

Abstract
Pseudomonas aeruginosa PAO1 is the most commonly used strain for research on this ubiquitous and metabolically versatile opportunistic pathogen. Strain PAO1, a derivative of the original Australian PAO isolate, has been distributed worldwide to laboratories and strain collections. Over decades discordant phenotypes of PAO1 sublines have emerged. Taking the existing PAO1-UW genome sequence (named after the University of Washington, which led the sequencing project) as a blueprint, the genome sequences of reference strains MPAO1 and PAO1-DSM (stored at the German Collection for Microorganisms and Cell Cultures [DSMZ]) were resolved by physical mapping and deep short read sequencing-by-synthesis. MPAO1 has been the source of near-saturation libraries of transposon insertion mutants, and PAO1-DSM is identical in its SpeI-DpnI restriction map with the original isolate. The major genomic differences of MPAO1 and PAO1-DSM in comparison to PAO1-UW are the lack of a large inversion, a duplication of a mobile 12-kb prophage region carrying a distinct integrase and protein phosphatases or kinases, deletions of 3 to 1,006 bp in size, and at least 39 single-nucleotide substitutions, 17 of which affect protein sequences. The PAO1 sublines differed in their ability to cope with nutrient limitation and their virulence in an acute murine airway infection model. Subline PAO1-DSM outnumbered the two other sublines in late stationary growth phase. In conclusion, P. aeruginosa PAO1 shows an ongoing microevolution of genotype and phenotype that jeopardizes the reproducibility of research. High-throughput genome resequencing will resolve more cases and could become a proper quality control for strain collections.
The metabolically versatile Pseudomonas aeruginosa is an opportunistic pathogen of plants, animals, and humans and is ubiquitously distributed in soil and aquatic habitats. The common reference strain is P. aeruginosa PAO1, a spontaneous chloramphenicol-resistant mutant of the original PAO strain (earlier called “P. aeruginosa strain 1”) that had been isolated in 1954 from a wound in Melbourne, Australia (9, 10). This PAO1 strain from Bruce Holloway's laboratory has become the reference strain for Pseudomonas genetics and functional analyses of the physiology and metabolism of this gammaproteobacterium. A genetic map of its chromosome was generated by exploiting the mechanisms of gene exchange in bacteria, i.e., transduction and conjugation (11). With the advent of pulsed-field gel electrophoresis (PFGE), a physical map of the PAO1 genome was constructed (32) and later merged with the genetic map information (12). By 2000 the PAO1 strain had been completely sequenced (36). Thereafter, the genome annotation has been continually updated and the database content and functionality have been expanded to facilitate accelerated discovery of P. aeruginosa drug targets and vaccine candidates (38). Two near-saturation libraries of transposon insertion mutants have been constructed in P. aeruginosa PAO1 as a global resource for the scientific community (14, 22).
Comparison of the genome sequence with the physical map revealed a large, 2.2-Mb inversion between the sequenced PAO1-UW strain (36) and the original PAO1 strain (9, 10), indicating that PAO1 sublines maintained worldwide in numerous laboratories and strain collections had diversified their genomic sequence. Mutational events were already reported in the 1970s (10), and more recently sequence variations of MexT, which regulates the MexEF-OprN multidrug efflux system, were described (18, 24). Furthermore, a PAO1 subline from a German strain collection (PAO1-D) and another, independent PAO1 subline from a Japanese strain collection (PAO1-J) that had been stored by research groups in Germany and Japan, respectively, were found to be quorum-sensing-negative mutants that carried point mutations in the regulatory gene lasR (6). In addition, spontaneous secretion-defective vfr mutants from a PAO1 population were observed after several cycles of static growth (2). Similarly, we noted a difference in virulence in a mouse infection model (see below) between the MPAO1 and PAO1-DSM sublines that had been utilized for the construction of the transposon library (14) and the physical map (32), respectively. PAO1-DSM was indistinguishable in its SpeI-DpnI-SwaI-PacI physical map from the PAO1 subline that had been stored in the Holloway laboratory (12). Hence, we decided to compare the genomic sequence of the initially sequenced PAO1 subline PAO1-UW (36) with that of MPAO1 and PAO1-DSM. Combined physical mapping and DNA sequencing-by-synthesis revealed numerous single-nucleotide polymorphisms (SNPs) and insertions-deletions (indels) in the chromosomes that were associated with differences in fitness, antimicrobial susceptibility, and virulence of the sublines.
MATERIALS AND METHODS
Bacterial strains.
PAO1-DSM was obtained in 1986 and again in 2004 from the German Collection of Microorganisms and Cell Cultures (DSMZ), Brunswick, Germany (catalogue no. DSM 1707). MPAO1 was received in 2003 from the distributor of the PAO1 mutant library of the University of Washington, Seattle (14). PAO1-UW was supplied in 1999 by A. L. Erwin, Pathogenesis Corporation, Seattle, WA, and again in 2005 by T. Rasmussen, DTU, Lyngby, Denmark. For long-term storage glycerol stocks were generated from freshly grown cultures and kept at −80°C.
Media and culturing.
P. aeruginosa PAO1 sublines were cultivated in either LB broth or M9 medium supplemented with 30 mM succinate and 10 μM FeSO4. Cells were grown at 37°C if used as a source for DNA preparation.
Preparation of genomic DNA.
Genomic DNA was prepared from bacterial cells following standard procedures (1). To improve DNA purity, additional phenol-chloroform extraction steps were included.
Physical mapping.
Regions suspicious for insertions or deletions in PAO1-DSM and MPAO1 were analyzed by partial or complete restriction digests of genomic DNA and Southern hybridization with digoxigenin-labeled PCR-generated probes (procedure performed according to previously described protocols [33]) to detect differences in restriction patterns between the tested PAO1 sublines and the PAO1 reference sequence (36). Potential deletions were interrogated by combinatorial PCR, taking the PAO1 sequence as a blueprint.
PFGE.
Agarose plugs containing high-molecular-weight DNA were prepared by embedding 2 × 109, 5 × 109, or 1 × 1010 bacterial cells/ml in low-melting-point agarose (Sigma), followed by proteinase K digestion at 56°C for 48 h. Restriction digests of these agarose plugs with SwaI, PacI, DpnI, and SpeI were done as described previously (33), using the respective restriction buffer recommended by the manufacturer (New England Biolabs). PFGE of restricted DNA was performed in a contour-clamped homogeneous electric field (CHEF) DR III apparatus with a 120° reorientation angle at 9.5°C with 0.5× TBE (45 mM Tris, 45 mM boric acid, 1 mM EDTA, pH 8.3) as electrophoresis buffer. SwaI and PacI digests were separated in 0.8% agarose gels for 60 h at 2.5 V/cm (single ramp with linearly increasing pulse times from 500 to 2,000 s). DpnI and SpeI digests were separated by CHEF in 1.5% agarose at 6 V/cm. Pulse times were linearly increased in three ramps from 8 to 50 s for 24 h, 12 to 25 s for 22 h, and 1 to 14 s for 14 h. Restriction fragments were visualized with ethidium bromide.
Illumina Genome Analyzer sequencing.
Illumina Genome Analyzer libraries were prepared following the manufacturer's instructions. Purified genomic DNA was fragmented by nebulization into fragments of less than 800 bp in size. The resulting 3′ and 5′ overhangs of the double-stranded DNA were converted into blunt ends, and a terminal “A” was then ligated to the 3′ end. The modified DNA fragments were ligated to adapters, purified, and amplified by PCR according to the manufacturer's manual. Thirty-six cycles of sequencing-by-synthesis were performed for each library in four lanes of a flow cell with the Genome Analyzer I. Illumina Genome Analyzer Pipeline version 0.2 software was used to qualify reads passing default signal quality filters.
Each position in a sequence read is associated with a quality score, Qsolexa, reflecting the quality of the respective base call (depending on the measured signal during the sequencing process). For a base X, the relation of its probability value p(X) to the Qsolexa score is given by the formula Qsolexa = 10 log [p(X)/1 − p(X)]. Therefore, a maximal Qsolexa value of 40 indicates a probability of 99.99% for the called base or an error probability of 0.01%, respectively. Lower Qsolexa values typically appear at the end of reads. If no decisive base call could be made, the respective position in the sequence read was designated “N” (with Qsolexa set to −0.5 automatically).
The sequence reads were mapped to the PAO1-UW reference sequence (NC_002516) using Illumina's ELAND aligner (efficient large-scale alignment of nucleotide databases). ELAND aligns a large number of short reads to reference sequences, allowing up to two mismatches in the first 32 bases of a read.
Thereafter, the frequency of coverage, base predictions, and “consensus qualities” were computed for each position of the reference sequence covered by aligned reads. The “consensus quality” Qconsensus for each position s in the reference sequence is defined by Qconsensus (s) = Qmax/[Q (A) + Q (C) + Q (G) + Q (T)], whereby Q (A), Q (C), Q (G), and Q (T) are the quality sums for each base and Qmax is the maximal value thereof. Thus, a Qconsensus (s) value of 1 indicates identical base calls for position s in all covering reads, while lower values are caused by heterogeneous results.
Assembly of sequence reads.
Mapping the sequence reads obtained from the PAO1-DSM and MPAO1 subline to the PAO1-UW reference sequence with ELAND allowed binning of the reads: 727 and 695 Mb of sequence were aligned to the reference (“in_ref” bins); 29.4/17.4 Mb of sequence (“not_in_ref” bins) contained putatively novel DNA. Obviously incorrect reads with homooligomers of >13 bases in length (not present in the P. aeruginosa genome) or an “N” base call in at least three positions were excluded from the analysis.
The de novo assembler Velvet version 0.7.11 (41) was then used with a k-mer size of 27 to generate contigs of novel DNA from the “not_in_ref” sequences. The resulting sequence contigs were extended using an in-house program, MarkerCounter, to locate reads from the “not_in_ref” bin. Assembly was completed by inclusion of reads from the “in_ref” bin. Thus, the novel DNA could be attributed to a new phage sequence, part of which shows 94% to 100% sequence identity with the PA0717 to PA0727 contig. The short read mapper Mosaik (The MarthLab, Boston College [http://bioinformatics.bc.edu/marthlab/Mosaik]) was used in tandem with EagleView (13) to verify the variable positions in the duplicated sequence. The assembly and the presence of duplicated DNA were confirmed by combinatorial PCR.
The exact size of the 1,006-bp deletion already detected in the initial assembly was determined by using MarkerCounter to identify reads covering the breakpoint.
SNPs and small deletions.
Analysis of the primary sequence data generated potential sequence variations (SNPs) defined as consensus bases differing from the published PAO1 reference. Potential SNPs in 41 positions with varying coverage and confidence of base prediction were selected for validation by diagnostic restriction digests. Sequences encompassing the potential SNP position were amplified by PCR (lengths, 163 to 292 bp). If the predicted nucleotide exchange resulted in gain or loss of a restriction site, the PCR products of PAO1-UW, PAO1-DSM, and MPAO1 DNA templates were digested with the respective enzyme. If no appropriate diagnostic enzyme could be found, artificial restriction sites were introduced by one or two mismatches in a PCR primer (5).
The threshold for a reliable SNP in the primary sequence data was set to be coverage of at least 10-fold and a Qconsensus of 0.9 or more (see above). Thirteen of 14 candidate SNPs in MPAO1 and PAO1-DSM that fulfilled these quality criteria were verified to be nucleotide substitutions by informative restriction digestions (see Table S1 in the supplemental material). In contrast, a nucleotide substitution was experimentally proven for only 1 of 32 tested candidate SNPs that did not pass the threshold. Hence, the chosen rigorous threshold criteria were appropriate to identify more than 95% of the SNPs with an acceptable rate of false positives of less than 10%, and therefore, they were applied to the entire sequencing data.
PAO1-UW regions that were not covered by MPAO1- or PAO1-DSM-derived sequence reads were candidates for deletions. These segments were queried for the number of deleted bases by direct blotting electrophoresis and subsequent chemiluminescence visualization of combinatorial biotinylated PCR products (26).
Antimicrobial susceptibility testing.
Susceptibility to antimicrobial agents was tested by serial twofold dilution using liquid cation-adjusted Mueller-Hinton (MH) medium. The panel included seven broad-spectrum penicillins, 10 cephalosporins, three carbapenems, aztreonam, fusidic acid, doxycycline, chloramphenicol, four aminoglycosides, colistin, and four quinolones. Precultures of the PAO1 sublines were diluted in 0.9% NaCl solution, and 2 × 105 viable cells thereof were inoculated into 2 ml MH medium containing the respective antimicrobial agent. In the case of fusidic acid 1 μg/ml of the sensitizing nonbactericidal polymyxin B nonapeptide (PMBN) was added to each culture (20, 37). MIC was determined after 24 h of growth at 37°C.
Growth competition experiments.
Five milliliters LB broth was inoculated from glycerol stocks of the respective PAO1 sublines and grown overnight at 37°C with shaking. An aliquot was transferred into 20 ml LB adjusted to an optical density at 578 nm (OD578) of 0.1 and cultivated for about 6 h until an OD578 of 2.5 was reached. An amount of 2 × 1010 CFU per PAO1 subline was mixed, and an aliquot thereof was added to 20 ml medium to give an OD578 of 0.1. The six different culturing conditions are described in Table S2 in the supplemental material. At the respective time points, samples were taken and genomic DNA was prepared following standard procedures (see above). All cultures were performed in duplicate. The relative contents of PAO1 sublines were determined by utilizing subline-discriminating markers: PAO1-DSM was identified via specific SNPs at positions 4769678 and 2807853. The portion of PAO1-UW was analyzed by semiquantitative PCR kinetics (16) for the presence of the nucleotides 5253694 to 5254699, which were deleted in both other sublines (see Results).
Murine airway infection model.
Bacteria were grown in LB broth overnight at 37°C (230 rpm), pelleted by centrifugation (5,000 × g, 10 min), and washed twice with sterile phosphate-buffered saline (PBS), and the optical density of the bacterial suspension was adjusted by spectrophotometry at 578 nm. The intended number of CFU was extrapolated from a standard growth curve, and appropriate dilutions with sterile PBS were made to prepare the inoculum for the mice. To verify the correct dilution, an aliquot was serially diluted on LB agar plates. Ten- to 12-week-old female mice of the inbred strain C3H/HeN (Charles River, Sulzfeld, Germany) were inoculated with 30 μl of this bacterial suspension containing 7.5 × 106 CFU of the different P. aeruginosa PAO1 sublines via view-controlled intratracheal instillation. In the case of 14-day infection experiments, the weight and rectal temperature of the mice were measured daily and their body condition was scored for the parameters vocalization, piloerection, attitude, locomotion, breathing, curiosity, nasal secretion, grooming, and dehydration (29).
During the experiments mice were maintained in microisolator cages with filter top lids at 21 ± 2°C and 50% ± 5% humidity and in a 12-h light-dark cycle. They were supplied with autoclaved, acidulated water and fed ad libitum with autoclaved standard diet. Mice were sacrificed after 48 h for the evaluation of lung histology or the determination of CFU in homogenized organs (lungs, liver, spleen, and brain).
Prior to the start of the experiments, animals were acclimatized for at least 7 days. All animal procedures were approved by the local district governments and carried out according to the guidelines of the German law for the protection of animal life.
Nucleotide sequence accession numbers.
The nucleotide sequence of the PAO1-RGP42 island described in this paper has been deposited in the GenBank database (accession number GQ141978). The SNPs listed in Table 1 have been submitted to the Database of Single Nucleotide Polymorphisms (dbSNP) (submission SNP accession numbers, ss138660650 to ss138660685).
TABLE 1.
List of SNPs in PAO1-DSM and MPAO1a
| Category | Position | dbSNP accession no.b | Locus | Encoded product | Nucleotide change | Amino acid change |
|---|---|---|---|---|---|---|
| SNPs in both strains | 169284 | ss138660650 | Interg. (PA0148-PA0149) | G → C | ||
| 183697 | ss138660651 | PA0159 | Prob. transcriptional regulator | T → G | C → W | |
| 413850 | ss138660652 | Interg. (PA0369-PA0370) | T → C | |||
| 1467483 | ss138660654 | Interg. (PA1352-PA1353) | C → G | |||
| 2239547 | ss138660657 | Interg. (PA2046-PA2047) | T → G | |||
| 4212201 | ss138660660 | PA3760 | Prob. phosphotransferase | A → G | H → R | |
| 4344266 | ss138660661 | PA3877 | Nitrite extrusion protein 1 NarK | A → G | Syn. | |
| 4448855 | ss138660662 | Interg. (PA3969-PA3970) | C → G | |||
| 4448856 | ss138660662 | Interg. (PA3969-PA3970) | G → C | |||
| 4869855 | ss138660663 | PA4341 | Prob. transcriptional regulator | T → G | E → N | |
| 4924552 | ss138660664 | PA4394 | Hypoth. protein | C → G | V → L | |
| 4924553 | ss138660664 | PA4394 | Hypoth. protein | G → C | Syn. | |
| 5036891c | ss138660665 | Interg. (PA4499-PA4500) | A → C | |||
| 5743461 | ss138660666 | PA5100 | Urocanate hydratase HutU | C → G | T → S | |
| 5743462 | ss138660666 | PA5100 | Urocanate hydratase HutU | G → C | T → S | |
| 6098781 | ss138660668 | PA5418 | Sarcosine oxidase subunit SoxA | G → C | Syn. | |
| SNPs in PAO1-DSM only | 447942 | ss138660670 | PA0406 | TonB-like periplasmic protein | T → C | R → G |
| 792696d | ss138660671 | PA0725 | Phage Pf1-like hypoth. protein | C → T | Syn. | |
| 792834d | ss138660672 | PA0725 | Phage Pf1-like hypoth. protein | C → T | Syn. | |
| 792882d | ss138660673 | PA0725 | Phage Pf1-like hypoth. protein | T → G | Syn. | |
| 793143d | ss138660674 | PA0726 | Phage Pf1-like hypoth. protein | T → C | Syn. | |
| 793242d | ss138660675 | PA0726 | Phage Pf1-like hypoth. protein | T → G | Syn. | |
| 793284d | ss138660676 | PA0726 | Phage Pf1-like hypoth. protein | A → G | Syn. | |
| 793347d | ss138660677 | PA0726 | Phage Pf1-like hypoth. protein | G → A | Syn. | |
| 793389d | ss138660678 | PA0726 | Phage Pf1-like hypoth. protein | T → C | Syn. | |
| 793410d | ss138660679 | PA0726 | Phage Pf1-like hypoth. protein | T → C | Syn. | |
| 2807853 | ss138660680 | PA2492 | Transcriptional regulator MexT | G → C | A → P | |
| 4769678 | ss138660681 | PA4266 | Elongation factor G (FusA1) | G → A | T → I | |
| 4939350 | ss138660682 | PA4407 | Cell division protein FtsZ | C → A | D → Y | |
| 5033102e | ss138660683 | Interg. (PA4496-PA4497) | T → C | |||
| 5035975 | ss138660684 | PA4498 | Prob. metallopeptidase | G → A | R → stopf | |
| 5744862 | ss138660685 | Interg. (PA5100-PA5101) | T → C | |||
| SNP in MPAO1 only | 1275767g | ss138660653 | PA1174 | Periplasmic nitrate reductase NapA | A → G | F → S |
| Base exchanges in both strains, also detected for PAO1-UW (Lyngby) | 1589438 | ss138660655 | PA1459 | Chemotaxis-specific methylesterase | G → C | G → A |
| 1835045 | ss138660656 | PA1685 | Enolase-phosphatase MtnC | G → C | S → T | |
| 2669175 | ss138660658 | PA2400 | Prob. nonribosomal peptide synthetase PvdJ | G → C | P → A | |
| 2807982 | ss138660659 | PA2492 | Transcriptional regulator MexT | T → A | F → I | |
| 6079222 | ss138660667 | PA5399 | Prob. ferredoxin | A → G | Syn. | |
| 6115455 | ss138660669 | PA5434 | Tryptophan permease (Mtr) | T → G | K → N |
SNPs/exchanges printed in bold have been validated by diagnostic restriction digest. Abbreviations: interg., intergenic; prob., probable; hypoth., hypothetical; syn., synonymous.
Submitted SNP accession number.
Occurs in predicted small RNA (35) (genome position 5036879 to 5036995; MultiZ-predicted locus 176, window 743).
Located within ORF of RGP5 prophage sequence.
Base at this position uncertain in MPAO1.
Would cause truncated protein of only 34 aa (in comparison to 405 aa in PAO1-UW).
Base at this position uncertain in PAO1-DSM.
RESULTS
Large genomic differences between PAO1 sublines.
PFGE of restriction digestions of PAO1-UW and PAO1-DSM revealed discordant PacI and SwaI fragment patterns (Fig. 1A; see also Table S3 and Fig. S1 in the supplemental material), which visualize the known inversion in PAO1-UW between the rRNA gene operons rrnA and rrnB (36). Please note that the ter locus is located in PAO1 sublines PAO1-DSM and MPAO1 opposite the origin of replication (Fig. 1B) as has been observed in all other sequenced P. aeruginosa chromosomes (21, 25, 39). Hence, the first sequenced PAO1-UW strain with its asymmetric localization of the terminus of replication has to be considered an exception to the global genome architecture of P. aeruginosa. The inversion in PAO1-UW must have occurred after the PAO1 strain had been deposited by the original investigator in public strain collections.
FIG. 1.

Genomic differences in PAO1 sublines. (A) PFGE of SwaI- and PacI-digested DNA of PAO1 sublines PAO1-DSM (lanes a) and PAO1-UW (lanes b). Restriction fragments that are altered due to the chromosomal inversion are labeled. (B) Circular maps of PAO1 sublines PAO1-DSM and PAO1-UW showing the localization of the ori and ter regions, SwaI (S) and PacI (P) restriction sites, and ribosomal operons. The part of the genome shown in gray is inverted in PAO1-UW in contrast to PAO1-DSM and other sublines. The inversion breakpoints (inv) are located within rrnA and rrnB. Genome coordinates are given in accordance with the published PAO1-UW sequence (36). The black dot indicates the genomic region with a complex insertion-deletion event shown in panels C and D. (C) PFGE of SpeI- and DpnI-digested DNA of PAO sublines. DpnI restriction patterns were not distinguishable for PAO1-DSM (a, left part) and PAO1-UW (b, right part). However, SpeI patterns show fragments SpZ and SpAF of PAO1-DSM replaced by SpZ′ in PAO1-UW. (D) Physical map of the genomic region with altered SpeI pattern, based on the PAO1-UW reference sequence. A 1,006-bp portion of the reference sequence (white box) in proximity to rrnC is missing in sublines PAO1-DSM and MPAO1. Instead, both these sublines harbor 12,066 bp of additional DNA (including a further SpeI restriction site) located next to the indicated tRNAMet PA4673.1. (E) Gene map of the 12,066-bp insertion. Homologs of phage-like ORFs PA0717 to PA0727 in PAO1-UW are shown with a mesh-like pattern; genes encoding typical phage proteins are colored in gray (integrase, int; coat proteins, coaAB). Black arrows indicate ORFs encoding serine/threonine phosphotransferases missing in PAO1-UW.
SpeI and DpnI restriction digestions yielded nearly indistinguishable fragment patterns of the PAO1-UW and PAO1-DSM chromosomes (Fig. 1C) because both SpeI and DpnI have recognition sites in the 23S rRNA gene sequence and hence do not visualize the inversion between the rrn operons. Closer inspection of the SpeI digest, however, showed a loss of a SpeI recognition site in PAO1-UW (Fig. 1C). Fragments SpZ and SpAF of the initial PAO1 subline kept in the Holloway laboratory were replaced by fragment SpZ′, which was smaller than the sum of the adjacent SpZ and SpAF fragments. Restriction mapping and sequencing of this region revealed a 12-kb insertion in the 3′ end of a tRNAMet (PA4673.1) in PAO1-DSM and MPAO1 accompanied by a 1-kb deletion about 12 kb downstream of the insertion (Fig. 1D). The 12-kb insertion harbors the SpeI recognition site that is missing in the PAO1-UW chromosome.
The genomic island at PA4673.1 classified as an RGP42 insertion (according to the nomenclature introduced by Mathee et al. [25]) encodes a putative protein phosphatase, two protein kinases, and a prophage that is highly homologous to part of the phage Pf4 inserted at the tRNAGly PA0729.1 (31). The region spanning from open reading frame 2 (ORF2) to ORF12 (Fig. 1E) is syntenic with PA0727 to PA0717 (RGP5) and shows 94% to 100% sequence identity between the individual homologs (see Table S4 in the supplemental material). ORF1 codes for the phage-specific integrase that is identical in sequence with integrases in other P. aeruginosa strains like 2192 that targeted the same tRNAMet gene. ORF14 to ORF16 are specific for the genomic island. ORF14 codes for a protein phosphatase, and ORF15 and ORF16 each code for a protein kinase. A homologous operon with the same organization of ORFs has to date been observed only in the genome of the Mono Lake deltaproteobacterium MLMS-1 (8). Due to the insertion, 46 bp of the 3′-terminal sequence of the tRNAMet gene and 36 bp downstream were duplicated and flank the insertion at both sites. The presence of flanking tRNA 3′ ends is typical for genomic islands in P. aeruginosa (15).
PAO1-DSM and MPAO1 lack the PAO1-UW sequence from 5253694 to 5254699. This in-frame deletion disrupts the conserved hypothetical sequences PA4684 (1,299 bp) and PA4685 (696 bp) and yields a chimeric ORF, PA4684′, with the initial 936 bp of PA4684 and the terminal 42 bp of PA4685. The PAO1 genomic gene arrays showed above-average cDNA hybridization signals for the PA4684′ fusion gene of PAO1-DSM during growth in LB broth in late exponential phase in the presence and absence of various exogenous stresses, indicating that PA4684′ is transcribed. Full-length PA4684 and PA4685 genes are present in all other P. aeruginosa strains sequenced so far, demonstrating that the in-frame deletion is a unique feature of a fraction of PAO1 sublines.
SNPs and small deletions.
Genome sequencing of the chromosomes of PAO1 sublines MPAO1 and PAO1-DSM by Genome Analyzer sequencing-by-synthesis technology uncovered 39 validated single-nucleotide deviations (SNPs) from the PAO1-UW database entries (Table 1; Fig. 2), six of which are pairs of adjacent nucleotides. Twenty-two SNPs are transversions, and 17 SNPs are transitions. Nine SNPs are located in intergenic regions, of which SNP 5036891 resides within one of the noncoding RNA loci described so far (3, 23, 35). Of the 30 SNPs in coding regions, 13 are synonymous substitutions, 16 cause an amino acid exchange, and one changes an arginine into a stop codon. Several amino acid exchanges are nonconservative and should influence structure and/or function of the protein. Examples include the change from phenylalanine to serine in the nitrate reductase NapA involved in anaerobiosis or the change from aspartate to tyrosine in the cell division protein FtsZ (Table 1).
FIG. 2.
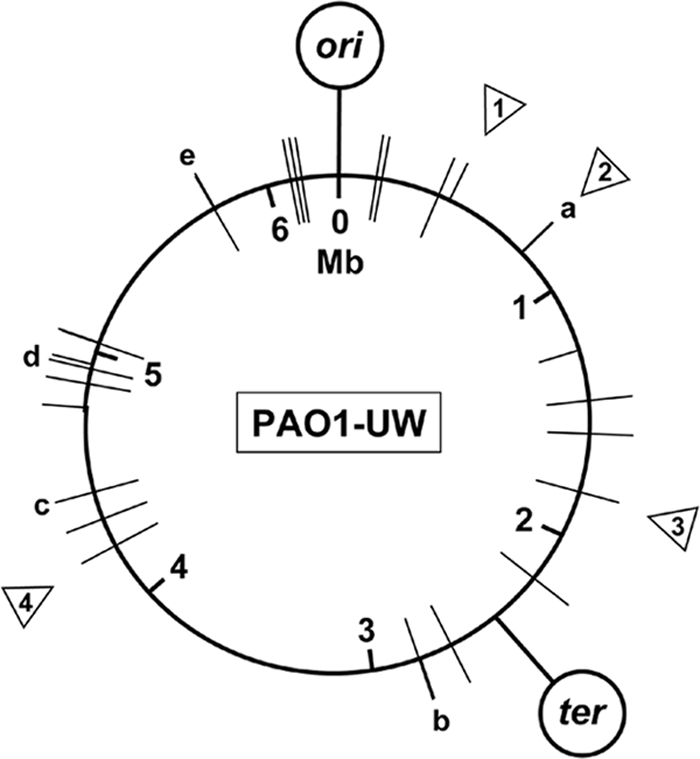
Genomic distribution of SNPs and indels. Positions of SNPs (bars) in P. aeruginosa sublines MPAO1 (inside the circle) and PAO1-DSM (outside) and of small deletions (triangles) in PAO1-DSM and the tested PAO1-UW subline. a, nine substitutions in PAO-DSM within 725 bp; b, one substitution in both sublines and another one in PAO1-DSM 130 bp upstream; c and d, two substitutions in both sublines at adjacent positions; e, two substitutions in both sublines at adjacent positions and one substitution in PAO1-DSM 1,400 bp downstream. Deletion 1, 3 bp deleted in PAO1-DSM; deletion 2, 2 of 18 heptanucleotides deleted in PAO1-DSM; deletion 3, deletion in PA1695 in tested PAO1-UW subline; deletion 4, deletion in PA3760 in tested PAO1-UW subline.
The initial analysis of the sequencing data pointed to the potential existence of 140 SNPs in PAO1-DSM and 153 SNPs in MPAO1 compared to the published PAO1 sequence. However, only a minority of them passed the rigorous threshold criteria applied to the primary data after validating several potential nucleotide substitutions by informative restriction digests (see Materials and Methods). Table 1 lists the nucleotide positions of the 39 validated SNPs, one of which was found only in MPAO1 and 16 of which were found only in PAO1-DSM. Nine strain-specific synonymous SNPs are located in ORFs PA0725 and PA0726 of the RGP5 prophage region that is duplicated in PAO1-DSM and MPAO1 but not in PAO1-UW (see above). Five nonsynonymous and one synonymous nucleotide substitution were also detected by diagnostic restriction digestions of PCR products in the PAO1-UW strain maintained in our laboratory. Figure 2 shows the genome map positions of the SNPs.
The initial data analysis did not predict an SNP within the lasR gene encoding the major transcriptional regulator for quorum sensing in P. aeruginosa (19, 34), a known hot spot of mutation in the P. aeruginosa genome (6). Closer inspection of the sequenced DNA sample of the PAO1-DSM strain, however, revealed a G-T transversion in 9 of 79 sequence reads, resulting in a change of arginine to leucine in about 10% of the population. This single amino acid substitution leads to pleiotropic changes of phenotype, including defective LasR function, overproduction of pyocyanin, impaired utilization of adenosine as carbon source, and higher resistance to cell lysis in stationary phase (6). The laboratory of one of us authors (S.H.) sequenced individual clones selected from PAO1-DSM stock cultures of two laboratories (Miguel Cámara, personal communication) and could identify the two lasR variants in both stocks.
The Genome Analyzer sequencing identified 17 regions that contained putative insertions or deletions. Subsequent combinatorial PCR and direct blotting electrophoresis verified four small deletions (Fig. 2). First, the PAO1-UW subline maintained in our laboratory showed intragenic deletions in PA1695 and PA3760 compared to the published PAO1-UW sequence. Second, the PAO1-DSM subline lacked 2 of the 18 heptanucleotide repeats in the terminal region of the RGP5 prophage downstream of PA0715 and carried a 3-bp deletion within PA0406. The latter deletion (447948 to 447950) affected two codons but resulted only in the loss of a glutamine codon while the neighbored lysine codon was restored.
Antimicrobial susceptibility.
Nonsynonymous SNPs in mexT and fusA1 of PAO1-DSM (Table 1) could affect antimicrobial susceptibility (17, 18, 24). Comparative MIC testing revealed indistinguishable susceptibility profiles for PAO1-UW and PAO1-DSM. The MPAO1 subline was fourfold more resistant to imipenem and ertapenem, up to fourfold more resistant to fluoroquinolones, and 16-fold more resistant to chloramphenicol than the PAO1-UW and PAO1-DSM sublines. Thus, no correlation was observed between divergent susceptibilities to antimicrobials in PAO1 sublines and the detected SNPs in loci mexT and fusA1 that are known to confer natural antibiotic resistance. The MPAO1 subline has retained the chloramphenicol resistance phenotype that had already occurred in the PAO1 strain in Bruce Holloway's laboratory. The loss of this marker in the two other sublines, however, should have been caused by other mutations.
Comparative fitness of PAO1 sublines in vitro and in vivo.
Next, we studied the global impact of the sequence diversity among sublines on competitive growth under various in vitro conditions (see Table S2 in the supplemental material) and on virulence in an acute murine airway infection model (28).
If the three PAO1 sublines were cultured in parallel in LB in separate flasks, the growth behavior and doubling times of the three strains were identical within experimental error. When equal CFU of the three PAO1 sublines were inoculated together into fresh medium, all sublines persisted under the six tested conditions (see Table S2 in the supplemental material). The proportion of sublines was retained in the majority of the analyzed samples. A variant growth behavior, however, was noted for subline PAO1-DSM in LB medium at 37°C (aerobic) or 42°C (microaerophilic) and in iron-supplemented M9 plus succinate medium (37°C, aerobic). PAO1-DSM was present in less than 10% during the first 2 h but returned to equal proportion thereafter and became the dominant member of the consortium in late stationary phase in LB broth of about 60% of all cells at 37°C (aerobic) and about 65% of cells at 42°C (microaerophilic), respectively. Moreover, subline PAO1-UW became the most prominent member among the three strains during stationary phase in microaerophilic cultures grown in LB (37°C) and aerobic cultures grown in M9 plus succinate (37°C). This subline accounted for approximately 60% of the cells or more than 75%, respectively. Thus, a differential fitness of PAO1 sublines was noted under several culturing conditions during onset of growth and stationary state.
Virulence of the PAO1 sublines was tested by intratracheal instillation of exponentially growing bacteria into the lungs of C3H/HeN mice (28). A dose of 106 CFU caused a moderate acute pneumonia by day 2 and led to the death of about half of the animals by day 5 postinfection (Fig. 3). A 10-fold-higher dose of 107 CFU had a similar outcome for infections with MPAO1 but led to a profound purulent pneumonia and subsequent death of all animals between days 2 and 3 for infections with PAO1-DSM or PAO1-UW. Thus, a differential pathogenic potential of PAO1 sublines became evident at fatal doses.
FIG. 3.

Murine airway infection. Pathohistological findings in C3H/HeN murine lungs 48 h after intratracheal infection with 106 CFU or 107 CFU of P. aeruginosa PAO1 sublines or vehicle control (hematoxylin-eosin staining; original magnification, ×100; scale bars, 100 μm). (Top row) Infection with 106 CFU of all three PAO1 strains has induced a moderate acute fibrinogenic pneumonia, mostly confined to the alveoli. Bronchi are almost free of inflammatory cells. Vehicle control shows normal lung parenchyma after PBS application. (Middle row) Animals infected with 107 CFU of PAO1-UW and PAO1-DSM show profound purulent pneumonia with numerous foci of inflammation (infiltration of neutrophils and alveolar macrophages) up to tissue necrosis. In contrast strain MPAO1 has generated only moderate inflammation comparable to the infection with 106 CFU. The right inset depicts the CFU recovered from murine organs (lu, lung; spl, spleen; li, liver; br, brain) 48 h after intratracheal instillation of 106 CFU (closed symbols) or of 107 CFU (open symbols) of PAO1-DSM (squares), MPAO1 (circles), or PAO1-UW (triangles). (Bottom row, left) Survival of mice whose airways had been inoculated with 106 CFU (closed symbols) or 107 CFU (open symbols) of PAO1-DSM (squares), MPAO1 (circles), or PAO1-UW (triangles). (Right) Body condition score at day 1 after infection (untroubled, u; slightly troubled, ▾; moderately troubled, ▾▾; profoundly troubled, ▾▾▾; moribund; dead).
DISCUSSION
The PAO1 strain remains the major reference strain for research on P. aeruginosa. Physical mapping and whole-genome resequencing of PAO1 sublines uncovered an extended genomic diversity between the two strains that were the basis for the initial sequencing project and the first genome-wide mutant library. Moreover, progeny of the strain PAO1 that had been identical in its physical map with the original strain stored in Bruce Holloway's laboratory (12) turned out to harbor numerous deviations from the published PAO1 sequence. The major differences of sublines MPAO1 and PAO1-DSM from the genomic sequence of the strain PAO1-UW (36) are a synteny of genes identical to that of all other sequenced P. aeruginosa strains (21, 25, 39), a duplication of an RGP prophage carrying a distinct integrase and putative protein serine/threonine phosphotransferases of unknown function as well as numerous SNPs and short indels. These variations that make up 0.2% of the whole-genome sequence were found to be associated with an impact on virulence and fitness. Under some growth conditions the PAO1-DSM subline outnumbered the two other sublines during stationary phase, indicating better adaptation and/or enhanced survival under these conditions.
The reported SNPs and small indels represent a conservative lower estimate of the actual sequence variation. The chosen threshold for the number and quality of sequence reads selected those sequence variations identified by the Illumina Genome Analyzer that represent real SNPs with more than 95% probability (see Results). According to our wet lab data up to 10 further SNPs may exist in the genome that were not detected with sufficient specificity to pass the quality criterion.
These estimates, however, apply only to homogeneous sequence variations in all cells of a PAO1 subline. One major objective of novel next-generation sequencing technologies is the identification of sequence variations that are present in a minority of molecules in a DNA sample. Ultradeep sequencing of loci that are diagnostic for minimum residual disease in human cancer would be a typical application (27). Sequencing of microbial genomes by the Genome Analyzer also pinpoints positions with more than one nucleotide in the different sequence reads. If we exclude the technical artifacts that are typically seen at the end of the short reads, the read counts of the various sequence variants represent the proportions of distinct sequence isoforms in the bacterial population. An instructive example within our project is the known hot spot of mutation in the P. aeruginosa genome, the gene lasR. The two described lasR variants coexisted in two analyzed stocks of PAO1-DSM (see Results), illustrating nicely the ongoing microevolution of the PAO1 reference strain that remains unnoticed by researchers until some unexpected changes in phenotype emerge.
The two variants of the PAO1-UW strain kept in our laboratory further substantiate the genotypic diversification of PAO1 during its worldwide distribution. The strain received in 1999 from the genome sequencing team showed the diagnostic features of the large inversion and the small 12-kb deletion of the island RGP42 (Fig. 1). On the other hand, the genome of the strain received in 2005 and also designated PAO1-UW harbored the complete PA4684-PA4685 genes as did the sequenced PAO1-UW strain but also, like PAO1-DSM or MPAO1, contained RGP42. This “PAO1-UW” strain, moreover, shared six sequence deviations from the published PAO1 sequence with PAO-DSM and MPAO1 (Table 1). The variable presence of RGP42 in PAO1-UW and “PAO1-UW” is consistent with our notion during physical mapping that the diagnostic SpeI fragment exhibited a variable intensity of the ethidium bromide stain in separate PFGE runs, implying that this prophage region is prone to deletion. Besides this inherent genomic plasticity of the PAO1 strains, the individual handling of the strain may drive microevolution. A member of our laboratory, for example, unintentionally generated a hypermutator variant of PAO1-DSM by continuous subculturing. In summary, intrinsic biological features of the strain and laboratory procedures have caused a broad spectrum of PAO1 genotypes during the last 50 years.
Some nucleotide substitutions affected genes such as fusA1 (PA4266) and ftsZ (PA4407) that encode basal functions of the bacterial cell. fusA1 encodes the elongation factor G (EF-G) that during translation is involved in the movement of peptidyl-tRNA from the A site to the P site and in the disassembly of the posttermination ribosomal complex (7, 40). The PAO1 variants carry either a threonine or an isoleucine at position 493 of the 706-amino-acid (aa)-long protein. Position 493 is located at the beginning of the functional domain 4 with a close to 100% conserved sequence of the next 10 amino acids in the N-terminal direction and a flexible region with numerous exchanges among the next 10 amino acids in the C-terminal direction. Of the 70 most closely related EF-G proteins in the NCBI database, threonine is the dominant amino acid at position 493, while isoleucine is not present. Consequently we hypothesize that the amino acid exchange is functionally not neutral, although for subline PAO1-DSM, in which this exchange occurred, no altered sensitivity to fusidic acid was observed. The binding of this antibiotic to FusA1/EF-G is apparently not affected by the change to isoleucine. A formally more drastic change of an aspartate to tyrosine was detected in the cell division protein FtsZ (PA4407) at position 379 close to its C terminus. The tubulin-like FtsZ forms long dynamic spirals that are reorganized into a ring structure at the onset of division (4). The amino acid substitution is located in the least conserved region of FtsZ and thus—despite the pronounced impact on the local environment—should not influence the global function of this core element of cell division. A further nonconservative phenylalanine-to-serine change affected the nitrate reductase NapA (PA1174). Nitrate reductases enable P. aeruginosa to grow under microaerophilic or anaerobic conditions by denitrification. However, it is the membrane-bound nitrate reductase NarG, but not the periplasmic enzyme NapA, that is essential to sustain anaerobic growth (30). Although NapA is more strongly expressed during anaerobiosis (unpublished transcriptome data), the sequence variations in the NapA protein should not significantly influence the global fitness of the PAO1 strains. In summary, these three amino acid substitutions can be classified as modulators or benign sequence variants.
Besides the loss-of-function mutation in the lasR gene of a subpopulation of PAO1-DSM bacteria, a further nonconservative change of alanine to proline was identified in MexT, a further hot spot of mutation in the P. aeruginosa genome (34). MexT activates the expression of the MexEF-OprN efflux pump and represses the expression of the outer membrane porin protein OprD (17). Both porin and efflux pump exhibit broad specificity, and hence, functionally nonequivalent MexT isoforms could promote pleiotropic effects on the transport of small molecules across the cell membrane and thus on the global transport of the cell. Correspondingly, mexT sequence variations were reported to affect antibiotic resistance and/or cell-to-cell signaling (18, 24). However, the nonsynonymous SNP in mexT of PAO1-DSM was not associated with a change in susceptibility to any of 33 tested antimicrobial agents, implying that the detected SNP did not significantly modulate MexT function.
The sequence variants in PA4498 encoded either a 405-amino-acid-long protein or a truncated version of 34 amino acids (Table 1). PA4498 mRNA transcript was expressed at very low levels in six tested P. aeruginosa strains. PAO1-DSM, however, which harbors the premature termination codon, displayed more-than-10-fold-higher mRNA transcript levels, indicating that the full-length protein is needed. Correspondingly, we classify this change of an arginine to a stop codon as a deleterious mutation in PAO1-DSM. Thus, sequence diversity in PA4498 could contribute to the diversification of phenotype among the PAO1 sublines.
In conclusion, the maintenance and propagation of P. aeruginosa PAO1 in laboratories throughout the world have entertained an ongoing microevolution of genotype and phenotype that jeopardizes the reproducibility of research. PAO1 is still the major reference strain for the P. aeruginosa field and hence—with the data of this study at hand—we now envisage a similar complexity as has been recognized in the past for E. coli K-12, the major workhorse for biochemists, molecular biologists, and bacterial geneticists. Standardization of genotype by accredited strain collections is advisable. High-throughput genome resequencing will resolve more cases and could become a proper quality control for strain collections.
Supplementary Material
Acknowledgments
This work was partially supported by grants provided by the Deutsche Forschungsgemeinschaft (SFB 587, project A9; GRK 653/3). C.F.D. was a member of the DFG-supported IRTG Pseudomonas: Pathogenicity and Biotechnology.
We thank Miguel Cámara and Jean Dubern for the thorough analysis of the lasR locus in PAO1-DSM stock cultures and Silke Hedtfeld for the verification of small indels by direct blotting electrophoresis. Supply of PAO1 sublines by Alice L. Erwin and Thomas Rasmussen is gratefully acknowledged.
Footnotes
Published ahead of print on 18 December 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidmann, J. A. Smith, and K. Struhl (ed.). 1994. Current protocols in molecular biology. Wiley, New York, NY.
- 2.Fox, A., D. Haas, C. Reimmann, S. Heeb, A. Filloux, and R. Voulhoux. 2008. Emergence of secretion-defective sublines of Pseudomonas aeruginosa PAO1 resulting from spontaneous mutations in the vfr global regulatory gene. Appl. Environ. Microbiol. 74:1902-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González, N., S. Heeb, C. Valverde, E. Kay, C. Reimmann, T. Junier, and D. Haas. 2008. Genome-wide search reveals a novel GacA-regulated small RNA in Pseudomonas species. BMC Genomics 9:167. doi: 10.1186/1471-2164-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graumann, P. L. 2007. Cytoskeletal elements in bacteria. Annu. Rev. Microbiol. 61:589-618. [DOI] [PubMed] [Google Scholar]
- 5.Grimes, E., M. Koob, and W. Szybalski. 1990. Achilles' heel cleavage: creation of rare restriction sites in lambda phage genomes and evaluation of additional operators, repressors and restriction/modification systems. Gene 90:1-7. [DOI] [PubMed] [Google Scholar]
- 6.Heurlier, K., V. Dénervaud, M. Haenni, L. Guy, V. Krishnapillai, and D. Haas. 2005. Quorum-sensing-negative (lasR) mutants of Pseudomonas aeruginosa avoid cell lysis and death. J. Bacteriol. 187:4875-4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokawa, G., N. Iwakura, A. Kaji, and H. Kaji. 2008. The role of GTP in transient splitting of 70S ribosomes by RRF (ribosome recycling factor) and EF-G (elongation factor G). Nucleic Acids Res. 36:6676-6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoeft, S. E., T. R. Kulp, J. F. Stolz, J. T. Hollibaugh, and R. S. Oremland. 2004. Dissimilatory arsenate reduction with sulfide as electron donor: experiments with mono lake water and isolation of strain MLMS-1, a chemoautotrophic arsenate respirer. Appl. Environ. Microbiol. 70:2741-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holloway, B. W. 1955. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 13:572-581. [DOI] [PubMed] [Google Scholar]
- 10.Holloway, B. W. 1975. Genetic organization of Pseudomonas, p. 133-161. In P. H. Clarke and M. H. Richmond (ed.), Genetics and biochemistry of Pseudomonas. John Wiley & Sons Ltd, London, United Kingdom.
- 11.Holloway, B. W., V. Krishnapillai, and A. F. Morgan. 1979. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 43:73-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holloway, B. W., U. Römling, and B. Tümmler. 1994. Genomic mapping of Pseudomonas aeruginosa PAO. Microbiology 140:2907-2929. [DOI] [PubMed] [Google Scholar]
- 13.Huang, W., and G. Marth. 2008. EagleView: a genome assembly viewer for next-generation sequencing technologies. Genome Res. 18:1538-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs, M. A., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klockgether, J., O. N. Reva, and B. Tümmler. 2006. Spread of genomic islands between clinical and environmental strains, p. 187-200. In N. A. Logan, H. M. Lapin-Scott, and P. C. F. Oyston (ed.), Prokaryotic diversity: mechanisms and significance. Cambridge University Press, Cambridge, United Kingdom.
- 16.Klockgether, J., D. Würdemann, L. Wiehlmann, and B. Tümmler. 2008. Transcript profiling of the Pseudomonas aeruginosa genomic islands PAGI-2 and pKLC102. Microbiology 154:1599-1604. [DOI] [PubMed] [Google Scholar]
- 17.Köhler, T., S. F. Epp, L. K. Curty, and J. C. Pechère. 1999. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 181:6300-6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köhler, T., C. van Delden, L. K. Curty, M. M. Hamzehpour, and J. C. Pechere. 2001. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183:5213-5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Köhler, T., A. Buckling, and C. van Delden. 2009. Cooperation and virulence of clinical Pseudomonas aeruginosa populations. Proc. Natl. Acad. Sci. U. S. A. 106:6339-6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon, D. H., and C. D. Lu. 2006. Polyamines increase antibiotic susceptibility in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50:1623-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, D. G., J. M. Urbach, G. Wu, N. T. Liberati, R. L. Feinbaum, S. Miyata, L. T. Diggins, J. He, M. Saucier, E. Déziel, L. Friedman, L. Li, G. Grills, K. Montgomery, R. Kucherlapati, L. G. Rahme, and F. M. Ausubel. 2006. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewenza, S., R. K. Falsafi, G. Winsor, W. J. Gooderham, J. B. McPhee, F. S. Brinkman, and R. E. Hancock. 2005. Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res. 15:583-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livny, J., A. Brencic, S. Lory, and M. K. Waldor. 2006. Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res. 34:3484-3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maseda, H., K. Saito, A. Nakajima, and T. Nakae. 2000. Variation of the mexT gene, a regulator of the MexEF-OprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 192:107-112. [DOI] [PubMed] [Google Scholar]
- 25.Mathee, K., G. Narasimhan, C. Valdes, X. Qiu, J. M. Matewish, M. Koehrsen, A. Rokas, C. N. Yandava, R. Engels, E. Zeng, R. Olavarietta, M. Doud, R. S. Smith, P. Montgomery, J. R. White, P. A. Godfrey, C. Kodira, B. Birren, J. E. Galagan, and S. Lory. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U. S. A. 105:3100-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mekus, F., U. Laabs, H. Veeze, and B. Tümmler. 2003. Genes in the vicinity of CFTR modulate the cystic fibrosis phenotype in highly concordant or discordant F508del homozygous sib pairs. Hum. Genet. 112:1-11. [DOI] [PubMed] [Google Scholar]
- 27.Morozova, O., and M. A. Marra. 2008. Applications of next-generation sequencing technologies in functional genomics. Genomics 92:255-264. [DOI] [PubMed] [Google Scholar]
- 28.Munder, A., S. Krusch, T. Tschernig, M. Dorsch, A. Lührmann, M. van Griensven, B. Tümmler, S. Weiss, and H. J. Hedrich. 2002. Pulmonary microbial infection in mice: comparison of different application methods and correlation of bacterial numbers and histopathology. Exp. Toxicol. Pathol. 54:127-133. [DOI] [PubMed] [Google Scholar]
- 29.Munder, A., A. Zelmer, A. Schmiedl, K. E. Dittmar, M. Rohde, M. Dorsch, K. Otto, J. H. Hedrich, B. Tümmler, S. Weiss, and T. Tschernig. 2005. Murine pulmonary infection with Listeria monocytogenes: differential susceptibility of BALB/c, C57BL/6 and DBA/2 mice. Microbes Infect. 7:600-611. [DOI] [PubMed] [Google Scholar]
- 30.Palmer, K. L., S. A. Brown, and M. Whiteley. 2007. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J. Bacteriol. 189:4449-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice, S. A., C. H. Tan, P. J. Mikkelsen, V. Kung, J. Woo, M. Tay, A. Hauser, D. McDougald, J. S. Webb, and S. Kjelleberg. 2009. The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J. 3:271-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Römling, U., D. Grothues, W. Bautsch, and B. Tümmler. 1989. A physical genome map of Pseudomonas aeruginosa PAO. EMBO J. 8:4081-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Römling, U., T. Heuer, and B. Tümmler. 1994. Bacterial genome analysis by pulsed field gel electrophoresis techniques. Adv. Electrophoresis 7:353-406. [Google Scholar]
- 34.Smith, E. E., D. G. Buckley, Z. Wu, C. Saenphimmachak, L. R. Hoffman, D. A. D'Argenio, S. I. Miller, B. W. Ramsey, D. P. Speert, S. M. Moskowitz, J. L. Burns, R. Kaul, and M. V. Olson. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U. S. A. 103:8487-8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonnleitner, E., T. Sorger-Domenigg, M. J. Madej, S. Findeiss, J. Hackermüller, A. Hüttenhofer, P. F. Stadler, U. Bläsi, and I. Moll. 2008. Detection of small RNAs in Pseudomonas aeruginosa by RNomics and structure-based bioinformatic tools. Microbiology 154:3175-3187. [DOI] [PubMed] [Google Scholar]
- 36.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 37.Vaara, M. 1992. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56:395-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winsor, G. L., T. Van Rossum, R. Lo, B. Khaira, M. D. Whiteside, R. E. Hancock, and F. S. Brinkman. 2009. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 37(database issue):D483-D488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winstanley, C., M. G. Langille, J. L. Fothergill, I. Kukavica-Ibrulj, C. Paradis-Bleau, F. Sanschagrin, N. R. Thomson, G. L. Winsor, M. A. Quail, N. Lennard, A. Bignell, L. Clarke, K. Seeger, D. Saunders, D. Harris, J. Parkhill, R. E. Hancock, F. S. Brinkman, and R. C. Levesque. 2009. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 19:12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu, H., Y. L. Chan, and I. G. Wool. 2009. The identification of the determinants of the cyclic, sequential binding of elongation factors tu and g to the ribosome. J. Mol. Biol. 386:802-813. [DOI] [PubMed] [Google Scholar]
- 41.Zerbino, D. R., and E. Birney. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.