manner (Figs. S1 and S2 in Supplementary data).
We then analyzed the specificity of G6 by performing a cell free kinase assay with c-Src (Fig. S4 in the Supplementary data) and TYK2 autophosphorylation assays within intact cells (Fig. S5 in the Supplementary data). We found that G6 had no effect on c-Src and TYK2 tyrosine kinase activity/autophosphorylation at 25 μM, a concentration that inhibited JAK2 kinase activity by >99%.
The specificity of G6 was further tested using four different cell lines. These included (i) the HEL cell line containing the JAK2-Val617Phe mutation, (ii) a human megakaryoblastic leukemia cell line harboring a JAK3-Ala572Val activating mutation (CMK), (iii) a Burkitt Lymphoma cell line possessing a translocation between the c-Myc gene on chromosome 8 and the heavy chain locus on chromosome 14 (Raji) and (iv) a green monkey kidney epithelial cell line (BSC-40) that have been immortalized with the SV-40 large T antigen (details of the assay are provided as Supplementary data). We found that G6 specifically reduced HEL cell numbers at lower concentrations (10−9–10−5 M) when compared to the CMK, Raji and BSC-40 cells, indicating that G6 is selective for JAK2-Val617Phe at these doses (Fig. 2). However, only the highest concentration of the inhibitor (10−4 M) resulted in non-specific cytotoxicity. These data demonstrate that in the 10−9–10−5 M range, G6 specifically suppresses JAK2-dependent pathologic cell growth.
Figure 2.
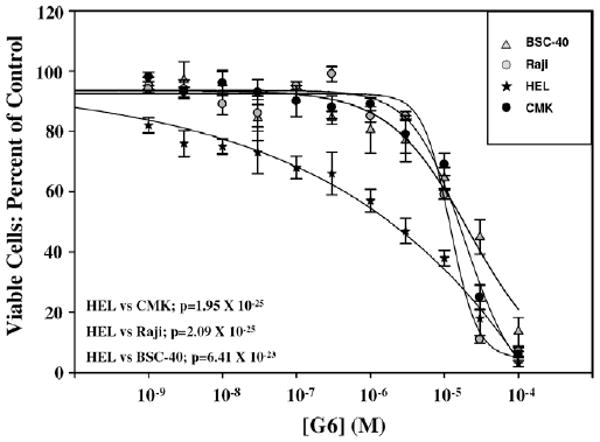
Growth inhibitory effect of G6 on HEL, CMK, Raji and BSC-40 cells.
To further demonstrate the efficacy of G6 in suppressing JAK2-dependent pathological cell growth, we obtained low density mononuclear cells from the bone marrow of a 60 year old woman diagnosed with polycythemia vera and who was JAK2-Val617Phe positive. Her hematopoietic progenitor cells were subsequently cultured in a semisolid colony assay medium in the presence or absence of G6. In addition, since hematopoietic progenitors taken from patients with polycythemia vera can be hypersensitive to cytokine stimulation,45 the cells were cultured in both the presence and the absence of human erythropoietin. We found that her hematopoietic progenitor cells exhibited a large degree of pathological, erythropoietin-independent growth (details of the assay are provided as Supplementary data). However, treatment of the cells with 2.5 μM G6 inhibited this pathological growth by greater than 50% (Fig. 3).
Figure 3.
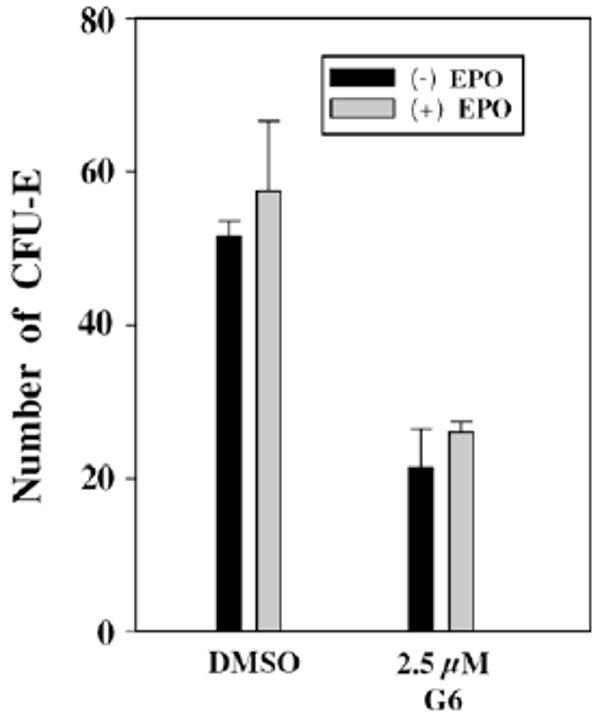
Growth inhibition of G6 on cells from polycythemia vera patient.
Collectively, the results demonstrate that G6 greatly reduces JAK2-Val617Phe human pathologic cell growth, ex vivo.
Finally, we analyzed the structural basis of G6 inhibition investigating its binding mode within the JAK2 active site. We found that one of its phenol OH groups forms strong H-bonds with the backbone NH group of Leu932 and the backbone C=O group of Glu930 (Fig. 4). These interactions seem to be highly desired for significant JAK2 affinity, since all the inhibitors in the available JAK2 crystal structures form interaction with these residues.26,37 Interestingly, G6 formed a salt bridge with Asp994, previously not recognized for JAK2 inhibitors. This aspartate is part of the DFG motif—a crucial triad of several kinase activation loops. Therefore, this new interaction can result in higher inhibitory potency and may be worth exploiting in the optimization of JAK2 inhibition. All crucial residues participating in G6 binding (Leu855, Glu856, Met929, Glu930, Tyr931, Leu932, Gly935 and Asp994) were found in the same orientation as in the available JAK2 crystal structures.
Figure 4.
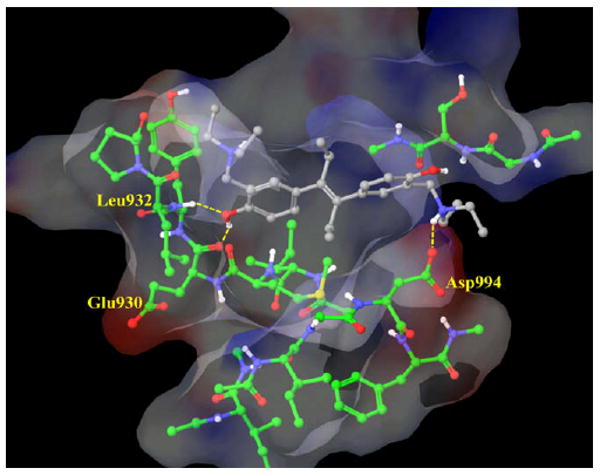
Binding mode of G6 (gray) at the JAK2 binding site (green). Van der Waals surface of the most important residues are shown and colored by their electrostatic potential. Picture was generated by MAESTRO.46
In conclusion, we have conducted a structure-based virtual screen on JAK2 successfully. Several compounds with significant HEL cell growth inhibition were identified. The most potent compound, G6, inhibited JAK2 tyrosine kinase with an IC50 of 60 nM. G6 also suppressed JAK2-dependent pathological cell growth in vitro and ex vivo. Finally, G6 was found to form a novel salt bridge with Asp994, a molecular interaction that may be of high importance in JAK2 inhibition.
Supplementary Material
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2009.04.138.
References and notes
- 1.Rane SG, Reddy EP. Oncogene. 2000;19:5662. doi: 10.1038/sj.onc.1203925. [DOI] [PubMed] [Google Scholar]
- 2.Harpur AG, Andres AC, Ziemiecki A, Aston RR, Wilks AF. Oncogene. 1992;7:1347. [PubMed] [Google Scholar]
- 3.Tanner JW, Chen W, Young RL, Longmore GD, Shaw AS. J Biol Chem. 1995;270:6523. doi: 10.1074/jbc.270.12.6523. [DOI] [PubMed] [Google Scholar]
- 4.Yan H, Piazza F, Krishnan K, Pine R, Krolewski JJ. J Biol Chem. 1998;273:4046. doi: 10.1074/jbc.273.7.4046. [DOI] [PubMed] [Google Scholar]
- 5.Leonard WJ, O'Shea JJ. Annu Rev Immunol. 1998;16:293. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 6.Ward AC, Touw I, Yoshimura A. Blood. 2000;95:19. [PubMed] [Google Scholar]
- 7.Verma A, Kambhampati S, Parmar S, Platanias LC. Cancer Metastasis Rev. 2003;22:423. doi: 10.1023/a:1023805715476. [DOI] [PubMed] [Google Scholar]
- 8.Klein B, Zhang XG, Lu ZY, Bataille R. Blood. 1995;85:863. [PubMed] [Google Scholar]
- 9.Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Blood. 2004;104:607. doi: 10.1182/blood-2004-01-0037. [DOI] [PubMed] [Google Scholar]
- 10.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Cancer Cell. 2005;7:387. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Dusa A, Staerk J, Elliott J, Pecquet C, Poirel HA, Johnston JA, Constantinescu SN. J Biol Chem. 2008;283:12941. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- 12.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Cell. 1998;93:385. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 13.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Cell. 1998;93:397. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 14.Wang K, Wang C, Xiao F, Wang H, Wu ZJ. Biol Chem. 2008;283:34029. doi: 10.1074/jbc.M803012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung JJ, Jeon J, Lee JJ, Kim CG. Biochem Biophys Res Commun. 2009;378:629. doi: 10.1016/j.bbrc.2008.11.116. [DOI] [PubMed] [Google Scholar]
- 16.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM. Nature. 1996;379:645. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 17.Kleinberger-Doran N, Shelah N, Capone R, Gazit A, Levitzki A. Exp Cell Res. 1998;241:340. doi: 10.1006/excr.1998.4061. [DOI] [PubMed] [Google Scholar]
- 18.Oda Y, Renaux B, Bjorge J, Saifeddnine M, Fujita DJ, Hollenberg MD. Can J Physiol Pharmacol. 1999;268:606. [PubMed] [Google Scholar]
- 19.Osherov N, Gazitk A, Gilon C, Levitzki A. J Biol Chem. 1993;268:11134. [PubMed] [Google Scholar]
- 20.Gu Y, Zou Y, Aikawa R, Hayashi D, Kudoh S, Yamauchi T, Uozumi H, Zhu W, Kadowaki T, Yazaki Y, Komuro I. Mol Cell Biochem. 2001;223:35. doi: 10.1023/a:1017941625858. [DOI] [PubMed] [Google Scholar]
- 21.van den Akker E, van Dijk TB, Schmidt U, Felida L, Beug H, Löwenberg B, von Lindern M. Biol Chem. 2004;385:409. doi: 10.1515/BC.2004.045. [DOI] [PubMed] [Google Scholar]
- 22.Burger R, Le Gouill S, Tai YT, Shringarpure R, Tassone P, Neri P, Podar K, Catley L, Hideshima T, Chauhan D, Caulder E, Neilan CL, Vaddi K, Li J, Gramatzki M, Fridman JS, Anderson KC. Mol Cancer Ther. 2009;8:26. doi: 10.1158/1535-7163.MCT-08-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verstovsek S, Manshouri T, Quintás-Cardama A, Harris D, Cortes J, Giles FJ, Kantarjian H, Priebe W, Estrov Z. Clin Cancer Res. 2008;14:788. doi: 10.1158/1078-0432.CCR-07-0524. [DOI] [PubMed] [Google Scholar]
- 24.Duan Z, Bradner J, Greenberg E, Mazitschek R, Foster R, Mahoney J, Seiden MV. Mol Pharmacol. 2007;72:1137. doi: 10.1124/mol.107.038117. [DOI] [PubMed] [Google Scholar]
- 25.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, Ko YD, Lee BH, Soll RM, Tefferi A, Hood JD, Gilliland DG. Cancer Cell. 2008;13:311. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Antonysamy S, Hirst G, Park F, Sprengeler P, Stappenbeck F, Steensma R, Wilson M, Wong M. Bioorg Med Chem Lett. 2009;19:279. doi: 10.1016/j.bmcl.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 27.Sandberg EM, Ma X, He K, Frank SJ, Ostrov DA, Sayeski PP. J Med Chem. 2005;48:2526. doi: 10.1021/jm049470k. [DOI] [PubMed] [Google Scholar]
- 28.Sayyah J, Magis A, Ostrov DA, Allan RW, Braylan RC, Sayeski PP. Mol Cancer Ther. 2008;7:2308. doi: 10.1158/1535-7163.MCT-08-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sudbeck EA, Liu XP, Narla RK, Mahajan S, Ghosh S, Mao C, Uckun FM. Clin Cancer Res. 1999;5:1569. [PubMed] [Google Scholar]
- 30.Boggon TJ, Li Y, Manley PW, Eck MJ. Blood. 2005;106:996. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acid Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thorton JM. J Appl Crystallogr. 1993;26:283. [Google Scholar]
- 34.Vriend G, Sander C. J Appl Cryst. 1993;26:47. [Google Scholar]
- 35.sybyl, version 7.0. Tripos Inc.; St. Louis, MO: [Google Scholar]
- 36.Halgren TA. J Am Chem Soc. 1990;112:4710. [Google Scholar]
- 37.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, Rossjohn J. Blood. 2006;107:176. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 38.Rarey M, Kramer B, Lengauer T, Klebe G. J Mol Biol. 1996;261:470. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 39.Kiss R, Kiss B, Könczöl A, Szalai F, Jelinek I, László V, Noszál B, Falus A, Keserű GM. J Med Chem. 2008;51:3145. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 40.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson PJ. J Chem Inf Comput Sci. 2004;44:793. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 41.The following criteria were used to filter out non-druglike compounds: 200 < Mw < 800, log P < 7, rotatable bonds<15. Inorganic compounds were also eliminated. 5354 compounds out of the remaining molecules were chosen based on a diversity selection (2D Unity Fingerprint) with a maximal Tanimoto index of 0.68.
- 42.Lyne PD, Kenny PW, Cosgrove DA, Deng C, Zabludoff S, Wendoloski JJ, Ashwell S. J Med Chem. 2004;47:1962. doi: 10.1021/jm030504i. [DOI] [PubMed] [Google Scholar]
- 43.Prous Science. http://www.integrity.com.
- 44.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Axelrad AA, Eskinazi D, Correa PN, Amato D. Blood. 2000;96:3310. [PubMed] [Google Scholar]
- 46.maestro, Version 8.5. Schrodinger; New York, NY: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.