

Abstract
Aims
One promising approach for treatment of Alzheimer’s disease (AD) is use of anti-amyloid therapies, based on the hypothesis that increases in amyloid-beta (Aβ) deposits in brain are a major cause of AD. Several groups have focused on Aβ immunotherapy with some success. Small molecules derivatives of Congo red have been shown to inhibit Aβ aggregation and protect against Aβ neurotoxicity in vitro. The agents described here are all small molecule Aβ-binding agents (SMAβBA’s) derivatives of Congo red.
Main Methods
Here, we have explored the anti-amyloid properties of these SMAβBA’s in mice doubly transgenic for human prensenilin-1 (PS1) and APP gene mutations that cause early-onset AD. Mice were treated with either methoxy-X04, X:EE:B34 and X:034-3-OMe1. After treatment, brains were examined for Aβ-deposition, using histochemistry, and soluble and insoluble Aβ levels were determined using ELISA.
Key Findings
A range of anti-amyloid activity was observed with these three compounds. PS1/APP mice treated with methoxy-X04 and X:EE:B34 showed decrease in total Aβ load, a decrease in Aβ fibril load, and a decrease in average plaque size. Treatment with methoxy-X04 also resulted in a decrease in insoluble Aβ levels. The structurally similar compound, X:034:3-OMe1, showed no significant effect on any of these measures. The effectiveness of the SMAβBA’s may be related to a combination of binding affinity for Aβ and entry into brain, but other factors appear to apply as well.
Significance
These data suggest that SMAβBA’s may significantly decrease amyloid burden in brain during the pathogenesis of AD and could be useful therapeutics alone, or in combination with immunotherapy.
Keywords: Alzheimer’s Disease, amyloid β, Congo Red, Methoxy-X04, transgenic mice
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia in mid to late life. Currently, most treatments for AD are symptomatic and do not address the underlying causes of AD. It is essential as the population ages, that treatments that target the cause(s) of this disease be developed. One promising approach are anti-amyloid therapies, based on the hypothesis that increased amyloid-beta (Aβ) deposits in the brain is a major underlying cause of the pathology of AD.
The “amyloid cascade hypothesis” argues dysfunctions of Aβ metabolism, through overproduction or failures in clearance, cause amyloid deposition, followed by neurofibrillary tangles, cell death and, symptoms of AD [1–3]. There is significant genetic support for this hypothesis: mutations in the amyloid precursor protein (APP) gene, lying in or near the Aβ peptide region, lead to early onset AD, perhaps the strongest evidence for the amyloid cascade hypothesis [3–5]. Further, mutations in the presenilin-1 (PS1) gene, implicated in the “γ-secretase” enzyme complex, which cleaves APP, lead to the most aggressive early-onset familial autosomal dominant AD [6].
Several groups have focused on Aβ immunotherapy as a clearance mechanism for Aβ. Schenk et al. [7] demonstrated that active immunization of transgenic mice with Aβ peptides leading to circulating anti-AB antibodies and prevention of Aβ deposition.
Another approach that may have merit alone, or in combination with immunotherapy is inhibition of aggregation of Aβ. We have developed small molecule Aβ-binding agents (SMAβBA’s). SMAβBA’s, derivatives of Congo Red (CR), may have therapeutic potential against Aβ deposition. CR and its derivatives display anti-aggregation effects on Aβ in vitro and significant anti-oxidant properties [8–10]. Others have explored anti-amyloid properties in curcumin, the yellow curry/turmeric pigment, which structurally resembles these CR derivatives. Curcumin was shown to label plaques in vivo, decrease Aβ aggregation and induce disaggregation of pre-aggregated Aβ. Curcumin also inhibited formation of soluble Aβ oligomers and toxicity of Aβ oligomer in vitro. Curcumin reduced synaptic marker loss after Aβ infusion into rats and decreased Aβ deposition in older APP transgenic mice [11, 12] Unfortunately, the bioavailability of curcumin is low and required high (50 µM) concentrations injected directly into the carotid artery to display in vivo plaque labeling. In vivo, curcumin is rapidly metabolized and many have concluded that the medicinal properties of curcumin are low due to poor bioavailability [13–15].
In these studies, we explored the anti-amyloid properties of CR derivatives in mice (PS1/APP) doubly transgenic for mutated Prensenilin-1 and APP that cause early-onset auto-somal dominant AD in humans. These CR derivatives included the Aβ imaging agent methoxy-X04 [16]. The mice examined in these studies were relatively young, treated with SMAβBA’s between 9 and 12 weeks of age, with their brain tissue examined at 12 weeks. This age was selected because it represents the earliest consistent appearance of amyloid deposition in the PS1/APP mouse [17]. We measured the anti-amyloid properties of the CR derivatives using ELISA and histological analytic techniques. These analyses measure related parameters, but have fundamental differences. For example, histological assays measure the percent area covered by plaques and do not take into account density of Aβ deposits within those plaques - a feature that may be detected by ELISA. The ELISA assay applied here measures all forms of insoluble Aβ, both fibrillar and disordered. Some of these insoluble forms may not be detectable by our histological techniques.
METHODS
Chemical Synthesis
Synthesis and characterization of (E,E)-1,4-bis(4’-hydroxystyryl)-2-methoxybenzene (Methoxy-X04),(E,E)-1-(3’,4’-dihydroxystyryl)-4-(3’-methoxy-4’hydroxystyryl)benzene (X:034-3-OMe1) and, 1,4-bis[(1E,3E)-4-(3’-carboxy-4’-hydroxyphenyl)-1,3-butadienyl]-benzene (X:EE:B34) (Table (1)) was previously described by our group in detail (Klunk et al. 2002; Wang et al. 2002). The salicylic acid derivative of Congo red, Chrysamine-G (CG), was synthesized [18]. [3H]CG was prepared by analogous synthetic schemes except [3,3’-3H]benzidine (specific activity ~30Ci/mmol, American Radiolabeled Chemicals, St. Louis, MO) was substituted for unlabeled benzidine and [3H]CG (28Ci/mmol) was purified to >97% radiochemical and chemical purity by HPLC. All other chemicals were obtained at highest purity available from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Table 1.
Chemical Structures and Properties of Study Compounds
| NAME | STRUCTURE | Ki (nM) | 2 min %IDI |
|---|---|---|---|
| Methoxy-X04 | 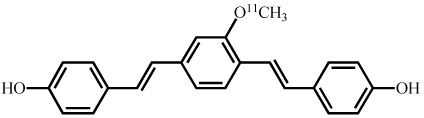 |
24 | 81 |
| X:034-3-OMe1 | 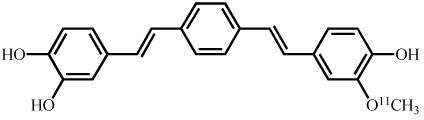 |
21 | 68 |
| X:EE:B34 | 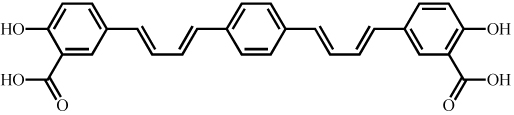 |
0.13 | n/a* |
| X:EE:B34-OMe1 | 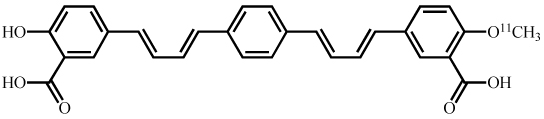 |
n/a* | 14 |
not applicable.
Synthetic Aβ Binding Assays
Preparation of Aβ Fibrils
Aβ(1–40) (Bachem Biosciences, King of Prussia, PA) was dissolved in phosphate-buffered saline (PBS) (137mM NaCl, 2.7mM KCl, 10mM phosphate; pH 7.4) at final concentrations of 433µg/ml (100µM). This solution was stirred (760rpm) for 3d at room temperature, after which it was visibly cloudy. Following stirring, approximately 2% of protein remained in the supernatant after centrifugation (28,000×g) for 15min. 100 µM PBS stock was then diluted 1:20 with 150mM Tris-HCl, pH 7.0 (5µM). Aggregated peptide was then stored at −80° C until prior to binding assays. Immediately prior to binding assays, 5µM stock was diluted 1:50 with binding buffer (150mM Tris-HCl, pH 7.0, containing 20% ethanol) to 100nM. Aggregated Aβ stock suspensions were stirred constantly to maintain homogenous suspensions.
Synthetic Aβ Binding Assay
Unlabeled test compounds were dissolved in DMSO at 400µM and diluted in binding buffer (to yield <1% DMSO in the assay after final dilution). Appropriate concentrations of non-radioactive SMAβBA’s were added to [3H]CG in a volume of 950µl of binding buffer. Assays were begun by addition of 50µl 100nM Aβ fibril stock to achieve a final concentration of 0.3nM [3H]CG (~19,000dpm/ml), 5nM Aβ fibrils, and appropriate concentrations of SMAβBA’s. Following 60min incubations at room temperature, binding mixtures were filtered through Whatman GF/B glass filters (Florham Park, New Jersey) via a Brandel M-24R cell harvester (Gaithersburg, Maryland) and rapidly washed twice with 3mls binding buffer. Filters were then counted in Cytoscint-ES (MP Biochemicals, Solon, Ohio) after sitting in cocktail overnight. Complete (100%) inhibition of binding was defined as number of counts displaced by 10 µM of unlabeled CG. Specific binding varied between 90–95% of total binding. All assays were done in at least triplicate.
Brain Entry Assays
Synthesis of Radiolabeled Compounds
The [11C]methylation of Methoxy-X04 and X:034-3-OMe1 were performed as previously described in detail, to produce X:EE:B34 itself can not be carbon-11 labeled because it does not contain an −OCH3 group. Therefore, to approximate the brain entry of X:EE:B34, a [11C]monomethoxy derivative, 1-[(1E,3E)-4-(3’-carboxy-4’-hydroxyphenyl)-1,3-butadienyl]-4-[(1E,3E)-4-(3’-carboxy-4’-methoxyphenyl)-1,3-butadienyl]-benzene (X:EE:B34-OMe1), was prepared by labeling the diethyl ester of X:EE:B34 according to previously described procedures [16]. The positions of the 11C-labels are shown in Table 1.
Brain Entry Studies in Mice
Female, Swiss Webster mice (Harlan, Indianapolis, Indiana) (n=3/time-point) were injected with 5–30µCi of [11C]Methoxy-X04 or [11C]X:034-3-OMe1 (specific activity ~1000Ci/mmol) contained in 0.1ml of isotonic saline solution (~5% ethanol) into lateral tail veins. Mice were anesthetized 2 or 30min post-injection and killed by cardiac excision in order to obtain a terminal blood sample. Brains were then rapidly removed and dissected into cerebellum and remaining whole brain fractions. Radioactivity content of samples were assayed using a gamma well counter (Packard Instruments Model 5003, Meridian, Connecticut), and counts were decay corrected to time of injection relative to carbon-11 standards prepared from injection solution to determine percent injected dose (% ID) in samples. Samples were weighed to determine the percent injected dose/g of tissue (% ID/g). This was then normalized to body weight and expressed as %IDI, where % IDI is defined as: (%ID in an organ)/(organ weight/total body weight).
Transgenic Mouse Colony
Pathogen-free mice of the PS1M148L line 6.2 homozygotes from a Swiss Webster (sw) background [19] were obtained from the University of South Florida. APPsw line Tg2576 [20] male breeders were obtained from Taconic Farms (Germantown, New York). All animals were maintained on 12h light/dark cycle with food (Purina lab chow, Richmond, Virginia) and water available ad-libitum. All procedures followed guidelines for the NIH Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. The Tg2576 males were bred with PS1 females to generate doubly transgenic PS1/APP mice. Mice were tail-clipped at 3 weeks of age and genotyping was performed to identify mice containing both PS1 and APP mutations.
Mouse Treatment with SMAβBA’s
Mice were treated beginning at 9wk of age, 3× week, totaling 9 treatments (N=14 vehicle group, N=9 methoxy-X04, N=7 X:EE:B34, N= 10 X:O34:3-OMe1). SMAβBA’s were prepared as 40mg/ml stock solutions in DMSO just prior to treatment. On each treatment day the SMAβBA stock solution was diluted to final concentrations of 2mg/ml with 9.5 volumes of propylene glycol followed by 9.5 volumes of PBS (pH 7.4). The vehicle was composed of 5% DMSO, 47.5% propylene glycol and 47.5% PBS. Treatment solution was administered at 20mg/kg by i.p. injection of 0.1mL/10g body weight. Following the final treatment, mice were deeply anesthetized with isoflurane (3–4% in 100% O2; Halocarbon, River Edge, NJ) followed by perfusion with phosphate buffer. The brain was then rapidly removed, bisected at the mid-sagittal plane and one half was frozen immediately at −80°C for ELISA analysis and the other half was immersion fixed in cold 4% paraformaldeyde for histological analysis (Sigma, St. Louis, Missouri).
ELISA Analysis of Aβ
Extraction of Aβ from Brain Tissue
Frozen mouse hemibrains were homogenized (150 mg tissue wet weight/ml homogenization buffer) in a Tissue Homogenization Buffer [250mM sucrose, 20mM Tris-base, 1mM EDTA, 1mM EGTA, plus protease inhibitors (Sigma #8340 protease inhibitor cocktail; 10µL/mL of buffer)] using a glass vessel and piston-type Teflon pestle and Aβ was extracted using described methods (Refolo et al. 2000; 2001). Briefly, soluble Aβ was extracted using cold 0.4% diethylamine in 100mM NaCl, spun at 135,000 × g for 1hr at 4°C and was neutralized by adding 0.5M Tris-HCl, pH 6.8. The pellet was further extracted using 70% cold formic acid, sonicated for 1min, spun as above and neutralized in 1M Tris-base, 0.5M Na2PO4, 0.05% NaN3.
ELISA for Soluble and Insoluble Aβ
The soluble and insoluble Aβ extracts were further diluted with ELISA buffer [20mM sodium phosphate, 2mM EDTA, 400mM NaCl, 0.2% BSA, 0.05% CHAPS, 0.4% Block Ace (Serotec, Raleigh, North Carolina), 0.05% NaN3, pH 7.0] and Aβ40 and Aβ42 concentrations were measured by sandwich ELISA previously described [21]. Briefly, 6E10 monoclonal antibody (Signet, Dedham, Massachusetts) was used as a capture antibody. Horseradish peroxidase-conjugated anti- Aβ40 and Aβ42, specific for Aβ ending at residue 40 and 42, respectively, were used as detection antibodies (Genetics Company, Schlieren, Switzerland). Aβ values, based on standard curves using Aβ40 and Aβ42 peptide standards (Bachem Biosciences, King of Prussia, Pennsylvania) were normalized to total protein and expressed as fold of vehicle.
Histochemical Analysis of Aβ Plaque Load
Staining Procedures
Brain hemispheres for histological/immunohistochemical (IHC) evaluation were immersion-fixed in cold 4% paraformaldehyde for 24hr, then cryoprotected by sequential immersion in 15% and 30% sucrose in 0.1M phosphate buffer (pH 7.4). Forty micron thick coronal brain sections were collected serially and stored in −20°C in cryprotectant (0.3g/mL sucrose, 0.01g/mL polyvinylpyrrolidone-40, 30% v/v ethylene glycol in 0.05M sodium phosphate buffer, pH 7.4) as previously described [22]. Throughout the anteroposterior length of the hippocampus, three evenly spaced sets of five adjacent tissue sections (see below) were processed for IHC using antibodies against the N-terminus of Aβ (6E10), or C-terminus specific Aβx-40 and Aβx-42 (Chemicon/Millipore Bioscience Research Reagents, Temecula, CA), or histofluorescence staining using thioflavin-S or X-34 as previously described in detail [23, 24].
Quantitative Plaque Load Analysis
For each staining technique, plaque load in the hippocampus and cortex was analyzed in three evenly spaced, coronal tissue sections 25 at the level of the rostral (1.00mm caudal to bregma), middle (−1.75mm) and caudal (−2.75mm) hippocampus using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image/). Sections processed for Aβ IHC and thioflavin-S/X-34 histofluorescence were analyzed by light and fluorescence microscopy, respectively, using an Olympus Vanoz AHBT3 microscope (Center Valley, PA). Grayscale digital images acquired at 10× magnification with a Sony CCD video camera, and montages of the entire surface of cerebral cortex and hippocampus in each tissue section were constructed using Adobe Photoshop 6.0 (Adobe Systems Inc., San Jose, CA). Percent plaque load in the cerebral cortex and hippocampus for each image were determined by standardized region of interest grayscale thresholding analysis [26] using NIH image, and modified algorithms to automate the procedure. Data was expressed as percent plaque load, corresponding to the total amount of area covered with plaques relative to the total brain area. On 6E10 immunostained sections (3/animal) frequencies of small (<6µm2), medium (6.5–9.5µm2), and large (>10µm2) plaques were determined as percents of all plaques.
Statistical Analysis
Data were analyzed with one-way ANOVA, using Bonferonni-corrected multiple comparisons for post hoc analysis.
RESULTS
Binding Affinity (Ki) for Aβ
Methoxy-X04, X:034-3-OMe1, X:EE:B34 all showed relatively high binding affinity for Aβ. Methoxy-X04 displayed a Ki of 24 nM, X:034-3-OMe1 displayed a Ki of 21 nM, and X:EE:B34 displayed a Ki of 0.13 nM (Table (1)).
Brain Entry
[11C]Methoxy-X04 and [11C]X:034-3-OMe1 showed very good and rapid brain entry, the %IDI values at 2 minutes were 81 and 68, respectively. X:EE:B34 itself was not tested because the 11C-labeling procedure modified the parent compound to generate the mono-methyl ether of the 4’-position phenol (i.e., 4’-monomethoxy). This [11C]X:EE:B34-OMe1 has many of the same properties as the parent X:EE:B34 and was believed to be informative for the brain entry of the salicylic acid compounds and so was tested. [11C]X:EE:B34-OMe1 did not enter brain well, achieving only 14% IDI at 2 min (Table (1)).
Effects on Aβ Levels in PS1/APP Mice
Treatment with Methoxy-X04 significantly (p<0.05) decreased insoluble Aβ40 and Aβ42 levels by ~64% when compared with vehicle treated PS1/APP mice (Fig. (1)). Conversely, both X:034-3-OMe1 and X:EE:B34 had no significant effect on insoluble Aβ levels in PS1/APP mice (p>0.05) (Fig. (1)). In addition, none of the SMAβBA’s tested here displayed any significant effect on soluble Aβ40 or Aβ42 levels in PS1/APP mice (data not shown).
Fig. (1).

Effect of study compounds on levels of insoluble Aβ42 and Aβ40 determined by ELISA (*- p<0.05). Aβ was extracted first by diethylamine (soluble Aβ) followed by formic acid extraction (insoluble, or FA-extracted Aβ) and quantified by ELISA. Values were normalized to total protein and expressed as fold of vehicle.
Effect on Aβ Plaque Load
A one way ANOVA revealed a significant (p=0.023) decrease in Aβ40 plaque load after treatment with Methoxy-X04 and X:EE:B34. A significant (p=0.001) decrease in total Aβ plaque load (measured by the 6E10 antibody) also was observed after Methoxy-X04 and X:EE:B34 treatment (Fig. (2) & Fig. (4)). No significant decrease was observed in Aβ42 load, although the trend was similar to that for Aβ40 (Fig. (2)). Additionally, a one way ANOVA revealed a significant (p=0.002) decrease in total Aβ plaque load measured by X-34 after treatment with Methoxy-X04 and X:EE:B34 (Fig. (2) & Fig. (4)). A non-significant (p= 0.064) decrease was also observed after Methoxy-X04 and X:EE:B34 treatment using the ThioS stain (Fig. (3)).
Fig. (2).

Effect of study compounds on plaque load determined by IHC with antibodies against Aβ40, Aβ42 and total Aβ (*- p<0.05, **- p<0.01).
Fig. (4).

Photomicrographs of coronal mouse brain sections, processed immunohistochemically using antibody clone 6E10 (A,C,E,G) and histochemically using the amyloid binding compound X-34 (B,D,F,H) are representative of experimental groups treated with vehicle (A,B), X:034-3-OMe1 (C,D), Methoxy-X04 (E,F), and X:EE:B34 (G,H). High power inserts are representative plaques in low-power images. Scale bar 250 µm (low power), 25 µm (insets).
Fig. (3).

Effect of study compounds on plaque load determined by ThioS and X-34 (+ p= 0.06, **- p<0.001).
Effect on Aβ Plaque Size
A one way ANOVA revealed a significant decrease in the number of medium (p=0.002) and large plaques (p=0.004) visualized by 6E10 antibody staining (i.e., total Aβ) after treatment with Methoxy-X04 and X:EE:B34 (Fig. (4) & Fig. (5a)). This was accompanied by a significant change in the distribution of plaque size after Methoxy-X04 and X:EE:B34 treatment since these SMAβBA’s increased the proportion of small plaques (not significant) and decreased the proportion of large plaques (p=0.006) (Fig. (4) & Fig. (5b)).
Fig. (5).

A) Small, medium and large plaques as percents of all plaques. Bars show mean value and SD. B) Frequencies of small (<6 µm2), medium (6.5–9.5 µm2), and large (>10 µm2) plaques. Plaques were visualized using 6E10 immunohistochemistry. (*- p<0.05).
DISCUSSION
Anti-amyloid therapies have become an important therapeutic target in AD research. The use of SMAβBA’s is one approach to anti-amyloid therapy. The SMAβBA’s discussed here are derivatives of CR, and display anti-Aβ properties (see Introduction). Methoxy-X04, X:034-3-OMe1, X:EE:B34 displayed good binding affinity for Aβ, in the range of 0.1–25 nM. Using the administered dose and the measured %injected dose/gram of brain at 2 min after injection, the peak brain concentration of Methoxy-X04 and X:034-3-OMe1 can be calculated at ~50 µM and it is possible that these compounds remain at concentrations above the Ki throughout the majority of the treatment course. However, we do not have information on brain levels of these compounds over the course of the study, so this remains speculative. While Methoxy-X04 and X:034-3-OMe1 both demonstrated very good brain entry, X:EE:B34 did not. As a point of reference, positron emission tomography tracers must have excellent and rapid brain uptake and typically show 2 min %IDI values in the range of 100–300 [16]. But because of the very high Aβ affinity of X:EE:B34 (100-fold better than the other compounds) it is possible that a lower level of brain entry may be required for this compound to produce equivalent effects, depending on the mechanism. That is, if binding to the same site on Aβ is the way these compounds exert their action, then X:EE:B34 can occupy the same number of Aβ binding sites as Methoxy-X04 at 100-fold lower brain levels.
PS1/APP mice treated with Methoxy-X04 displayed significant decreases in insoluble Aβ levels, measured by ELISA and significantly lower Aβ40 and total Aβ loads, measured by IHC and X-34. A similar, but non-significant, decrease was seen for Aβ42. Treatment with X:EE:B34 produced no significant decrease in insoluble Aβ levels using ELISA, but did cause a significant decrease in total Aβ fibril load measured by IHC and X-34. Conversely, X:034-3-OMe1, while displaying significant binding affinity for Aβ and good brain entry, had no effect on any measure of Aβ deposition. The fact that X:034-3-OMe1 had little effect despite brain entry and affinity characteristics similar to Methoxy-X04 suggests either this compound was more rapidly metabolized or something other than simple occupancy of binding sites on Aβ fibrils is required for anti-amyloid effects. This compound is a catechol and these compounds are known to be susceptible to oxidation. While no specific toxicity studies have been performed with these compounds we did monitor weight and general health of the animals treated with SMAβBA’s and there were no changes in any of these measures as compared to vehicle-treated animals (data not shown).
It is important to note that not only did animals treated with Methoxy-X04, and X:EE:B34 often show reductions in Aβ content and plaque load (% area), but remaining plaques were typically smaller as well (Fig. (4), (5a & b)). That is, the number of larger plaques was reduced more than the number of smaller plaques, resulting in distribution of plaque size that was skewed towards smaller plaques. Since the treatments were begun at the earliest stage of plaque deposition, this suggests that the presence of the SMAβBA’s in the extracellular milieu of the CNS tends to retard formation of plaques in general and of larger plaques in particular. Although there is currently no data regarding the effect of these compounds on Aβ synthesis, it seems more likely that these compounds would cause reductions in plaque size and number via general aggregation-retarding and clearance-promoting effects. It is important to note, however, that shifting the size of plaques towards smaller species may not have beneficial effects on brain function. Several have demonstrated that soluble dimers of Aβ have toxic effects on synaptic plasticity and long term potentiation in cell culture models and in vivo [27–30]. This suggests the possibility that shifting Aβ plaques to a smaller species could exacerbate the toxicity of Aβ. However, further studies including behavioral analyses, will have to be completed in order for this issue to be addressed.
The distinctions between ELISA and IHC discussed in the Introduction, could explain the differences in the results obtained by the two techniques. The difference is most apparent in the decreased Aβ42 load detected by fibril stains, Thio-S and X-34, after X:EE:B34 treatment that was not reflected in decreased amounts of insoluble Aβ as detected by ELISA. This could imply that X:EE:B34 treatment changed the way Aβ is deposited without changing total amounts of Aβ in the brain. This is consistent with the fact that there were more prominent decreases in Aβ42 when measured by fibrillar stains than when measured by anti-Aβ IHC or ELISA. The X:EE:B34 effect also is consistent with the finding that X:EE:B34 shifted the size distribution of plaques from large to small (determined by IHC). The fluorescent fibril stains may be less sensitive to smaller plaques and would not detect insoluble Aβ that was present in disordered (i.e., non-fibrillar) forms.
While it is clear that these SMAβBA’s bind to Aβ well, it is not clear to which species of Aβ these SMAβBAs must bind in order to exert their anti-amyloid effects. It is clear from observations made during the development of the CR method for the detection of Aβ fibril formation that the CR class of compounds does not bind monomeric Aβ, but binds well to large, insoluble, fibrillar Aβ [31, 32]. However, the exact oligomeric stage at which CR derivatives can bind to Aβ is not known. This is important as the evidence that the toxic species of Aβ in vivo is a relatively small aggregate [i.e., an oligomer, protofibril or Aβ-derived diffusible ligand (ADDL)] is increasingly convincing [33]. Recent data suggests that CR and X-34 derivatives can bind to Aβ oligomers with affinity in the 3–20 µM range [34]. Basic principles of chemical equilibrium would predict that soluble forms of Aβ oligomers can be regenerated from the ~100-fold larger pool of insoluble forms. Therefore, for there to be a long-lasting lowering of soluble Aβ oligomers, it is likely that effective anti-amyloid therapy will need to have a significant impact on both soluble and insoluble “reservoirs” of brain Aβ. SMAβBA’s that enhance intermolecular association could cause a shift toward fibrils and may lead to decreasing of soluble forms of Aβ. However, this would increase a potential source for new oligomers and would create a potentially dangerous situation if the compound driving this equilibrium was removed. SMAβBAs that inhibit intermolecular association would cause a shift away from fibrils and oligomers towards monomeric species, and this could result in higher proportions of more easily cleared monomeric forms, and an overall net increase in Aβ clearance. This would result in a lowering of not only the insoluble pool, but also the soluble oligomeric pool over time. While none of the SMAβBA’s tested here appeared to increase the soluble Aβ pool (data not shown), it will be important to further elucidate the species of Aβ being bound by these SMAβBA’s. The presence of equilibrium between pools of soluble Aβ in the brain and periphery could explain why we did not observe changes in soluble Aβ, despite reductions in insoluble Aβ. In our model, the portion of insoluble Aβ that was reduced by SMAβBA’s may have been converted to soluble Aβ that in turn was rapidly cleared from the brain (or degraded).
Methoxy-X04 has been used previously with two-photon microscopy to image amyloid deposits in living animals, as it is highly fluorescent and binds well to amyloid plaques [16, 35–37]. It is important to note that in the studies presented here, Methoxy-X04 was used at a dose that is 4–10 times that used for two-photon imaging with Methoxy-X04. In addition, in these studies animals were treated nine times over a three week period in contrast to the more acute use of Methoxy-X04 for imaging. Finally, as discussed below, the mice used here in these studies were relatively young, compared with the 7–18 month old mice typically used for imaging of plaques. Therefore, this study should not be taken to indicate that the typical use of Methoxy-X04 in two-photon studies will have any impact on the natural history of amyloid deposition under those conditions.
While the SMAβBA’s investigated were effective in this early age range, this may not be the case in animals with more advanced amyloid deposition. Indeed, it has been reported that chronic treatment of PDAPP mice with the m266 anti-Aβ antibody, from 4 to 9 months of age, reduces Aβ burden. In the subsequent study in 24 month-old PDAPP mice, they found that a “subchronic” six-week course of m266 immunotherapy reversed the cognitive deficits measured, but did not decrease Aβ immunohistochemicaly [38]. These data would indicate that timing of treatment and duration of Aβ deposition may be critical to the efficacy of anti-amyloid treatments.
Overall, at early stages of amyloid deposition, the SMAβBA’s tested here display significant affinity for and clearance of amyloid. Taken together, these data suggest that SMAβBA’s may significantly decrease amyloid burden in brain during the pathogenesis of AD. These data also bring up the possibility that the combination of a SMAβBA that could shift the equilibrium of Aβ aggregates towards smaller forms, with agents that might augment the rate of clearance of these more soluble forms of Aβ such as anti-Aβ antibodies could result in synergistic anti-amyloid effects.
ACKNOWLEDGMENTS
This work was supported by NIH R01 AG020226 and U01 AG028526 to WEK.
REFERENCES
- 1.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends in Pharmacological Sciences. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nature Neuroscience. 1998;1(5):355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 4.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiology of Disease. 1996;3(3):159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 5.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annual Review of Neuroscience. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 6.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J. Neuroimmunol. 2000;108(1–2):227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 7.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 8.Pollack SJ, Sadler II, Hawtin SR, Tailor VJ, Shearman MS. Sulfonated dyes attenuate the toxic effects of beta-amyloid in a structure-specific fashion. Neuroscience Letters. 1995;95(3):211–214. doi: 10.1016/0304-3940(95)11939-t. [DOI] [PubMed] [Google Scholar]
- 9.Pollack SJ, Sadler IIJ, Hawtin SR, Tailor VJ, Shearman MS. Sulfated glycosaminoglycans and dyes attenuate the neurotoxic effects of β-amyloid in rat PC12 cells. Neuroscience Letters. 1995;184:113–116. doi: 10.1016/0304-3940(94)11182-i. [DOI] [PubMed] [Google Scholar]
- 10.Kanski J, Sultana R, Klunk W, Butterfield DA. Antioxidant activity of X-34 in synaptosomal and neuronal systems. Brain Res. 2003;988(1–2):173–179. doi: 10.1016/s0006-8993(03)03369-9. [DOI] [PubMed] [Google Scholar]
- 11.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001;21(21):8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005;280(7):5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 13.Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998;64(4):353–356. doi: 10.1055/s-2006-957450. [DOI] [PubMed] [Google Scholar]
- 14.Garcea G, Berry DP, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, Steward WP, Gescher AJ. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol. Biomarkers Prev. 2005;14(1):120–125. [PubMed] [Google Scholar]
- 15.Garcea G, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, Steward WP, Gescher AJ, Berry DP. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br. J. Cancer. 2004;90(5):1011–1015. doi: 10.1038/sj.bjc.6601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J. Neuropathol. Exp. Neurol. 2002;61(9):797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- 17.Kurt MA, Davies DC, Kidd M, Duff K, Rolph SC, Jennings KH, Howlett DR. Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp. Neurol. 2001;171(1):59–71. doi: 10.1006/exnr.2001.7717. [DOI] [PubMed] [Google Scholar]
- 18.Klunk WE, Debnath ML, Pettegrew JW. Development of small molecule probes for the beta-amyloid protein of Alzheimer's disease. Neurobiol Aging. 1994;15(6):691–698. doi: 10.1016/0197-4580(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 19.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 20.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 21.Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J. Biol. Chem. 2005;280(6):4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 22.Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7(1):155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- 23.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 2004;190(1):192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J. Histochem. Cytochem. 2000;48(9):1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 25.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- 26.Fishman CE, Cummins DJ, Bales KR, DeLong CA, Esterman MA, Hanson JC, White SL, Paul SM, Jordan WH. Statistical aspects of quantitative image analysis of beta-amyloid in the APP(V717F) transgenic mouse model of Alzheimer's disease. J. Neurosci. Methods. 2001;108(2):145–152. doi: 10.1016/s0165-0270(01)00381-8. [DOI] [PubMed] [Google Scholar]
- 27.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-[beta] protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 29.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid [beta] protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 30.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-[beta] protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klunk WE, Pettegrew JW, Abraham DJ. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J. Histochem. Cytochem. 1989;37(8):1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 32.Klunk WE, Jacob RF, Mason RP. Quantifying amyloid by Congo red spectral shift assay. Methods Enzymol. 1999;309:285–305. doi: 10.1016/s0076-6879(99)09021-7. [DOI] [PubMed] [Google Scholar]
- 33.Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33(Pt 5):1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 34.Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, Kung HF, Lam KS, Oddo S, Laferla FM, Jin LW. Congo red and thioflavin-T analogs detect Abeta oligomers. J. Neurochem. 2008;104(2):457–468. doi: 10.1111/j.1471-4159.2007.04972.x. [DOI] [PubMed] [Google Scholar]
- 35.Sadowski M, Pankiewicz J, Scholtzova H, Tsai J, Li Y, Carp RI, Meeker HC, Gambetti P, Debnath M, Mathis CA, Shao L, Gan WB, Klunk WE, Wisniewski T. Targeting prion amyloid deposits in vivo. J. Neuropathol. Exp. Neurol. 2004;63(7):775–784. doi: 10.1093/jnen/63.7.775. [DOI] [PubMed] [Google Scholar]
- 36.Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J. Clin. Invest. 2005;115(2):428–433. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451(7179):720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nature Neuroscience. 2002;5(5):452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]