

Abstract
We report the design, synthesis and biological evaluation of a series of novel HIV-1 protease inhibitors. The inhibitors incorporate stereochemically defined flexible cyclic ethers/polyethers as the high affinity P2-ligands. Inhibitors containing small ring 1,3-dioxacycloalkanes have shown potent enzyme inhibitory and antiviral activity. Inhibitors 3d and 3h are the most active inhibitors. Inhibitor 3d maintains excellent potency against a variety of multi-PI-resistant clinical strains. Our structure-activity studies indicate that the ring size, stereochemistry, and position of oxygens are important for the observed activity. Optically active synthesis of 1,3-dioxepan-5-ol along with the syntheses of various cyclic ether and polyether ligands have been described. A protein-ligand X-ray crystal structure of 3d-bound HIV-1 protease was determined. The structure revealed that the P2-ligand makes extensive interactions including hydrogen bonding with the protease backbone in the S2-site. In addition, the P2-ligand in 3d forms a unique water-mediated interaction with the NH of Gly-48.
Introduction
The introduction of protease inhibitors (PIs) into highly active antiretroviral therapy (HAART), a combination therapy based on co-administration of PIs with reverse-transcriptase inhibitors, marked the beginning of a new era in HIV/AIDS chemotherapy. HAART treatment regimens have led to a significant decline in the number of deaths due to HIV infection in the developed World.1 Unfortunately there are a number of factors that severely limit current HAART treatment regimens. High frequency of dosing, heavy pill burden and issues of tolerability and toxicity can lead to poor adherence to treatment.2 The need for more potent, less toxic drug regimens is quite apparent.
It is the rapid emergence of drug resistance however, that is proving to be the most formidable problem. Mutations causing drug resistance are thought to occur spontaneously, through the recombination of mixed viral populations, and also due to drug pressure, particularly when administered at sub-standard doses.3–6 A growing number of patients are developing multi-drug-resistant HIV-1 variants.7,8 There is ample evidence that these viral strains can be transmitted. Thus, the development of antiretroviral agents able to maintain potency against resistant HIV strains has become an urgent priority.
Darunavir (TMC-114, 1, Figure 1) is a new nonpeptidic PI recently approved by the FDA for the treatment of antiretroviral therapy-experienced patients.9 Inhibitor 1, and its related analogue 2, are exceedingly active against both wild-type and multi-drug resistant HIV strains. Both PIs demonstrated potent in vitro activity against viral isolates resistant to currently licensed PIs.10–12 Our structure-based design strategies for these PIs are based on the presumption that maximizing active site interactions with the inhibitor, particularly hydrogen bonding with the protein backbone would give rise to potent inhibitors retaining activity against mutant strains.13,14 Indeed, side chain amino acid mutations cannot easily disrupt inhibitor-backbone interactions, because the active site backbone conformation of mutant proteases is only minimally distorted compared to the wild-type HIV-1 protease.15–17 In this context, the fused bis-tetrahydrofuran (bis-THF) urethane of compounds 1 and 2 was demonstrated to be a privileged P2-ligand, being able to engage in a number of hydrogen bonding interactions with the backbone atoms of amino acids at the protease S2-site.
Figure 1.

Structure of inhibitors 1, 2, and 3c,d.
We are continuing our efforts toward the development of novel PIs characterized by a high activity against both wild-type HIV-1 and resistant strains. We further speculated that an inhibitor interacting strongly with the protein backbone, while being able to accommodate amino acid side chain variations by means of repacking with a flexible ring, would maintain significant affinity against both wild-type and mutant enzymes. With this goal in mind, we designed a series of PIs based on the (R)-(hydroxyethylamino)sulfonamide isostere and bearing flexible cyclic ethers and polyethers as P2-ligands (inhibitors 3a–m, Table 1). Starting from compound 3c, incorporating a (1R)-3,5-dioxacyclooctan-1-yl urethane which can be considered as the flexible counterpart of the bis-THF moiety, we designed a series of structural variants of this inhibitor. These inhibitors contain polyether-based P2-ligands ranging from 6- to 13-membered rings coupled to a p-methoxyphenylsulfonamide as the P2'-ligand. Herein we report the structure-based design, synthesis, and preliminary biological evaluation of inhibitors 3a–m. Among these inhibitors, 3d (Figure 1) is the most potent with an impressive enzyme inhibitory and antiviral activity (Ki = 26 pM, IC50 = 4.9 nM). Furthermore, a protein-ligand X-ray structure of 3d-bound HIV-1 protease has revealed important molecular insight regarding ligand-binding site interactions.
Table 1.
Enzyme Inhibitory and Antiviral Activity of Inhibitors 3a–m
| Entry | Inhibitor | Ki (nM) | IC50 (nM)a |
|---|---|---|---|
| 1 | 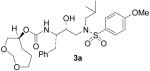 |
0.15 ± 0.019 | ndb |
| 2 | 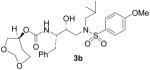 |
0.16 ± 0.04 | 30 ± 1 |
| 3 | 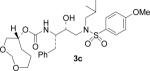 |
0.16 ± 0.011 | nd |
| 4 | 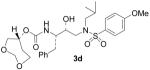 |
0.026 ± 0.012 | 4.9 ± 0.3 |
| 5 | 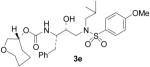 |
0.81 ± 0.12 | nd |
| 6 | 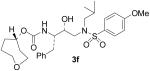 |
0.74 ± 0.15 | nd |
| 7 | 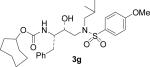 |
27 ± 0.81 | nd |
| 8 | 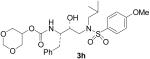 |
0.041 ± 0.002 | 3.4 ± 0.7 |
| 9 | 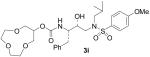 |
16 ± 2.2 | nd |
| 10 | 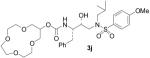 |
33 ± 1.9 | nd |
| 11 | 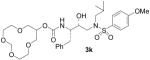 |
6.3 ± 0.57 | >1000 |
| 12 | 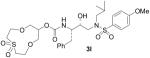 |
1.9 ± 0.2 | >1000 |
| 13 | 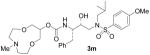 |
19 ± 0.76 | >1000 |
| SQVc | - | - | 16 ± 3 |
| APVd | - | - | 27 ± 6 |
MT-2 human T-lymphoid cells exposed to HIV-1LAI
nd = not determined
SQV = saquinavir
APV = amprenavir.
Chemistry
The syntheses of seven and eight membered 1,3-dioxacycloalkanes 8a–d for the corresponding inhibitors 3a–d, are shown in Scheme 1. Protected diol 6a was prepared by a two step procedure starting from (S)-hydroxyglutaric acid 4, obtained by following a known protocol.18 The hydroxyl group of 4 was protected as a tert-butyldiphenylsilylether 5 in quantitative yield. LiBH4 reduction of both ester groups afforded 6a in good yield.19
Scheme 1.

Optically active synthesis of 1,3-dioxacycloalkanes
Compounds 6a and 6b20 were converted to cyclic derivatives by exposure to paraformaldehyde and BF3·OEt2 21 to afford cyclic ethers 7a and 7b in 51% and 82% yield, respectively. Deprotection of compounds 7a to 8a was carried out by using n-Bu4N+F in THF. Benzylether of 7b was removed by a catalytic hydrogenation over 10% Pd-C to furnish 8b. Mitsunobu inversion of the secondary hydroxyl groups of 8a,b was accomplished by using p-nitrobenzoic acid, triphenylphosphine and diisopropylazodicarboxylate in benzene at 23 °C. Saponification of the resulting esters provided 8c and 8d.
For the synthesis of compounds 8e and 8f, that represent the monoxygenated analogues of 8d, a synthetic strategy based on a ring-closing metathesis reaction as the key step was planned (Schemes 2 and 3). Accordingly, secondary alcohol 922 (Scheme 2) was protected as the corresponding methoxyethoxymethyl (MEM)-ether 10 in 90% yield using an excess of MEM-Cl in the presence of DIPEA in CH2Cl2.
Scheme 2.

Synthesis of cyclic ether 8e
Scheme 3.

Synthesis of cyclic ether 8f
Subsequent n-Bu4N+F−-promoted deprotection of the TBDMS-group afforded the corresponding primary alcohol which was treated with sodium hydride and alkylated with allyl bromide in the presence of a catalytic amount of n-Bu4N+I− to afford olefin 11 in 78% yield (2 steps). A 0.01 M solution of 11 in CH2Cl2 was then treated with a catalytic amount (5 mol%) of 2nd generation Grubbs catalyst and heated to 45 °C to afford the cyclooxepane 12 in 94% yield. The double bond of 12 was finally reduced by catalytic hydrogenation using 10% Pd-C as the catalyst and the MEM-ether was removed by acidic hydrolysis in a 1:1 THF/H2O mixture to obtain the target alcohol 8e in good overall yield.
For the synthesis of alcohol 8f (Scheme 3), compound 13 was used as the starting material. It was in turn prepared following a described procedure starting from acrolein and tert-butylacetate.23 Alkylation of the primary hydroxyl group of 13 with allyl bromide and n-Bu4N+I− using sodium hydride as the base furnished the ring closing metathesis precursor 14. The cyclization reaction was performed by using 2nd generation Grubbs catalyst (5 mol%) in CH2Cl2 and afforded olefin 15 in good yield. Subsequent hydrogenation of the double bond and n-Bu4N+F−-mediated removal of TBDMS-ether finally afforded the target alcohol 8f.
Alcohols 8h–j required for the preparation of inhibitors 3h–j were synthesized starting from the common intermediate 2-benzyloxypropane-1,3-diol 17 as shown in Scheme 4.
Scheme 4.

Synthesis of polyethers 8h–j
Compound 17 was prepared by alkylation of commercially available benzylidene acetal 16 with benzyl chloride in the presence of sodium hydride and a catalytic amount of n-Bu4N+I− in THF at 23 °C.
The benzylidene group was subsequently removed by hydrolysis with 6 N HCl in a mixture (1:1) of THF and water to give 2-benzyloxy-1,3-propanediol 17 in quantitative yield. Treatment of 17 with paraformaldehyde and BF3·OEt2 as described above, followed by hydrogenolysis of the resulting O-benzylether afforded 8h in 78% overall yield.
Treatment of diol 17 with an excess of sodium hydride in refluxing THF followed by addition of di(ethyleneglycol)dimesylate or tri(ethyleneglycol)dimesylate afforded macrocycles 18 and 19 in 19% and 29% yield, respectively. Dilution of the reaction mixture to assist the intramolecular cyclization reaction did not result in a significant improvement of the reaction yields. Given the poor enzymatic inhibitory activity observed for the corresponding final compounds 3i, 3j, no further attempts were made to improve the cyclization yield for the preparation of these 10- and 13-membered polyether rings. Compounds 18 and 19 were subsequently deprotected by hydrogenolysis to obtain alcohols 8i and 8j.
We planned to investigate the effect of heteroatom functionalities in the polyether rings. In this context, we prepared the compounds 8k, 8l and 24 from known diols 2024 as shown in Scheme 5. Thus, exposure of 20 to paraformaldehyde in the presence of BF3·OEt2 furnished the corresponding cyclic polyether product, which, upon hydrogenolysis, gave alcohol 8k. Bromination of 20 using carbon tetrabromide and triphenylphosphine afforded dibromide 21.24 This dibromide was used for the synthesis of sulfone 8l and protected amine 24. Thus, compound 21 was reacted with one equivalent of benzylamine in refluxing MeCN in the presence of sodium carbonate, as reported by Calverley and Dale25 to provide 23 in 24% yield. Dimerization is the main side product in this reaction and one can reduce such dimerization by using an excess of LiClO4.26 Benzylamine 23 was hydrogenated over 10% Pd-C in the presence of di-t-butyl dicarbonate to provide N-Boc protected alcohol 24. Sulfone 22 was obtained by cyclization of 21 with lithium sulfide followed by oxidation of the corresponding sulfide with an excess of m-CPBA in CH2Cl2 at 23 °C. Benzyl derivative 22 was converted to 8l by a catalytic hydrogenation over 10% Pd-C.
Scheme 5.

Synthesis of alcohols 8k,l and 24
Scheme 6 depicts the conversion of various P2-ligands to the corresponding active carbonates for urethane formation. Accordingly, alcohols 8a–h,j–l were reacted with p-nitrophenylchloroformate and N-methylmorpholine in THF at 23 °C to provide corresponding carbonates 25a–h,j–l in 67–89% yields. Alcohol 8i was converted to succinimidylcarbamate 25i by treatment with N,N'-succinimidylcarbonate in the presence of Et3N in MeCN in 37% isolated yield.
Scheme 6.

Synthesis of various active carbonates
The synthesis of designed inhibitors 3a–l is shown in Scheme 7. Methoxysulfonamide derivative 27 was prepared from commercially available epoxide 26 as described previously.27 The Boc group in 27 was removed by exposure to a 30% solution of TFA in CH2Cl2 at 23 °C. The resulting amine 28 was reacted with the suitable mixed activated carbonates 25a–l in THF at 23 °C for 2 to 4 days to furnish inhibitors 3a–l in 36–89% yield.
Scheme 7.

Synthesis of inhibitors 3a–l
The synthesis of inhibitor 3m is shown in Scheme 8. Alcohol 24 was converted to active carbonate 29 as described above in Scheme 6. Reaction of 29 with amine 28 provided urethane 30 in good yield. Removal of Boc group of 30 by exposure to 30% TFA in CH2Cl2 furnished amine 31. The resulting secondary amine was subjected to a reductive amination reaction using 37% aqueous formaldehyde and sodium cyanoborohydride in 1% acetic acid in MeOH to furnish N-methyl derivative 3m in 87% yield.
Scheme 8.

Synthesis of inhibitor 3m
Results and Discussion
All inhibitors contain a (R)-hydroxyethylamine sulfonamide isostere with a p-methoxysulfonamide as the P2'-ligand and various designed cyclic ethers and polyethers as the P2-ligands. These inhibitors were first evaluated in an enzyme inhibitory assay utilizing a protocol described by Toth and Marshall.28 Compounds that showed potent enzymatic Ki values were then further evaluated in an antiviral assay. The results are shown in Table 1. The Ki-values denote the mean values of at least four determinations.
As it can be seen, introduction of the 8-membered (S)- or (R)-1,3-dioxacyclooctan-5-yl urethanes as P2-ligands (inhibitors 3a and 3c) resulted in subnanomolar inhibitors. However, these inhibitors are significantly less potent than inhibitor 2 that contains the bis-THF ligand. Interestingly, incorporation of a (5R)-1,3-dioxacycloheptan-5-yl urethane as the P2-ligand resulted in the most potent inhibitor 3d in this series with a Ki value of 26 pM. We speculated that the 7-membered 1,3-dioxepanyl-ligand with R-configuration may bind to residues in the S2-site similar to bis-THF ligand of inhibitor 2. Inhibitor 3d exhibited more than 6-fold potency increase relative to epimeric (5S)-1,3-dioxacycloheptan-5-yl urethane 3b, suggesting an important role for the ring stereochemistry. Inhibitors 3e–g were prepared to assess the role played by both oxygen atoms of 3d on the binding mode of this latter compound. As shown in Table 1, a dramatic drop in enzymatic inhibitory activity was observed when the cycloheptanol was introduced as the P2-ligand (3g). Moreover, nearly 30-fold reduction in enzymatic inhibitory potency of both 3e and 3f with respect to 3d clearly demonstrated that both oxygen atoms are crucial for the interaction with the enzyme at the S2-subsite. It appears that both oxygen atoms engage in strong hydrogen bonding which equally contribute to the binding affinity for the enzyme. This result was further confirmed by the determination of the X-ray crystal structure of 3d-bound HIV-1 protease.
Further reduction of the ring size of the P2-ligand resulted into the design of inhibitor 3h, bearing a 6-membered 1,3-dioxan-5-yl urethane. This inhibitor also showed an impressive enzymatic Ki value of 41 pM. This result suggested that the 1,3-dioxane ring could be nicely accommodated by the S2-site. Furthermore, both oxygens may be involved in specific interactions with the amino acid residues in this region.
Subsequently, we tested compounds 3i–m, presenting larger polyether rings but all compounds showed Ki values in the high nM range (Kis ranging from 6.3 to 33 nM), proving that large rings could not be easily accommodated at the S2-site.
However, subtle differences in the activity among these compounds suggested that not only the ring size, but also the position of the oxygen atoms within the polyether structure, could be important for inhibitory activity. In fact, compound 3k, presenting a 12-membered ring bearing a methylenedioxy unit instead of the ethylenedioxy of 3j, exhibited 5-fold potency enhancement compared to inhibitor 3j. It is also more than 2-fold more potent compared to 3i, which contains a smaller 10-membered ring. Substitution of a ring oxygen in 3i by a N-Me group provided inhibitor 3m with no change in inhibitory activity. However, replacement of ring oxygen with a SO2 moiety provided inhibitor 3l with a 9-fold improvement in potency. The sulfone oxygens may be involved in specific interactions with the amino acid residues at the S2 site.
In MT-2 human T-lymphoid cells exposed to HIV-1LAI, inhibitors 3d and 3h have shown antiviral IC50 values of 4.9 nM and 3.4 nM, respectively (Table 1). Consistent with its enzymatic potency, compound 3b showed an antiviral activity of 30 nM in the same assay system, while compounds 3k–m did not exhibit appreciable antiviral properties at doses up to 1 μM. We have examined two selected compounds, 3d, and 3h, for their activity against HIV-1 using a human CD4+ T-cell line (MT-2 cells) and human peripheral blood mononuclear cells (PBMCs) as target cells. We employed two endpoints for the activity against HIV-1: (i) the inhibition of the HIV-1-elicited cytopathic effect for MT-2 cells and (ii) the inhibition of HIV-1 p24 production for PBMCs.14a As examined in MT-2 cells as target cells, the two compounds, 3d and 3h exerted extremely potent antiviral activity against an X4-HIV-1 isolate (HIV-1LAI) with IC50 values of 4.9 and 3.4 nM, respectively (Table 1). Such anti-HIV-1 potency was generally parallel to the potency in enzymatic inhibition of the compounds. We further examined the two compounds in PBMCs against a clinical wild-type X4-HIV-1 isolate (HIV-1ERS104pre) along with various multi-drug-resistant clinical X4- and R5-HIV-1 isolates (Table 2).14 The activity of 3d and 3h against HIV-1ERS104pre was more potent or at least comparable as compared to those of currently available protease inhibitors, APV, IDV, and RTV. It is interesting to note that the values of 3d were greater than those with MT-2 cells by factors of about 4. With regard to this difference, considering that 3d was highly potent as examined in human T cells (MT-2 cells) but its activity was slightly less in PBMCs, it is possible that relatively higher concentrations of 3d are required to suppress HIV-1 production in chronically infected macrophages.42 Two currently available protease inhibitors (IDV and RTV) were not capable of efficiently suppressing the replication of most of the multi-drug-resistant clinical isolates examined (HIV-1MDR-B, HIV-1MDR-G, HIV-1MDR-TM, HIV-1MDR-JSL, and HIV-1MDR-MM) with IC50 values of >1.0 μM. Although the two selected compounds were also less potent against the multi-drug-resistant clinical isolates examined, their IC50 values were quite low with 0.22 − 0.54 μM (Table 2). During testing of the anti-HIV-1 activity of compounds 3b, 3d, 3h, and 3k–m, we examined 4 concentrations (1, 0.1, 0.01, and 0.001 μM) in the antiretroviral assay, conducted on three independent occasions (each assay was performed in duplicate). As noted, no cytotoxicity was observed for any of the compounds examined. Thus, it was deemed that the CC50 values were greater than the highest concentration, 1 μM.
Table 2.
Antiviral activity (IC50) of inhibitors 3d and 3h against clinical HIV-1 isolates in PBMC cells (nM).
| Virus | IC50 (nM) valuesa |
|||||
|---|---|---|---|---|---|---|
| 3d | 3h | DRV | RTV | APV | IDV | |
| ERS104pre (wild-type) | 20 | 6 | 3.5 | 34 | 33 | 26 |
| MDR/TM | 220 (11) | 64 (10) | 4 (1) | >1000 (>29) | 290 (9) | >1000 (>38) |
| MDR/MM | 250 (13) | 110 (5) | 17 (5) | >1000 (>29) | 300 (9) | >1000 (>38) |
| MDR/JSL | 500 (25) | 330 (55) | 26 (7) | >1000 (>29) | 430 (13) | >1000 (>38) |
| MDR/B | 340 (17) | 230 (38) | 26 (7) | >1000 (>29) | 320 (10) | >1000 (>38) |
| MDR/C | 210 (11) | 160 (27) | 7 (2) | >1000 (>29) | 230 (7) | >1000 (>38) |
| MDR/G | 360 (18) | 300 (50) | 7 (2) | >1000 (>29) | 340 (10) | 290 (11) |
| MDR/A | 20 (1) | 13 (2) | 3 (1) | >1000 (>29) | 100 (3) | >1000 (>38) |
Amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database include L63P in HIV-1ERS104pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1MDR-B; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90M in HIV-1MDR-G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, I93L in HIV-1MDR-TM; L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, and V82A in HIV-1MDR-JSL; and L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, and Q92K in HIV-1MDR-MM. HIV-1ERS104pre served as a source of wild-type HIV-1. The IC50 values were determined by employing PHA-PBMC (phytohemaglutinin-activated peripheral blood mononuclear cells) as target cells and the inhibition of p24Gag protein production as the endpoint. All values were determined in triplicate. DRV (Darunavir), SQV (Saquinavir), APV (Amprenavir), IDV (Indinavir).
X-ray crystallography
The mode of binding of the inhibitor was determined by analyzing the atomic resolution crystal structure of HIV-1 protease with 3d. The crystal structure was solved and refined to an R factor of 14.9% at 1.00 Å resolution. The inhibitor binds with extensive interactions from P2 to P2' to the protease atoms, and most notably the favorable polar interactions including hydrogen bonds, weaker C–H…O and C–H…pi interactions, as shown in Figure 2. The central hydroxyl group forms hydrogen bonds to the side chain carboxylate oxygen atoms of the catalytic Asp25 and Asp25' residues. The inhibitor hydrogen bonds with protease main chain atoms of the amide of Asp29, the carbonyl oxygen of Gly 27, and the water-mediated interactions with the amides of Ile50 and 50' which conserved in the majority of protease complexes with inhibitors29 and substrate analogs30. Inhibitor 3d has retained the water-mediated interaction with the pi system of the P2' aromatic ring, which was observed for darunavir (1) and GRL-98065.31 The P2' methoxy group forms a hydrogen bond with the amide of Asp30'. Interestingly, the P2 group forms a water-mediated interaction with the amide of Gly48, similar to the interactions described for several peptide substrate analogs.30
Figure 2.

Stereoview of compound 3d bound to the active site of wild-type HIV-1 protease.
Conclusions
In summary, a series of novel and highly potent HIV-1 protease inhibitors were designed, synthesized and evaluated. The inhibitors incorporate a variety of flexible cyclic ethers/polyethers as the P2-ligand. Inhibitors containing small size 1,3-dioxacycloakanes have shown potent inhibitory properties. In particular, inhibitors 3d and 3h have shown remarkable enzyme inhibitory and antiviral potency. Inhibitors incorporating medium-size cyclic polyethers or polyethers containing a sulfone or amine functionality were significantly less potent in antiviral assays. For inhibitor 3d, we have carried out an optically active synthesis of (R)-1,3-dioxepan-5-ol using (S)-malic acid as the starting material. Syntheses of various cyclic ethers/polyethers were developed albeit in moderate yields. Inhibitor 3d has shown excellent activity against multi-PI-resistant variants compared to other FDA approved inhibitors. A protein-ligand X-ray structure of 3d-bound HIV-1 protease was determined at 1.0 Å resolution. One of the oxygens of the 1,3-dioxepane ligand is involved in hydrogen bonding with Asp29 and Asp30 NH's. The other oxygen is involved in a unique interaction with Gly-48 NH through a water molecule. One goal of our inhibitor design strategy is to combat drug resistant HIV. The design of inhibitor using the concept of maximizing `backbone binding' has led to the development of PIs characterized by high potency against both wild-type and multi-drug-resistant HIV-1 strains. Further design of inhibitors utilizing this molecular insight is in progress.
Experimental Section
General
All moisture sensitive reactions were carried out under nitrogen or argon atmosphere. Anhydrous solvents were obtained as follows: THF, diethyl ether and benzene, distilled from sodium and benzophenone; dichloromethane, pyridine, triethylamine, and diisopropylethylamine, distilled from CaH2. All other solvents were HPLC grade. Column chromatography was performed with Whatman 240–400 mesh silica gel under low pressure of 5–10 psi. TLC was carried out with E. Merck silica gel 60-F-254 plates. 1H and 13C NMR spectra were recorded on Varian Mercury 300 and Bruker Avance 400 and 500 spectrometers. Optical rotations were measured using a Perkin-Elmer 341 polarimeter.
(S)-2-(tert-Butyldiphenylsilyloxy)pentanedioic acid dimethyl ester (5)
A mixture of (2S)-hydroxypentadienoic acid dimethyl ester 418 (0.39 g, 2.2 mmol), imidazole (0.45 g, 6.6 mmol) and tert-butyldiphenylsilyl chloride (1.2 mL, 4.4 mmol) in dry DMF (4 mL) was stirred at 23 °C for 4 h. Subsequently, the reaction mixture was poured into water and the aqueous phase was extracted with Et2O, the organic extracts were washed with 1 N HCl and brine, dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography (1:10 EtOAc/Hex) to furnish 0.89 g (90%) of 5 as a colourless oil: [α]D20 = − 21.1 (c 9.0, CHCl3); 1H NMR (CDCl3) δ 7.69–7.62 (m, 4H), 7.46–7.33 (m, 6H), 4.31 (t, J = 5.4 Hz, 1H), 3.64 (s, 3H), 3.45 (s, 3H), 2.57–2.34 (m, 2H), 2.14–2.04 (m, 2H), 1.11 (s, 9H); 13C NMR (CDCl3) δ 173.4, 172.9, 135.9, 135.7, 133.0, 132.9, 129.9, 129.8, 127.7, 127.5, 71.4, 51.6, 51.5, 29.9, 28.9, 26.9, 19.4.
(S)-2-(tert-Butyldiphenylsilyloxy)pentan-1,5-diol (6a)
Compound 5 (0.8 g, 1.8 mmol) was dissolved in dry Et2O (8.5 mL) and the solution was cooled to 0 °C, afterward lithium borohydride (0.12 g, 5.4 mmol) and dry methanol (0.22 mL, 5.4 mmol) were sequentially added. The resulting suspension was stirred at 23 °C for 24 h, then a few drops of 6 N HCl were added and the salts were filtered off. The filtrate was concentrated under reduced pressure and the residue was purified by flash-chromatography (1:1 EtOAc/Hex) to furnish 0.61 g (93%) of 6a as a colourless oil: [α]D20 = − 15.6 (c 3.1, CHCl3); 1H NMR (CDCl3) δ 7.70–7.65 (m, 4H), 7.44–7.32 (m, 6H), 3.82–3.77 (m, 1H), 3.53–3.48 (m, 2H), 3.45–3.41 (m, 2H), 1.65ȃ1.47 (m, 4H), 1.05 (s, 9H); 13C NMR (CDCl3) δ 135.9, 135.7, 133.8, 133.7, 130.1, 129.8, 127.7, 127.6, 73.6, 65.7, 62.7, 29.7, 28.0, 27.0, 19.3.
(S)-1-(tert-Butyldiphenylsilyloxy)-3,5-dioxacyclooctane (7a)
To a mixture of 6a (0.55 g, 1.5 mmol) and paraformaldehyde (46 mg, 1.5 mmol) in EtOAc (30 mL), boron trifluoride etherate (195 μL, 1.5 mmol) was added and the resulting mixture was stirred at 23 °C for 4 h. The organic phase was washed with a saturated solution of NaHCO3, dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography (1:4 EtOAc/Hex) to afford 0.29 g (51%) of 7a as a colourless oil: [α]D20 = − 8.7 (c 1.9, CHCl3); 1H NMR (CDCl3) δ 7.67–7.63 (m, 4H), 7.45–7.34 (m, 6H), 4.69 (d, J = 6.2 Hz, 1H), 4.45 (d, J = 6.2 Hz, 1H), 4.03–3.95 (m, 1H), 3.70–3.61 (m, 1H), 3.59–3.48 (m, 3H), 1.93–1.80 (m, 1H), 1.77–1.61 (m, 2H), 1.47–1.34 (m, 1H), 1.12 (s, 9H); 13C NMR (CDCl3) δ 135.7, 134.2, 129.5, 127.5, 95.6, 72.2, 71.9, 69.0, 33.2, 27.0, 26.7, 19.2.
(S)-O-Benzyl-3,5-dioxacycloheptan-1-ol (7b)
Compound 6b20 (50 mg, 0.26 mmol) was reacted as described for compound 6a to afford 44 mg (82%) of 7b after chromatographic purification (1:9 EtOAc/Hex): [α]D20 = + 64.6 (c 1.2, CHCl3); 1H NMR (CDCl3) δ 7.35–7.26 (m, 5H), 4.81–4.77 (m, 2H), 4.58 (s, 2H), 3.95–3.73 (m, 3H), 3.73–3.62 (m, 2H), 1.98-1.91 (m, 2H); 13C NMR (CDCl3) δ 138.3, 128.3, 127.5, 126.2, 94.9, 75.8, 70.7, 68.8, 62.6, 35.0.
(S)-3,5-Dioxacyclooctan-1-ol (8a)
Compound 7a (0.27 g, 0.74 mmol) was dissolved in dry THF (5 mL) and TBAF (1.0 M solution in THF, 0.81 mL, 0.81 mmol) was added. The resulting mixture was stirred at 23 °C overnight, afterward a saturated solution of NaHCO3 was added, the solvent was removed and the aqueous phase was extracted with EtOAc. The organic extracts were dried and evaporated and the residue was purified by flash-chromatography (EtOAc) to afford 76 mg (77%) of 8a as a colourless oil: [α]D20 = − 12.6 (c 1.6, CHCl3); 1H NMR (CDCl3) δ 4.65 (d, J = 6.0 Hz, 1H), 4.57 (d, J = 6.0 Hz, 1H), 4.92–3.81 (m, 2H), 3.75–3.60 (m, 2H), 3.55 (dd, J = 3.4, 12.1 Hz, 1H), 2.96 (bs, 1H), 1.95–1.69 (m, 3H), 1.65–1.53 (m, 1H); 13C NMR (CDCl3) δ 94.9, 73.7, 69.3, 68.2, 30.2, 24.7.
(S)-3,5-Dioxacycloheptan-1-ol (8b)
To a solution of 7b (38 mg, 0.18 mmol) in EtOAc (3 mL), 10% Pd/C was added and the resulting suspension was stirred at 23 °C under a hydrogen atmosphere. After 12 h, the catalyst was filtered off, the filtrate was evaporated in vacuo and the residue (19 mg, 91%) was used in the next step without further purification: [α]D20 = + 12.9 (c 0.9, CHCl3); 1H NMR (CDCl3) δ 4.78–4.74 (m 2H), 3.93–3.91 (m, 1H), 3.81–3.75 (m, 4H), 2.51 (bs, 1H), 1.93–1.83 (m, 2H); 13C NMR (CDCl3) δ 94.4, 69.5, 68.4, 62.3, 37.8.
(R)-3,5-Dioxacyclooctan-1-ol (8c)
To a mixture of (S)-8a (46 mg, 0.35 mmol), p-nitrobenzoic acid (86 mg, 0.52 mmol), and triphenylphosphine (181 mg, 0.69 mmol), diisopropylazodicarboxylate (135 μL, 0.69 mmol) was added dropwise and the resulting mixture was stirred at 23 °C overnight. The solvent was removed under reduced pressure and the residue was purified by flash-chromatography (1:3 EtOAc/Hex). The resulting ester was dissolved in a 3:2:1 mixture of THF, methanol and water (4 mL) and LiOH·H2O (72 mg, 1.7 mmol) was added. The yellow mixture was stirred at 23 °C overnight and then the solvent was removed in vacuo, the residue was diluted with water and the aqueous phase was extracted with ether. The organic extracts were dried (Na2SO4) and the solvent evaporated. Purification of the residue by flash-chromatography (EtOAc) afforded 20 mg (44%) of (R)-8c as a colourless liquid. [α]D = + 12.1 (c 1.4, CHCl3). 1H and 13C NMR are consistent with those reported for the (S)-enantiomer 8a
(R)-3,5-Dioxacycloheptan-1-ol (8d)
The title compound was obtained from 8b as described for (S)-8c in 73% yield. Flash-chromatography was performed using a 1:1 mixture of EtOAc and CHCl3 as the eluant: [α]D20 = − 12.6 (c 1.3, CHCl3). 1H and 13C NMR are consistent with those reported for the (S)-enantiomer 8b
(R)-1-(tert-Butyldimethylsilyloxy)-2-[(2-methoxyethoxy)methoxy]pent-4-ene (10)
To a mixture of 9 (350 mg, 1.6 mmol) and diisopropylethylamine (1.2 mL, 7.2 mmol) in CH2Cl2 (8 mL), cooled to 0 °C, MEM-Cl (550 μL, 4.8 mmol) was added and the resulting mixture was stirred at 23 °C for 56 h. The organic phase was washed with 0.1 N HCl, brine and dried (Na2SO4). The solvent was removed and the residue was purified by flash-chromatography (1:10 EtOAc/Hex) to afford 440 mg (90%) of 10 as a colourless oil: [α]D20 = + 12.0 (c 1.1, CHCl3); 1H NMR (CDCl3) δ 5.88–5.74 (m, 1H), 5.11–5.01 (m, 2H), 4.82 (d, J = 6.9 Hz, 1H), 4.74 (d, J = 6.9 Hz, 1H), 3.76–3.63 (m, 3H), 3.60–3.51 (m, 4H), 3.37 (s, 3H), 2.38–2.19 (m, 2H), 0.86 (s, 9H), 0.02 (s, 6H); 13C NMR (CDCl3) δ 134.6, 117.0, 94.8, 77.4, 71.6, 66.7, 65.0, 58.9, 36.0, 25.7, 18.2, –5.5.
(R)-1-Allyloxy-2-[(2-methoxyethoxy)methoxy]pent-4-ene (11)
A mixture of 10 (440 mg, 1.4 mmol) and TBAF (1.0 M solution in THF, 4.7 mL, 4.7 mmol) in THF (3 mL) was stirred at 23 °C for 3 h, afterward a saturated solution of NaHCO3 was added, the solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography to afford 237 mg (87%) of (R)-2-[(2-methoxyethoxy)methoxy]pent-4-en-1-ol as a colourless oil: [α]D20 = − 55.0 (c 1.3, CHCl3); 1H NMR (CDCl3) δ 5.85–5.71 (m, 1H), 5.11–5.02 (m, 2H), 4.81 (d, J = 7.5 Hz, 1H), 4.75 (d, J = 7.5 Hz, 1H), 3.87–3.80 (m, 1H), 3.71–3.61 (m, 3H), 3.59–3.46 (m, 3H), 3.37 (s, 3H), 3.22 (bs, 1H), 2.36–2.19 (m, 2H); 13C NMR (CDCl3) δ 134.1, 117.3, 95.4, 81.0, 71.5, 67.3, 64.8, 58.9, 36.2. To a mixture of the above compound (240 mg, 1.25 mmol), allyl bromide (225 μL, 1.9 mmol) and a catalytic amount of TBAI in THF (12 mL), at 0 °C, sodium hydride (60% dispersion in oil, 102 mg, 2.5 mmol) was added in small portions. After 30 min, the reaction mixture was allowed to warm to 23 °C and was stirred at the same temperature for 18 h. Subsequently, the reaction was quenched with a saturated solution of NH4Cl, the organic solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was evaporated. The residue was purified by flash-chromatography (10:1 CHCl3/EtOAc) to afford 229 mg (80%) of 11 as a colourless oil. [α]D20 = − 5.2 (c 3.1, CHCl3); 1H NMR (CDCl3) δ 5.87–5.79 (m, 2H), 5.34 (dd, J = 1.3, 19.1 Hz, 1H), 5.16–5.03 (m, 3H), 4.81 (d, J = 7.0 Hz, 1H), 4.77 (d, J = 7.0 Hz, 1H), 3.98–3.97 (m, 2H), 3.84–3.81 (m, 1H), 3.72 (t, J = 5.0 Hz, 2H), 3.54 (t, J = 5.0 Hz, 2H), 3.46–3.44 (m, 2H), 3.38 (s, 3H), 2.35–2.31 (m, 2H); 13C NMR (CDCl3) δ 134.6, 134.3, 117.3, 116.7, 94.6, 75.3, 72.1, 71.9, 71.6, 66.7, 58.9, 36.3.
(R,Z)-3-[(2-Methoxyethoxy)methoxy]-2,3,4,7-tetrahydrooxepine (12)
A mixture of 11 (100 mg, 0.43 mmol) and 2nd generation Grubbs catalyst (18 mg, 0.02 mmol) in CH2Cl2 (10 mL) was heated to 45 °C for 1 h. After this time, the solvent was removed and the residue was purified by flash-chromatography (5:1 CHCl3/EtOAc) to afford 83 mg (94%) of 12 as a colourless oil: 1H NMR (CDCl3) δ 5.87–5.66 (m, 2H), 4.77–4.18 (m, 2H), 4.18–4.14 (m, 2H), 4.01–3.89 (m, 2H), 3.75–3.68 (m, 3H), 3.56–3.53 (m, 2H), 3.38 (s, 3H), 2.54–2.51 (m, 2H); 13C NMR (CDCl3) δ 130.6, 125.9, 94.3, 75.6, 75.1, 71.6, 70.3, 66.8, 58.9, 31.8.
(R)-Oxepan-3-ol (8e)
A mixture of 12 (90 mg, 0.44 mmol) and a catalytic amount of 10% Pd/C in EtOAc (3 mL) was stirred at 23 °C under a hydrogen atmosphere for 3 h. After this time, the catalyst was filtered off through a pad of Celite and the filtrate was concentrated under reduced pressure to afford (R)-3-[(2-methoxyethoxy)methoxy]oxepane (83 mg, 92%) as a colourless oil: 1H NMR (CDCl3) δ 4.70 (d, J = 7.2 Hz, 1H), 4.67 (d, J = 7.2 Hz, 1H), 3.83–3.58 (m, 7H), 3.50 (t, J = 4.6 Hz, 2H), 3.34 (s, 3H), 1.72–1.67 (m, 1H), 1.46–1.44 (m, 4H), 1.22–1.19 (m, 1H); 13C NMR (CDCl3) δ 93.9, 76.5, 73.7, 71.8, 71.6, 66.7, 58.8, 32.6, 30.7, 20.9. A mixture of the above compound (50 mg, 0.24 mmol) and 6 N HCl (0.5 mL) in THF (2 mL) was stirred at 23 °C for 16 h. The solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were washed with a saturated solution of NaHCO3, dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography (1:4 EtOAc/CHCl3) to afford 8e (24 mg, 84%) as a colourless oil: [α]D20 = − 4.2 (c 0.8, CHCl3); 1H NMR (CDCl3) δ 3.87–3.85 (m, 1H), 3.76–3.62 (m, 4H), 2.37 (bs, 1H), 1.78–1.65 (m, 5H), 1.54–1.52 (m, 1H); 13C NMR (CDCl3) δ 73.2, 70.7, 70.4, 36.4, 30.0, 20.2.
(R)-3-(tert-Butyldimethylsilyloxy)-5-(allyloxy)pent-1-ene (14)
A mixture of 13 (50 mg, 0.23 mmol), allyl bromide (30 μL, 0.35 mmol) and a catalytic amount of TBAI was cooled to 0 °C and sodium hydride (60% in mineral oil, 11 mg, 0.28 mmol) was added. The resulting mixture was allowed to warm to 23 °C and stirred for 18 h. The reaction was quenched by adding a saturated solution of NH4Cl, the solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography (1:20 EtOAc/Hex) to afford 57 mg (97%) of 14 as a colorless oil: 1H NMR (CDCl3) δ 5.96–5.87 (m, 1H), 5.85–5.78 (m, 1H), 5.29–5.24 (m, 1H), 5.19–5.13 (m, 2H), 5.04–5.00 (m, 1H), 4.31–4.26 (m, 1H), 3.96–3.94 (m, 2H), 3.55–3.42 (m, 2H), 1.84–1.67 (m, 2H), 0.90 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3) δ 141.5, 134.9, 116.6, 113.6, 71.8, 70.6, 66.5, 38.0, 25.8, 18.1, −4.5, −5.1.
(R,Z)-4-(tert-Butyldimethysilyloxy)-2,3,4,7-tetrahydrooxepine (15)
The title compound was obtained from 14 as described for 12 in 80% yield. Flash-chromatography was performed using a 1:10 mixture of EtOAc and Hex as the eluant: 1H NMR (CDCl3) δ 5.79–5.75 (m, 1H), 5.63–5.60 (m, 1H), 4.64–4.62 (m, 1H), 4.14–4.12 (m, 2H), 3.91–3.85 (m, 1H), 3.80–3.74 (m, 1H), 2.11–2.05 (m, 1H), 1.96–1.91 (m, 1H), 0.90 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (CDCl3) δ 138.4, 127.8, 69.9, 68.2, 67.4, 38.8, 25.8, 18.3, −4.8.
(S)-Oxepan-4-ol (8f)
Hydrogenolysis of 15 was carried out as described for 8e to afford (S)-4-(tert-butyldimethylsilyloxy)oxepane in 95% yield as a colourless oil: 1H NMR (CDCl3) δ 4.03–3.96 (m, 1H), 3.79–3.57 (m, 4H), 1.98–1.69 (m, 5H), 1.64–1.51 (m, 1H), 0.88 (s, 9H), 0.044 (s, 3H), 0.038 (s, 3H); 13C NMR (CDCl3) δ 70.2, 69.4, 64.2, 40.1, 34.5, 25.7, 23.7, 18.0, −4.9. Deprotection of the above compound was performed as described for compound 11 and afforded the title compound in 75% yield as a colorless oil: 1H NMR (CDCl3) δ 4.03–3.98 (m, 1H), 3.82–3.59 (m, 4H), 2.02–1.98 (m, 1H), 1.89–1.80 (m, 4H), 1.66–1.64 (m, 1H); 13C NMR (CDCl3) δ 70.5, 69.6, 64.7, 38.9, 34.9, 24.3.
2-(Benzyloxy)propane-1,3-diol (17)
To a solution of 16 (2.5 g, 13.8 mmol) in dry THF (20 mL), cooled to 0 °C, NaH (60% in mineral oil, 0.56 g, 14 mmol) was added portionwise. After 30 min, tetra-n-butylammonium iodide (51 mg, 0.14 mmol) and a solution of benzyl bromide (1.65 mL, 13.9 mmol) in THF (5 mL) were added. The reaction mixture was stirred at 23 °C for 3 h, afterward it was poured into ice. The organic solvent was removed in vacuo and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was removed. The crude 5-(benzyloxy)-2-phenyl-1,3-dioxane thus obtained was dissolved in a 1:1 mixture of THF and H2O (60 mL) and to the resulting solution, 6 N HCl was slowly added to the resulting solution. After stirring at 23 °C, the reaction mixture was brought to pH 8 by addition of a saturated solution of NaHCO3, the solvent was removed and the aqueous phase was extracted with diethyl ether. The organic extracts were dried and evaporated and the residue was purified by flash-column chromatography (EtOAc 2: Hex 1) to afford the title compound as a colourless oil in quantitative yield. Physical and spectroscopic data are consistent with those reported in the literature.32
1,3-Dioxan-5-ol (8h)
To a mixture of 17 (100 mg, 0.55 mmol) and paraformaldehyde (17 mg, 0.55 mmol) in EtOAc (10 mL), boron trifluoride etherate (70 μL, 0.55 mmol) was added and the reaction mixture was stirred at 23 °C for 4 h. The organic phase was washed with a saturated solution of NaHCO3, dried and the solvent was removed. The residue was purified by flash-chromatography eluting with a 1:4 mixture of EtOAc and hexanes to afford 84 mg (78%) of O-benzyl-1,3-dioxan-5-ol as a colourless oil. The above compound was dissolved in EtOAc (3 mL), Pd/C was added and the resulting suspension was stirred at rt under a hydrogen atmosphere. After 12 h, the catalyst was filtered off, the filtrate was evaporated in vacuo and the residue (39 mg, 100%) was used in the next step without further purification: 1H NMR (CDCl3) δ 4.93 (d, J = 6.3 Hz, 1H), 4.76 (d, J = 6.3 Hz, 1H), 3.94–3.84 (m, 4H), 3.64–3.61 (m, 1H), 2.78 (bs, 1H). 13C NMR (CDCl3) δ 94.0, 71.7, 64.1.
O-Benzyl-3,6,9-trioxacyclodecan-1-ol (18)
To a refluxing suspension of sodium hydride (60% in mineral oil, pre-washed with hexane, 84 mg, 2.1 mmol) in dry THF (5 mL), a solution of 17 (182 mg, 1.0 mmol) and di(ethyleneglycol)dimethanesulfonate (260 mg, 1.0 mmol) in dry THF (5 mL) was added dropwise. The resulting mixture was heated under reflux for 20 h, afterward was cooled to 23 °C and H2O (2 mL) was added. The solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were washed several times with water, dried (Na2SO4) and evaporated. The residue was purified by flash-chromatography (2:3 CH2Cl2/EtOAc) to afford 49 mg (19%) of 18 as a colourless oil: 1H NMR (CDCl3) δ 7.36–7.26 (m, 5H), 4.66 (s, 2H), 3.75–3.57 (m, 13H); MS (ESI) m/z 275 [M+Na]+.
O-Benzyl-3,6,9,12-tetraoxacyclotridecan-1-ol (19)
Compound 19 was obtained as described for 18 starting from 17 and tri(ethyleneglycol)dimethanesulfonate in 29% yield. 1H NMR (CDCl3) δ 7.39–7.26 (m, 5H), 4.72 (s, 2H), 3.83–3.58 (m, 17 H); MS (ESI) m/z 319 [M+Na]+.
3,6,9-Trioxacyclodecan-1-ol (8i)
A mixture of 18 (34 mg, 0.13 mmol) and a catalytic amount of 10% Pd/C in methanol (2 mL) was stirred at 23 °C under a hydrogen atmosphere. After 18 h the catalyst was filtered off and the filtrate was evaporated to afford 22 mg (99%) of 8i as a colourless oil: 1H NMR (CDCl3) δ 3.74–3.53 (m, 13H), 2.73 (bs, 1H).
3,6,9,12-Tetraoxacyclotridecan-1-ol (8j)
Starting from 19, compound 8j was obtained as described for 8i in quantitative yield: 1H NMR (CDCl3) δ 3.81–3.60 (m, 17 H), 2.95 (bs, 1H).
3,6,8,11-Tetraoxa-1-cyclododecanol (8k)
To a mixture of 2024 (78 mg, 0.29 mmol) and paraformaldehyde (8.7 mg, 0.29 mmol) in EtOAc (4 mL), boron trifluoride etherate (37 μL, 0.29 mmol) was added and the resulting mixture was stirred at 23 °C for 2 h. Subsequently, a saturated solution of NaHCO3 was added and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried (Na2SO4) and the solvent was removed in vacuo. The residue was purified by flash-chromatography to afford 31 mg (37%) of O-benzyl-3,6,8,11-tetraoxacyclododecan-1-ol as a colourless oil: 1H NMR (CDCl3) δ 7.35–7.27 (m, 5H), 4.67 (s, 2H), 3.88 (s, 2H), 3.86–3.81 (m, 2H), 3.77–3.61 (m, 11H); 13C NMR (CDCl3) δ 133.6, 128.3, 127.7, 126.2, 94.6, 75.8, 71.5, 69.6, 65.3, 64.6; MS (ESI) m/z 305 [M+Na]+. A mixture of the above compound and a catalytic amount of 10% Pd/C in EtOAc (2 mL) was stirred at 23 °C under a hydrogen atmosphere. After 18 h the catalyst was filtered off and the filtrate was evaporated to afford 21 mg (99%) of 8k as a colorless oil: 1H NMR (CDCl3) δ 4.67 (s, 2H), 3.85–3.63 (m, 11H), 3.54 (dd, J = 6.4, 8.2 Hz, 2H), 2.22 (d, J = 8.7 Hz, 1H).
O-Benzyl-3,9-dioxa-6-thiacyclodecan-1-ol 6,6-dioxide (22)
A solution of lithium sulfide (11 mg, 0.23 mmol) in water (0.3 mL) was added dropwise within 30 min to a solution of 2124 (60 mg, 0.15 mmol) in refluxing ethanol (15 mL). The resulting mixture was heated under reflux for 3 h and then was cooled to 23 °C. The solvent was removed and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was removed. Flash-chromatography of the residue (1:4 EtOAc/hexanes) afforded 16 mg (38%) of O-benzyl-3,9-dioxa-6-thiacyclodecan-1-ol as a colorless oil: 1H NMR (CDCl3) δ 7.36–7.27 (m, 5H), 4.59 (s, 2H), 3.90–3.85 (m, 2H), 3.82–3.48 (m, 7H), 2.91–2.74 (m, 4H); 13C NMR (CDCl3) δ 133.5, 128.3, 127.7, 126.0, 75.9, 71.9, 71.6, 68.0, 33.4. MS (ESI) m/z 291 [M+Na]+, 286 [M+H+NH3]+ 269 [M+H]+. To a solution of the above compound (11 mg, 0.040 mmol) in CH2Cl2 (2 mL), cooled to 0 °C, m-chloroperbenzoic acid (77%, 22 mg, 0.09 mmol) was added in small portions. After 18 h, a 1% solution of sodium bisulfite was added, the layers were separated and the organic phase was washed with a saturated solution of NaHCO3. The organic extracts were dried (Na2SO4) and evaporated. The residue was purified by flash-chromatography (1:4 EtOAc/CHCl3) to afford 11 mg (93%) of 22 as a brown oil: 1H NMR (CDCl3) δ 7.37–7.29 (m, 5H), 4.57 (s, 2H), 4.01–3.96 (m, 4H), 3.77–3.72 (m, 1H), 3.66–3.60 (m, 4H), 3.40–3.38 (m, 2H), 3.34–3.23 (m, 2H); 13C NMR (CDCl3) δ 133.4, 128.4, 127.9, 127.7, 74.7, 71.8, 66.4, 64.6, 52.4.
3,9-Dioxa-6-thiacyclodecan-1-ol 6,6-dioxide (8l)
A mixture of 22 (25 mg, 0.083 mmol) and a catalytic amount of 10% Pd/C in EtOAc (3 mL) was stirred at 23 °C under a hydrogen atmosphere. After 48 h the catalyst was filtered off and the filtrate was evaporated to afford 16 mg (92%) of 8l as a colorless oil: 1H NMR (CDCl3) δ 4.08–3.95 (m, 4H), 3.67 (dd, J = 4.2, 9.9 Hz, 2H), 3.61–3.59 (m, 1H), 3.51 (dd, J = 5.7, 9.9 Hz, 2H), 3.38–3.23 (m, 4H); 13C NMR (CDCl3) δ 69.0, 68.3, 64.5, 52.6.
O,6-Dibenzyl-3,9-dioxa-6-azocyclodecan-1-ol (23)
A mixture of 2124 (150 mg, 0.37 mmol), benzylamine (41 μL, 0.37 mmol), lithium perchlorate (340 mg, 3.7 mmol) and sodium carbonate (200 mg, 1.9 mmol) in acetonitrile (7.5 mL) was heated under reflux for 48 h. After cooling to 23 °C, the solvent was removed, the residue was suspended in CHCl3 and the organic phase was washed with water and dried (Na2SO4). Flash-chromatography of the residue (2:1 EtOAc/CHCl3) afforded 31 mg (24%) of 23 as a colourless oil: 1H NMR (CDCl3) δ 7.36–7.20 (m, 10H), 4.59 (s, 2H), 3.87–3.78 (m, 4H), 3.69 (s, 2H), 3.67–3.49 (m, 5H), 2.91–2.72 (m, 4H); MS (ESI) m/z 342 [M+1]+.
N-(tert-Butoxycarbonyl)-3,9-dioxa-6-azocyclodecan-1-ol (24)
A mixture of 23 (40 mg, 0.12 mmol), Boc2O (26 mg, 0.12 mmol) and a catalytic amount of 10% Pd/C in EtOAc (3 mL) was stirred at 23 °C under a hydrogen atmosphere. After 18 h the catalyst was filtered off and the filtrate was evaporated to afford 26 mg (95%) of 24 as a colorless oil: 1H NMR (CDCl3) δ 3.83–3.70 (m, 7H), 3.65–3.59 (m, 2H), 3.49–3.29 (m, 4H), 1.75 (bs, 1H), 1.46 (s, 9H); 13C NMR (CDCl3) δ 155.7, 79.8, 71.4, 71.0, 70.3, 69.9, 50.5, 50.2, 28.5.
(S)-1-(4-Nitrophenoxycarbonyloxy)-3,5-dioxacyclooctane (25a)
To a solution of 8a (15 mg, 0.11 mmol) and N-methylmorpholine (38 μL, 0.34 mmol) in dry THF (3 mL), p-nitrophenylchloroformate (70 mg, 0.28 mmol) was added and the resulting mixture was stirred at 23 °C for 1 h. To the reaction mixture was added water, the solvent was removed under reduced pressure and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent was removed. The residue was purified by flash-chromatography (1:4 EtOAc CHCl3) to afford 28 mg (81%) of 25a as a pale yellow solid. 1H NMR (CDCl3) δ 8.27 (d, J = 9.3 Hz, 2H), 7.38 (d, J = 9.3 Hz, 2H), 5.09–5.01 (m, 1H), 4.72–4.66 (m, 2H), 3.94–3.82 (m, 3H), 3.64–3.56 (m, 1H), 2.18–2.04 (m, 1H), 2.03–1.93 (m, 2H), 1.90–1.71 (m, 1H).
(S)-1-(4-Nitrophenoxycarbonyloxy)-3,5-dioxacycloheptane (25b)
The title compound was obtained from (S)-8b as described for 25a in 72% yield. Flash-chromatography was performed using a 1:5 mixture of EtOAc and CHCl3 as the eluant. 1H NMR (CDCl3) δ 8.28 (d, J = 9.3 Hz, 2H), 7.40 (d, J = 9.3 Hz, 2H), 5.00–4.98 (m, 1H), 4.84 (d, J = 4.5 Hz, 1H), 4.79 (d, J = 4.5 Hz, 1H), 4.11 (dd, J = 4.7, 13.1 Hz, 1H), 3.99–3.90 (m, 2H), 3.85–3.78 (m, 1H), 2.19–2.04 (m, 2H).
(R)-1-(4-Nitrophenoxycarbonyloxy)-3,5-dioxacyclooctane (25c)
The title compound was obtained from (R)-8c as described for 25a in 87% yield after flash-chromatography (1:4 EtOAc/CHCl3). 1H NMR data are consistent with those reported for the (S)-enantiomer 25a
(R)-1-(4-Nitrophenoxycarbonyloxy)-3,5-dioxacycloheptane (25d)
The title compound was obtained from (R)-8d as described for 25a in 70% yield after flash-chromatography (1:5 EtOAc/CHCl3). 1H data are consistent with those reported for the (S)-enantiomer 25b.
(R)-3-(4-Nitrophenoxycarbonyloxy)oxepane (25e)
The title compound was obtained from 8e as described for 25a in 86% yield. Flash-chromatography was performed using a 1:20 mixture of EtOAc and CHCl3 as the eluant; 1H NMR (CDCl3) δ 8.26 (d, J = 9.3 Hz, 2H), 7.38 (d, J = 9.3 Hz, 2H), 5.02–4.95 (m, 1H), 3.98–3.83 (m, 3H), 3.71–3.63 (m, 1H), 2.15–1.74 (m, 5H), 1.65–1.53 (m, 1H).
(S)-4-(4-Nitrophenoxycarbonyloxy)oxepane (25f)
The title compound was obtained from 8f as described for 25a in 77% yield. Flash-chromatography was performed using a 1:20 mixture of EtOAc and CHCl3 as the eluant; 1H NMR (CDCl3) δ 8.27 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.8 Hz, 2H), 5.05–5.01 (m, 1H), 3.84–3.62 (m, 4H), 2.18–1.86 (m, 5H), 1.78–1.63 (m, 1H).
1-(4-Nitrophenoxycarbonyloxy)cycloheptane (25g)
The title compound was obtained from commercially available cycloheptanol as described for 25a in 89% yield. Flash-chromatography was performed using a 1:10 mixture of EtOAc and CHCl3 as the eluant. 1H NMR (CDCl3) δ 8.26 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 4.96–4.89 (m, 1H), 2.08–2.02 (m, 2H), 1.86–1.78 (m, 2H), 1.71 (m, 2H), 1.59 (m, 4H), 1.40–1.36 (m, 2H).
5-(4-Nitrophenoxycarbonyloxy)-1,3-dioxane (25h)
The title compound was obtained from 8h as described for 25a in 72% yield. Flash-chromatography was performed using a 1:4 mixture of EtOAc and CHCl3 as the eluant: 1H NMR (CDCl3) δ 8.30 (d, J = 8.7 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 5.03 (d, J = 6.3 Hz, 1H), 4.87 (d, J = 6.3 Hz, 1H), 4.71 (t, J = 2.8 Hz, 1H), 4.19–4.06 (m, 4H).
3,6,9-Trioxa-1-cyclodecanol succinimidylcarbonate (25i)
To a solution of 8i (18 mg, 0.11 mmol) in dry acetonitrile (1 mL), N,N'-disuccimidyl carbonate (43 mg, 0.17 mmol) and triethylamine (32 μL, 0.23 mmol) were added and the resulting mixture was stirred at 23 °C. After 8 h the solvent was removed, the residue was taken-up in a saturated solution of NaHCO3 and the aqueous phase was extracted with EtOAc. The organic extracts were dried (Na2SO4) and the solvent was removed in vacuo. Purification of the residue (10:1 EtOAc/MeOH) afforded 17b (13 mg) in 37% yield: 1H NMR (CDCl3) δ 5.12–5.03 (m, 1H), 3.96–3.65 (m, 12H), 2.81 (s, 4H).
12-(4-Nitrophenoxycarbonyloxy)-1,4,7,10-tetraoxacyclotridecane (25j)
The title compound was obtained from 8j as described for 25a in 70% yield after flash-chromatography (EtOAc): 1H NMR (CDCl3) δ 8.27 (d, J = 9.3 Hz, 2H), 7.39 (d, J = 9.3 Hz, 2H), 5.15–5.08 (m, 1H), 3.92 (dd, J = 6.3, 10.2 Hz, 2H), 3.82 (dd, J = 4.5, 10.2 Hz, 2H), 3.74–3.60 (m, 12H).
9-(4-Nitrophenoxycarbonyloxy)-1,7-dioxa-4-thiacyclodecane 4,4-dioxide (25k)
The title compound was obtained from 8k as described for 25a in 73% yield after flash-chromatography (1:4 EtOAc/CHCl3): 1H NMR (CDCl3) δ 8.28 (d, J = 9.0 Hz, 2H), 7.37 (d, J = 9.0 Hz, 2H), 5.10–5.03 (m, 1H), 4.13–4.06 (m, 4H), 3.83–3.73 (m, 4H), 3.43–3.22 (m, 4H).
11-(4-Nitrophenoxycarbonyloxy)-1,4,6,9-tetraoxacyclododecane (25l)
The title compound was obtained from 8l as described for 25a in 67% yield after flash-chromatography (EtOAc): 1H NMR (CDCl3) δ 8.27 (d, J = 8.7 Hz, 2H), 7.38 (d, J = 8.7 Hz, 2H), 5.01–4.95 (m, 1H), 4.70 (s, 2H), 3.91–3.76 (m, 12H).
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (1S)-3,5-dioxacyclooctan-1-yl ester (3a)
A solution of 27 (25 mg, 0.05 mmol) in a mixture of 30% trifluoracetic acid in CH2Cl2 (5 mL) was stirred at 23 °C for 40 min and then the solvent was removed under reduced pressure. Compound 28 thus obtained was dissolved in CH2Cl2 (4 mL) and a solution of 25a (16 mg, 0.05 mmol) in THF (2 mL) were added followed by diisopropylethylamine. After 48 h the organic phase was washed with water, dried (Na2SO4) and evaporated. The residue was purified by flash-chromatography eluting with a 1:4 mixture of EtOAc and hexane to afford 3a in 63% yield after flash-chromatography (1:4 EtOAc/CHCl3) as a foam: [α]D20 = + 8.6 (c 1.1, CHCl3); 1 H NMR (CDCl3) δ 7.70 (d, J = 9.0 Hz, 2H), 7.31–7.21 (m, 5H), 6.97 (d, J = 9.0 Hz, 2H), 4.83–4.78 (m, 2H), 4.65–4.59 (m, 2H), 3.87 (s, 3H), 3.83–3.81 (m, 3H), 3.68 (dd, J = 4.9, 12.1 Hz, 1H), 3.55–3.48 (m, 2H), 3.14–2.90 (m, 5H), 2.78 (dd, J = 6.8, 12.6 Hz, 1H), 1.85–1.80 (m, 5H), 0.90 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 163.0, 153.4, 137.6, 129.8, 129.6, 129.5, 128.4, 126.5, 114.3, 95.7, 73.9, 72.6, 69.2, 68.6, 58.7, 55.6, 55.0, 53.7, 35.4, 29.2, 27.2, 26.1, 20.1, 29.8. HRMS-ESI (m/z): (M + Na)+ calcd for C28H40N2NaO8S, 587.2403; found, 587.2380.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (1S)-3,5-Dioxacycloheptan-1-yl ester (3b)
The title compound was obtained from 27 and 25b as described for 3a in 69% yield after flash-chromatography (1:4 EtOAc/CHCl3) as an amorphous solid: [α]D20 = + 10.5 (c 1.2, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 8.7 Hz, 2H), 7.31–7.19 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 4.93 (d, J = 8.4 Hz, 1H), 4.77–4.71 (m, 3H), 3.87 (s, 3H), 3.81–3.69 (m, 6H), 3.09–2.90 (m, 5H), 2.77 (dd, J = 6.9, 13.2 Hz, 1H), 1.98–1.95 (m, 1H), 1.85–1.76 (m, 2H), 0.90 (d, J = 6.9 Hz, 3H), 0.85 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 162.9, 155.5, 137.5, 129.7, 129.5, 129.4, 128.4, 126.5, 114.3, 94.9, 72.5, 71.9, 68.8, 62.3, 58.9, 55.6, 55.2, 53.7, 35.3, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C27H38N2NaO8S, 573.2247; found, 573.2260.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (1R)-3,5-dioxacyclooctan-1-yl ester (3c)
The title compound was obtained from 27 and 25c as described for 3a in 50% yield after flash-chromatography (1:4 EtOAc/CHCl3) as an amorphous solid [α]D20 = + 9.8 (c 1.1, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 8.7 Hz, 2H), 7.31–7.21 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 4.80–4.79 (m, 2H), 4.65–4.61 (m, 2H), 3.87 (s, 3H), 3.82–3.80 (m, 2H), 3.71–3.62 (m, 2H), 3.56–3.48 (m, 2H), 3.12–2.85 (m, 5H), 2.77 (dd, J = 6.3, 13.2 Hz, 1H), 1.83–1.74 (m, 4H), 1.71–1.66 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H). HRMS-ESI (m/z): (M + Na)+ calcd for C28H40N2NaO8S, 587.2403; found, 587.2405.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (1R)-3,5-dioxacycloheptan-1-yl ester (3d)
The title compound was obtained from 27 and 25d as described for 3a in 59% yield after flash-chromatography (1:4 EtOAc/CHCl3) as a foam: [α]D20 = + 15.9 (c 0.6, CHCl3); 1H NMR (CDCl3) δ 7.71 (d, J = 9.0 Hz, 2H), 7.30–7.18 (m, 5H), 6.98 (d, J = 9.0 Hz, 2H), 4.88 (d, J = 8.7 Hz, 1H), 4.77–4.71 (m, 3H), 3.87 (s, 3H), 3.81–3.61 (m, 6H), 3.18–3.07 (m, 2H), 3.04–2.92 (m, 2H), 2.86–2.74 (m, 2H), 1.90–1.77 (m, 3H), 0.92 (d, J = 6.3 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 162.8, 155.5, 137.6, 129.7, 129.5, 129.4, 128.4, 126.4, 114.3, 94.8, 72.6, 71.9, 68.6, 62.3, 58.8, 55.6, 55.1, 53.8, 35.8, 35.2, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C27H38N2NaO8S, 573.2247; found, 573.2254.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (R)-oxepan-3-yl ester (3e)
The title compound was obtained from 27 and 25e as described for 3a in 72% yield after flash-chromatography (1:2 EtOAc/Hex) as an amorphous solid: 1H NMR (CDCl3) δ 7.70 (d, 8.8 Hz, 2H), 7.30–7.19 (m, 5H), 6.97 (d, J = 8.8 Hz, 2H), 4.81 (d, J = 8.2 Hz, 1H), 4.77–4.74 (m, 1H), 3.87 (s, 3H), 3.81 (m, 3H), 3.70–3.69 (m, 2H), 3.61–3.57 (m, 1H), 3.12 (dd, J = 8.2, 14.7 Hz, 1H), 3.05–3.84 (m, 4H), 2.77 (dd, J = 6.6, 13.2 Hz, 1H), 1.86–1.60 (m, 6H), 1.49–1.41 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.86 (d, J = 6.7 Hz, 3H); 13C NMR (CDCl3) δ 162.9, 155.8, 137.5, 129.6, 129.5, 129.4, 128.4, 126.4, 114.2, 74.5, 73.6, 72.5, 72.4, 58.7, 55.5, 54.8, 53.7, 35.6, 31.9, 30.9, 27.1, 21.0, 20.0, 19.8. HRMS-ESI (m/z): (M + Na)+ calcd for C28H40N2NaO7S, 571.2454; found, 571.2458.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid (S)-oxepan-4-yl ester (3f)
The title compound was obtained from 27 and 25f as described for 3a in 68% yield after flash-chromatography (1:2 EtOAc/Hex) as an amorphous solid: 1H NMR (CDCl3) δ 7.71 (d, J = 8.8 Hz, 2H), 7.29–7.21 (m, 5H), 6.98 (d, J = 8.8 Hz, 2H), 4.78–4.76 (m, 2H), 3.94–3.81 (m, 5H), 3.71–3.60 (m, 3H), 3.56–3.50 (m, 1H), 3.12 (dd, J = 8.0, 15.2 Hz, 1H), 3.04–2.86 (m, 4H), 2.79 (dd, J = 6.4, 13.1 Hz, 1H), 1.94–1.64 (m, 7H), 0.91 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H). HRMS-ESI (m/z): (M + Na)+ calcd for C28H40N2NaO7S, 571.2454; found, 571.2452.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid Cycloheptanyl ester (3g)
The title compound was obtained from 27 and 25g as described for 3a in 84% yield after flash-chromatography (1:6 EtOAc/CHCl3) as an amorphous solid: [α]D20 = + 16.0 (c 0.9, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 8.7 Hz, 2H), 7.31–7.22 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 4.69–4.68 (m, 2H), 3.87 (s, 3H), 3.82–3.78 (m, 2H), 3.05–2.77 (m, 6H), 1.83–1.73 (m, 4H), 1.60–1.45 (m, 8H), 1.22–1.20 (m, 1H), 0.90 (d, J = 6.3 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H). HRMS-ESI (m/z): (M + Na)+ calcd for C29H42N2NaO6S, 569.2661; found, 569.2663.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 1,3-dioxan-5-yl ester (3h)
The title compound was obtained from 25h and 27 as described for 3a in 67% yield after flash-chromatography (1:6 EtOAc/CHCl3): [α]D20 = + 7.9 (12.3 mg/mL CH2Cl2); 1H NMR (CDCl3) δ 7.71 (d, J = 9.3 Hz, 2H), 7.32–7.22 (m, 5H), 6.98 (d, J = 9.3 Hz, 2H), 5.06 (d, J = 8.4 Hz, 1H), 4.92 (d, J = 6.2 Hz, 1H), 4.75 (d, J = 6.2 Hz, 1H), 4.51–4.49 (m, 1H), 3.95–3.74 (m, 9H), 3.14 (dd, J = 8.1, 15.0 Hz, 1H), 3.06–2.84 (m, 4H), 2.77 (dd, J = 6.7, 13.3 Hz, 1H), 1.86–1.77 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) δ 162.9, 155.4, 137.3, 129.7, 129.6, 129.5, 128.5, 126.5, 114.3, 93.6, 72.3, 68.7, 66.3, 58.8, 55.6, 55.2, 53.8, 35.7, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C26H36N2NaO8S, 559.2090; found, 559.2094.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 3,6,9-trioxacyclodecan-1-yl ester (3i)
The title compound was obtained from 25i and 27 as described for 3a in 37% yield after flash-chromatography (1:1 EtOAc/CHCl3) as a white solid: mp 60–62 °C; [α]D20 = + 6.2 (c 0.3, CHCl3); 1H NMR (CDCl3) δ 7.69 (d, J = 8.7 Hz, 2H), 7.33–7.18 (m, 5H), 6.96 (d, J = 8.7 Hz, 2H), 5.33 (d, J = 8.1 Hz, 1H), 4.84–4.82 (m, 1H), 3.86 (s, 3H), 3.79–3.75 (m, 2H), 3.68–3.55 (m, 12H), 3.07–2.78 (m, 6H), 1.84–1.81 (m, 1H), 0.89 (d, J = 7.2 Hz, 3H), 0.85 (d, J = 7.2 Hz, 3H). HRMS-ESI (m/z): (M + Na)+ calcd for C29H42N2NaO9S, 617.2509; found, 617.2501.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 3,6,9,12-tetraoxacyclotridecan-1-yl ester (3j)
The title compound was obtained from 27 and 25j as described for 3a in 30% yield after flash-chromatography (EtOAc) as a foam: [α]D20 = + 17.0 (c 0.9, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 9.0 Hz, 2H), 7.29–7.19 (m, 5H), 6.97 (d, J = 9.0 Hz, 2H), 4.96 (d, J = 8.0 Hz, 1H), 4.85–4.83 (m, 1H), 3.87 (s, 3H), 3.83–3.81 (m, 2H), 3.80–3.60 (m, 15H), 3.52 (dd, J = 3.5, 9.5 Hz, 1H), 3.13 (dd, J = 9.0, 15.5 Hz, 1H), 3.02–2.86 (m, 4H), 2.77 (dd, J = 6.5, 13.5 Hz, 1H), 1.83–1.76 (m, 1H), 0.90 (d, J = 6.5 Hz, 3H), 0.85 (d, J = 6.5 Hz, 3H); 13C NMR (CDCl3) (500 MHz) δ 163.0, 155.4, 137.6, 129.7, 129.6, 129.5, 128.5, 126.5, 114.4, 72.4, 71.7, 70.2, 70.1, 69.9, 67.8, 58.7, 55.6, 55.1, 53.7, 35.5, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C31H46N2NaO10S, 661.2771; found, 661.2788.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 3,6,8,11-tetraoxacyclododecan-1-yl ester (3k)
The title compound was obtained from 27 and 25k as described for 3a in 47% yield after flash-chromatography (EtOAc) as a foam: [α]D20 = + 6.5 (c 0.5, CHCl3); 1H NMR (CDCl3) 7.70 (d, J = 8.7 Hz, 2H), 7.30–7.18 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 4.92 (d, J = 8.1 Hz, 1H), 4.81–4.76 (m, 1H), 4.66 (s, 2H), 3.87 (s, 3H), 3.78–344 (m, 14H), 3.13 (dd, J = 8.4, 15.3 Hz, 1H), 3.06–2.82 (m, 4H), 2.75 (dd, J = 6.9, 13.5 Hz, 1H), 1.83–1.74 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 163.0, 155.6, 137.5, 129.8, 129.6, 129.5, 128.5, 126.5, 114.4, 94.7, 72.4, 71.5, 69.7, 64.9, 64.5, 58.8, 55.7, 55.1, 53.8, 35.6, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C30H44N2NaO10S, 647.2615; found, 647.2590.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 3,9-dioxa-6-thiacyclodecan-1-yl 6,6-dioxide ester (3l)
The title compound was obtained from 27 and 25l as described for 3a in 36% yield after flash-chromatography (1:1 EtOAc/CHCl3) as an amorphous solid: [α]D20 = + 5.5 (c 0.7, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 9.0 Hz, 2H), 7.31–7.20 (m, 5H), 6.98 (d, J = 9.0 Hz, 2H), 4.97 (d, J = 8.4 Hz, 1H), 4.85 (t, J = 4.5 Hz, 1H), 4.01–3.96 (m, 4H), 3.88 (s, 3H), 3.85–3.83 (m, 2H), 3.71–3.69 (m, 1H), 3.61 (dd, J = 3.9, 9.3 Hz, 1H), 3.54–3.47 (m, 2H), 3.61–3.27 (m, 4H), 3.13 (dd, J = 8.4, 15.0 Hz, 1H), 3.00–2.82 (m, 4H), 2.75 (dd, J = 6.6, 13.5 Hz, 1H), 1.83–1.75 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 163.0, 155.1, 137.4, 131.1, 129.6, 129.4, 128.4, 126.5, 114.3, 72.4, 70.2, 66.0, 64.6, 58.8, 55.7, 55.1, 53.7, 52.2, 35.4, 27.3, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C29H42N2NaO10S2, 665.2179; found, 665.2191.
N-(tert-Butoxycarbonyl)-9-(4-nitrophenoxycarbonyloxy)-1,7-dioxa-4-azocyclodecane (29)
The title compound was obtained from 24 as described for 25a in 73% yield after flash-chromatography (1:4 EtOAc/CHCl3): 1H MR (CDCl3) δ 8.27 (d, J = 9.0 Hz, 2H), 7.37 (d, J = 9.0 Hz, 2H), 5.02–4.96 (m, 1H), 3.98–3.76 (m, 8H), 4.52–3.23 (m, 4H), 1.47 (s, 9H).
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid N-(tert-butoxycarbonyl)-1,7-dioxa-4-azocyclodecan-9-yl ester (30)
The title compound was obtained from 27 and 29 as described for 3a in 74% yield after flash-chromatography (1:1 EtOAc/CHCl3) as a white solid: mp 71–73 °C; [α]D20 = + 4.7 (c 1.7, CHCl3); 1H NMR (CDCl3) 7.70 (d, J = 9.0 Hz, 2H), 7.30–7.20 (m, 5H), 7.0 (d, J = 9.0 Hz, 2H), 4.92–4.90 (m, 1H), 4.81 (t, J = 4.0 Hz, 1H), 3.86 (s, 3H), 3.79–3.66 (m, 6H), 3.62–3.57 (m, 2H), 3.49–3.42 (m, 2H), 3.40–3.28 (m, 4H), 3.12 (dd, J = 7.8, 15.3 Hz, 1H), 3.01–2.82 (m, 4H), 2.75 (dd, J = 6.3, 13.2 Hz, 1H), 1.83–1.74 (m, 1H), 1.44 (s, 9H), 0.90 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) 163.0, 155.6, 155.4, 137.4, 129.7, 129.5, 129.4, 128.4, 126.5, 114.3, 79.9, 72.4, 71.9, 71.0, 68.6, 68.1, 58.7, 55.6, 55.0, 53.7, 50.3, 35.6, 28.5, 27.3, 20.2, 19.9.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid 1,7-dioxa-4-azocyclodecan-9-yl ester (31)
A solution of 30 (13 mg, 0.02 mmol) in a mixture of 30% trifluoracetic acid in CH2Cl2 (1 mL) was stirred at 23 °C for 30 min and then the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2 and the organic phase was washed with a saturated solution of NaHCO3, dried (Na2SO4) and evaporated to afford 11 mg (100%) of 31 as a white solid: mp 65–66 °C; [α]D20 = + 13.8 (c 0.7, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 8.7 Hz, 2H), 7.30–7.18 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 5.20 (d, J = 8.4 Hz, 1H), 4.82–4.79 (m, 1H), 3.87 (s, 3H), 3.84–3.80 (m, 2H), 3.75–3.64 (m, 7H), 3.54 (dd, J = 5.4, 10.2 Hz, 1H), 3.13 (dd, J = 8.4, 15.3 Hz, 1H), 3.04–2.84 (m, 8H), 2.77 (dd, J = 6.9, 13.5 Hz, 1H), 2.38 (bs, 1H), 1.85–1.76 (m, 1H), 0.90 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) δ 162.9, 155.3, 137.5, 129.8, 129.5, 129.4, 128.4, 126.4, 114.3, 72.4, 71.8, 68.6, 58.7, 55.6, 55.1, 53.6, 53.4, 48.2, 35.6, 27.2, 20.2, 19.9.
(1S,2R)-{1-Benzyl-2-hydroxy-3-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl}carbamic acid N-methyl-1,7-dioxa-4-azocyclodecan-9-yl ester (3m)
To a solution of 31 (9.0 mg, 0.015 mmol) in a mixture of 1% acetic acid in methanol (0.5 mL), formaldehyde (37% solution in H2O, 12 μL, 0.15 mmol) and sodium cyanoborohydride (2.0 mg, 0.03 mmol) were added. After 18 h a saturated solution of NaHCO3 was added, the solvent was removed and the aqueous phase was extracted with CH2Cl2. The organic extracts were dried (Na2SO4), evaporated and the residue was purified by flash-chromatography eluting with a 10:1 mixture of CHCl3 and MeOH to afford 8.0 mg (87%) of 3m as an amorphous solid: [α]D20 = + 8.1 (c 0.6, CHCl3); 1H NMR (CDCl3) δ 7.70 (d, J = 8.7 Hz, 2H), 7.3–7.18 (m, 5H), 6.98 (d, J = 8.7 Hz, 2H), 4.99 (d, J = 8.1 Hz, 1H), 4.80–4.77 (m, 1H), 3.87 (s, 3H), 3.83–3.74 (m, 4H), 3.70–3.56 (m, 6H) 3.14 (dd, J = 8.1, 14.7 Hz, 1H), 3.02–2.69 (m, 9H), 2.40 (s, 3H), 1.83–1.74 (m, 1H), 0.90 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) δ 162.9, 155.5, 137.4, 129.9, 129.4 (× 2C), 128.4, 126.4, 114.3, 77.2, 72.3, 69.6, 67.6, 59.0, 55.6, 55.1, 53.6, 44.0, 35.6, 29.7, 27.2, 20.2, 19.9. HRMS-ESI (m/z): (M + Na)+ calcd for C30H46N3O8S, 608.3006; found, 608.3009.
Determination of X-ray structure of 3d-bound HIV protease
The HIV-1 protease construct with the substitutions Q7K, L33I, L63I, C67A and C95A to optimize protein stability33 was expressed and purified as described.34 Crystals were grown by the hanging drop vapor diffusion method using 1:15 molar ratio of protease at 2.0 mg/mL and inhibitor dissolved in dimethylsulfoxide. The reservoir contained 0.1 M sodium acetate buffer (pH = 4.2) and 1.5 M NaCl. Crystals were transferred into a cryoprotectant solution containing the reservoir solution and 20–30% (v/v) glycerol, mounted on a nylon loop and flash-frozen in liquid nitrogen. X-ray diffraction data were collected on the SER-CAT beamline of the Advanced Photon Source, Argonne National Laboratory. Diffraction data were processed using HKL200035 resulting in an Rmerge value of 8.0% (41.1%) for 110,362 unique reflections between 50 and 1.00 Å resolution with a completeness of 88.4% (52.6%), where the values in parentheses are for the final highest resolution shell. Data were reduced in space group P21212 with unit cell dimensions of a = 57.96 Å, b = 86.41 Å, c = 46.03 Å with one dimer in the asymmetric unit. The structure was solved by molecular replacement using the CPP4i suite of programs36,37, with the structure of the D30N mutant of HIV protease in complex with GRL-98065 (2QCI)34 as the starting model. The structure was refined using SHELX9738 and refitted manually using the molecular graphics programs O39 and COOT40. Alternate conformations were modeled for the protease residues when obvious in the electron density maps. Anisotropic atomic displacement parameters (B-factors) were refined for all atoms including solvent molecules. Hydrogen atoms were added at the final stages of the refinement. The identity of ions and other solvent molecules from the crystallization conditions was deduced from the shape and peak height of the 2Fo-Fc and Fo-Fc electron density, the hydrogen bond interactions and interatomic distances. The solvent structure was refined with two sodium ions, three chloride ions, and 216 water molecules including partial occupancy sites. The final Rwork was 14.9 % and Rfree was 17.5% for all data between 10 and 1.00 Å resolution. The rmsd values from ideal bonds and angle distances were 0.017 Å and 0.034 Å, respectively. The average B-factor was 11.4 and 16.5 Å2 for protease main chain and side chain atoms, respectively, 12.9 Å2 for inhibitor atoms and 22.6 Å2 for solvent atoms. The X-ray crystal structure of the 3d-bound HIV-1 protease has been deposited in the Protein Databank (PDB)41 with accession code 3DJK.
Supplementary Material
Acknowledgements
The research was supported by grants from the National Institutes of Health (GM53386, AKG and GM62920, IW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and in part by a Grant-in-aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho: H15-AIDS-001), and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re emerging Infectious Diseases (Renkei Jigyo: No. 78, Kumamoto University) of Monbu Kagakusho. The work was also supported in part by the Georgia State University Molecular Basis of Disease Program, the Georgia Research Alliance, the Georgia Cancer Coalition. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We also thank Ms. Sofiya Leshchenko-Yashchuk for her assistance in HIV-1 protease inhibitory assay.
Footnotes
The PDB accession code for 3d-bound HIV-1 protease X-ray structure is 3DJK.
Supporting Information Available. HPLC and HRMS data of inhibitors 3a–m. This material is available free of charge via the Internet at http://pubs./acs.org.
References
- (1).Sepkowitz KA. AIDS — The first 20 years. Engl. J. Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- (2).Waters L, Nelson M. Why do patients fail HIV therapy? Int. J. Clin. Pract. 2007;61:983–990. doi: 10.1111/j.1742-1241.2007.01383.x. [DOI] [PubMed] [Google Scholar]
- (3).Pillay D, Bhaskaran K, Jurriaans S, Prins M, Masquelier B, Dabis F, Gifford R, Nielsen C, Pedersen C, Balotta C, Rezza G, Ortiz M, de Mendoza C, Kücherer C, Poggensee G, Gill J, Porter K. The impact of transmitted drug resistance on the natural history of HIV infection and response to first-line therapy. AIDS. 2006;20:21–28. doi: 10.1097/01.aids.0000196172.35056.b7. [DOI] [PubMed] [Google Scholar]
- (4).Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. AIDS. 2000;14:141–149. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- (5).Wainberg MA, Friedland G. Public health implications of antiretroviral therapy and HIV drug resistance. J. Am. Med. Ass. 1998;279:1977–1983. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- (6).Harrigan PR, Hogg RS, Dong WW, Yip B, Wynhoven B, Woodward J, Brumme CJ, Brumme ZL, Mo T, Alexander CS, Montaner JS. Predictors of HIV drug-resistance mutations in a large antiretroviral naive cohort initiating triple antiretroviral therapy. J. Infect. Dis. 2005;191:339–347. doi: 10.1086/427192. [DOI] [PubMed] [Google Scholar]
- (7).Hertogs K, Bloor S, Kemp SD, Van den Eynde C, Alcorn TM, Pauwels R, Van Houtte M, Staszewski S, Miller V, Larder B. A Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS. 2000;14:1203–1210. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- (8).Yerly S, Kaiser L, Race E, Bru JP, Clavel F, Perrin L. Transmission of antiretroviral drug resistant HIV-1 variants. Lancet. 1999;354:729–733. doi: 10.1016/S0140-6736(98)12262-6. [DOI] [PubMed] [Google Scholar]
- (9).On June 23, 2006, FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.html.
- (10).Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg. Med. Chem. Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]
- (11).Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. A novel bis-tetrahydrofuranylurethane-containing non-peptide protease inhibitor (PI) UIC-94017 (TMC-114) potent against multi-PI-resistant HIV in vitro. Antimicrob. Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC114, a next generation of HIV-1 protease inhibitor. J. Med. Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- (13).Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active Site. J. Virol. 2002;76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Kohl Y, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Louis JM, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranyl-urethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) potent against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2003;2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Pretzer E, Cho H, Hussain KA, Duzgunes N. Antiviral activity of UIC-PI, a novel inhibitor of the human immunodeficiency virus type 1 protease. Antiviral Res. 2002;54:29–36. doi: 10.1016/s0166-3542(01)00209-1. [DOI] [PubMed] [Google Scholar]
- (15).Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High-resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- (16).Hong L, Zhang XC, Hartsuck JA, Tang J. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: Insights into the mechanisms of drug resistance. Protein Sci. 2000;9:1989–1904. doi: 10.1110/ps.9.10.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Laco GS, Schalk-Hihi C, Lubkowski J, Morris G, Zdanov A, Olson A, Elder JH, Wlodawer A, Gustchina A. Crystal Structures of the Inactive D30N Mutant of Feline Immunodeficiency Virus Protease Complexed with a Substrate and an Inhibitor. Biochemistry. 1997;36:10696–10708. doi: 10.1021/bi9707436. [DOI] [PubMed] [Google Scholar]
- (18).Winitz M, Bloch-Frankenthal L, Izumiya N, Birnbaum SM, Baker CG, Greenstein JP. Studies on Diastereoisomeric α-Amino Acids and Corresponding α-Hydroxy Acids. VII. Influence of β-Configuration on Enzymic Susceptibility. J. Am. Chem. Soc. 1956;78:2423–2430. [Google Scholar]
- (19).Soai K, Ookawa A. Mixed solvents containing methanol as useful reaction media for unique chemoselective reductions with lithium borohydride. J. Org. Chem. 1986;51:4000–4005. [Google Scholar]
- (20).Gmeiner P, Junge D. Regioselective transformation of malic acid: a practical method for the construction of enantiomerically pure indolizidines. J. Org. Chem. 1995;60:3910–3915. [Google Scholar]
- (21).Le Merrer Y, Gauzy L, Gravier-Pelletier C, Depezay J-C. Synthesis of C2-symmetric guanidino-sugars as potent inhibitors of glycosidases. Bioorg. Med. Chem. 2000;8:307–320. doi: 10.1016/s0968-0896(99)00294-1. [DOI] [PubMed] [Google Scholar]
- (22).Pospíšil J, Markó IE. Total synthesis of (R)-(+)-goniothalamin and (R)-(+)-goniothalamin oxide: first application of the sulfoxide-modified Julia olefination in total synthesis. Tetrahedron Lett. 2007;47:5933–5937. [Google Scholar]
- (23).Lu C-D, Zakarian A. Studies toward the synthesis of pinnatoxins: The B,C,D-dispiroketal fragment. Org. Lett. 2007;9:3161–3163. doi: 10.1021/ol071266e. [DOI] [PubMed] [Google Scholar]
- (24).Kasireddy K, Ahmad MU, Ali SM, Ahmad I. Synthesis of novel cationic cardiolipin analogues for the optimal delivery of therapeutic agents. Tetrahedron Lett. 2004;45:2743–2746. [Google Scholar]
- (25).Calverley MJ, Dale J. 1,4,7-Trioxa-10-azacyclododecane and some N-substituted derivatives; synthesis and cation complexing. Acta Chem. Scand. 1982;B36:241–247. [Google Scholar]
- (26).Sakamoto H, Kimura K, Koseki Y, Matsuo M, Shono T. Lipophilic bis(monoaza crown ether) derivatives: synthesis and cation-complexing properties. J. Org. Chem. 1986;51:4974–4979. [Google Scholar]
- (27).Ghosh AK, Fidanze S. Transition-state mimetics for HIV protease inhibitors: stereocontrolled synthesis of hydroxyethylene and hydroxyethylamine isosteres by ester-derived titanium enolate syn and anti-aldol reactions. J. Org. Chem. 1998;63:6146–6152. doi: 10.1021/jo980159i. [DOI] [PubMed] [Google Scholar]
- (28).Toth MV, Marshall GR. A simple, continuous fluorometric assay for HIV protease. Int. J. Pep. Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- (29).Gustchina A, Sansom C, Prevost M, Richelle J, Wodak S, Wlodawer A, Weber I. Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- (30).Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 Angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hronowski LJJ, Szarek WA, Hay GW, Krebs A, Depew WT. Synthesis and characterization of 1-O-β-lactosyl-(R,S)-glycerols and 1,3-di-O-β-lactosylglycerol. Carbohydrate Res. 1989;190:203–218. [Google Scholar]
- (33).Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- (34).Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/substrate analog complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- (35).Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- (36).Collaborative Computational Project, Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- (37).Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta. Cryst. 2003;D59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- (38).Sheldrick GM, Schneider TR. SHELXL: High resolution refinement. Methods in Enzymology. 1997;277:319–343. [PubMed] [Google Scholar]
- (39).Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- (40).Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (41).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).(a) Shirasaka T, Yarchoan R, O'Brien MC, Husson RN, Husson BD, Kojima E, Broder S, Mitsuya H. Changes in drug sensitivity of human immunodeficiency virus type 1 during therapy with azidothymidine, dideoxycytidine, and dideoxyinosine: An in vitro comparative study. Proc. National Acad. Sci. USA. 1993;90:562–566. doi: 10.1073/pnas.90.2.562. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Perno CF, Yarchoan R, Cooney DA, Hartman NR, Webb DSA, Hao Z, Mitsuya H, Johns DG, Broder S. Replication of HIV in monocytes: Granulocyte-macrophage colony stimulating factor (GM-CSF) potentiates viral production yet enhances the antiviral effect mediated by AZT and other dideoxynucleoside congeners of thymidine. J. Exp. Med. 1989;169:933–951. doi: 10.1084/jem.169.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.